

Abstract
The separation of sister chromatids in anaphase is followed by spindle disassembly and cytokinesis. These events are governed by the anaphase-promoting complex (APC), which triggers the ubiquitin-dependent proteolysis of key regulatory proteins: anaphase requires the destruction of the anaphase inhibitor Pds1, whereas mitotic exit requires the destruction of mitotic cyclins and the inactivation of Cdk1. We find that Pds1 is not only an inhibitor of anaphase, but also blocks cyclin destruction and mitotic exit by a mechanism independent of its effects on sister chromatid separation. Pds1 is also required for the mitotic arrest and inhibition of cyclin destruction that occurs after DNA damage. Even in anaphase cells, where Pds1 levels are normally low, DNA damage stabilizes Pds1 and prevents cyclin destruction and mitotic exit. Pds1 blocks cyclin destruction by inhibiting its binding partner Esp1. Mutations in ESP1 delay cyclin destruction; overexpression of ESP1 causes premature cyclin destruction in cells arrested in metaphase by spindle defects and in cells arrested in metaphase and anaphase by DNA damage. The effects of Esp1 are dependent on Cdc20 (an activating subunit of the APC) and on several additional proteins (Cdc5, Cdc14, Cdc15, Tem1) that form a regulatory network governing mitotic exit. We speculate that the inhibition of cyclin destruction by Pds1 may contribute to the ordering of late mitotic events by ensuring that mitotic exit is delayed until after anaphase is initiated. In addition, the stabilization of Pds1 after DNA damage provides a mechanism to delay both anaphase and mitotic exit while DNA repair occurs.
Keywords: APC, cyclin, DNA damage, Esp1, mitotic exit, Pds1
Successful cell division requires that cell cycle events occur in the correct order. In mitosis, for example, anaphase is not initiated until the mitotic spindle is fully assembled, and cytokinesis does not commence until anaphase is complete. Although intrinsic timing mechanisms can initiate events in the correct sequence, the order of events also depends on checkpoint mechanisms that block the onset of events until preceding events are completed (Hartwell and Weinert 1989; Murray 1994; Elledge 1996). Checkpoints also help to enforce the correct sequence of events after environmental insults such as DNA damage or spindle damage. These mechanisms arrest cell cycle progression in response to damage, allowing the cell time to repair DNA damage or to complete spindle assembly before cell cycle progression resumes.
In late mitosis, the timing and coordination of events are controlled by the ubiquitin-dependent proteolytic destruction of two major classes of regulatory molecules: anaphase inhibitors and mitotic cyclins (Murray 1995; Hershko 1997; Morgan 1999). In the budding yeast Saccharomyces cerevisiae, the destruction of the anaphase inhibitor Pds1 is required for sister chromatid separation (Cohen-Fix et al. 1996; Yamamoto et al. 1996a). Pds1 normally binds and inhibits Esp1, a protein that destroys sister chromatid cohesion; therefore, Pds1 destruction promotes anaphase by liberating Esp1 (Funabiki et al. 1996; Ciosk et al. 1998). After anaphase, destruction of mitotic cyclins, leading to inactivation of mitotic cyclin-dependent kinase (Cdk1/Cdc28) activity, causes spindle disassembly and cytokinesis (Holloway et al. 1993; Surana et al. 1993). In budding yeast, up-regulation of the inhibitor Sic1 in late mitosis also contributes to Cdk1 inactivation (Donovan et al. 1994; Schwab et al. 1997; Visintin et al. 1998).
Proteolysis of late mitotic regulators is controlled by a large, multisubunit ubiquitin–protein ligase known as the anaphase-promoting complex (APC) or cyclosome, which catalyzes the final step in the ubiquitination of substrates containing a specific sequence termed the destruction box (Glotzer et al. 1991; Hershko et al. 1994; Irniger et al. 1995; King et al. 1995; Peters 1998; Morgan 1999). APC activity is regulated in the cell cycle and increases in late mitosis, primarily because of changes in its association with the activating subunits Cdc20 and Hct1/Cdh1 (King et al. 1995; Lahav-Baratz et al. 1995; Sudakin et al. 1995; Zachariae and Nasmyth 1996; Charles et al. 1998; Fang et al. 1998; Zachariae et al. 1998). In yeast, these subunits may also confer substrate specificity on the APC; genetic evidence has led to the hypothesis that Cdc20 promotes degradation of Pds1, whereas Hct1 directs destruction of the major mitotic cyclin Clb2 (Schwab et al. 1997; Visintin et al. 1997). This simple model is unlikely to be correct, however, as some cyclins, such as Clb3 and Clb5, may be targeted by Cdc20 (Schwab et al. 1997; Alexandru et al. 1999). In addition, Cdc20 is capable of inducing some Clb2 destruction when overproduced in metaphase-arrested cells (Visintin et al. 1997). Nevertheless, although Cdc20 possesses some cyclin-targeting activity, Hct1–APC activation appears essential for the complete destruction of Clb2 (Schwab et al. 1997; Visintin et al. 1997).
The activity of Hct1 is controlled by its phosphorylation state. From S phase through early mitosis, phosphorylation by Cdk1 restrains Hct1 activity; dephosphorylation of Hct1 by the phosphatase Cdc14 then initiates Hct1 activation in late mitosis (Zachariae et al. 1998; Jaspersen et al. 1999). The inhibitor Sic1 is regulated by similar mechanisms: Cdk1-dependent phosphorylation inhibits its synthesis and stability, and Cdc14-dependent dephosphorylation reverses these effects and leads to its accumulation (Moll et al. 1991; Toyn et al. 1996; Skowyra et al. 1997; Verma et al. 1997; Visintin et al. 1998). These regulatory relationships generate a bistable switch in which a slight decrease in Cdk activity (or an increase in Cdc14 activity) can rapidly trigger complete Cdk inactivation. The signal that triggers this switch is not known, although there is evidence that Hct1 activation and Sic1 accumulation involve a complex ‘mitotic exit network’ comprising several signaling proteins, including the Ras-like protein Tem1, the Polo-like kinase Cdc5, and the protein kinase Cdc15 (Schweitzer and Philippsen 1991; Wan et al. 1992; Kitada et al. 1993; Shirayama et al. 1994, 1996; Taylor et al. 1997; Jaspersen et al. 1998). These proteins are all required for late mitotic destruction of Clb2 but not that of Pds1 (Jaspersen et al. 1998), but their mechanisms of action remain unclear.
Hct1 activation is also dependent on previous activation of Cdc20, apparently because Cdc20 triggers the destruction of proteins that inhibit Hct1 activation (Yamamoto et al. 1996b; Lim et al. 1998). One of these proteins may be Pds1; overexpression of a nondegradable mutant form of Pds1 (Pds1Δdb) blocks not only sister chromatid separation but also delays cytokinesis, suggesting that Pds1 may be capable of inhibiting cyclin destruction (Cohen-Fix et al. 1996). One explanation for the effects of Pds1Δdb is that cyclin destruction is dependent on the completion of sister chromatid separation. However, esp1-1 mutants are thought to undergo mitotic exit and cytokinesis without separating sister chromatids (McGrew et al. 1992; Surana et al. 1993; Ciosk et al. 1998), favoring the possibility that Pds1 controls cyclin destruction independently of sister chromatid separation. As yet, however, there is no direct evidence for this possibility.
Cells lacking both PDS1 and CDC20 arrest after anaphase with stable Clb2 (Yamamoto et al. 1996b; Lim et al. 1998), suggesting that Cdc20 is required for cyclin destruction even in the absence of Pds1. Thus, activation of the Hct1–APC may require the Cdc20-dependent destruction of at least two proteins: Pds1 and an unidentified inhibitor of cyclin destruction.
Cdc20-dependent proteolysis appears to be inhibited by checkpoint mechanisms that cause a metaphase arrest in cells with spindle defects or DNA damage (Elledge 1996; Rudner and Murray 1996; Weinert 1998). The spindle checkpoint component Mad2 binds and inhibits directly the Cdc20–APC, presumably inhibiting the destruction of cyclins as well as Pds1 (Li et al. 1997; Hwang et al. 1998; Kim et al. 1998; Alexandru et al. 1999; Fesquet et al. 1999; Li 1999). There is also evidence suggesting that Cdc20 is a target of the DNA damage response; overexpression of Cdc20 overrides the metaphase arrest caused by DNA damage (Lim and Surana 1996; Hwang et al. 1998). However, there is no evidence for a direct regulation of Cdc20 activity by the DNA damage response pathway.
Deletion of PDS1 allows cells to proceed through anaphase and mitotic exit in the presence of DNA damage, but allows only anaphase to occur in the presence of spindle defects (Yamamoto et al. 1996). In addition, Pds1 is hyperphosphorylated in response to DNA damage but not in response to spindle damage (Cohen-Fix and Koshland 1997). Therefore, it has been hypothesized that the phosphorylation of Pds1 after DNA damage protects it from Cdc20-dependent degradation. Stabilization of Pds1 would lead not only to inhibition of anaphase, but might also block mitotic exit (as seen in studies of the Pds1Δdb mutant) (Cohen-Fix et al. 1996). Once again, however, it is not clear whether DNA damage (or Pds1Δdb) blocks cytokinesis indirectly through the inhibition of sister separation or by a direct effect on the cyclin destruction machinery.
We analyzed the role of Pds1 in the direct control of cyclin destruction, both in normal cells and in cells arrested in mitosis by DNA damage. We found that Pds1Δdb and DNA damage both inhibit cyclin destruction not only during metaphase but also in cells arrested after sister separation in anaphase, where DNA damage leads to Pds1 stabilization. Interestingly, our experiments also suggest that Pds1 acts by inhibiting its partner Esp1, which is capable of promoting cyclin destruction.
Results
Pds1 is required for the DNA damage response in metaphase and late anaphase
X-irradiation is known to delay exit from mitosis in cells arrested transiently in metaphase by nocodazole treatment (Weinert and Hartwell 1988). We assessed the role of Pds1 in this response by analyzing the effects of DNA damage in wild-type and pds1Δ cells. We performed these experiments in cells lacking the RAD52 gene, which is required for recombinational double-stranded break repair (Kaytor and Livingston 1994; New et al. 1998); because damage cannot be repaired in these cells, only mutants defective in the DNA damage checkpoint are able to resume cell division after irradiation.
Metaphase-arrested rad52Δ cells were irradiated, nocodazole was removed by washing, and cells were incubated for 3 hr at 37°C in medium containing α factor to prevent entry into the next cell cycle. Clb2 levels and associated kinase activity remained high throughout the experiment (Fig. 1A), and cells remained large-budded with unseparated nuclei (data not shown). Deletion of PDS1 led to a dramatic reduction in the DNA damage response, although a low level of Clb2 protein and activity remained (Fig. 1A). A similar reduction in the response was observed in cells lacking the checkpoint gene RAD9 (data not shown). Deletion of both PDS1 and RAD9 led to a complete abolition of the response (Fig. 1A), resulting in Clb2 destruction and mitotic exit at the same rate as that seen in undamaged cells (data not shown). Thus, Pds1 is required for the inhibition of cyclin destruction that occurs after DNA damage, although it is probably not the sole target of the response (see Discussion section).
Figure 1.




Pds1 is required for the inhibition of cyclin destruction after DNA damage, and DNA damage acts in late anaphase to stabilize Pds1. (A) The indicated strains were arrested in metaphase with nocodazole at 19°C and γ-irradiated at 2.5 krads. Strains were then released into 37°C media containing α factor, and cell lysates were prepared from samples taken every 60 min. Clb2 and Cdc28/Cdk1 (an internal loading control) were detected by Western blotting. Clb2-associated histone H1 kinase activity (HH1) was measured in anti-Clb2 immunoprecipitations as described in Materials and Methods. (B) cdc15-2 and cdc15-2 rad9Δ strains were arrested in anaphase by shifting to 37°C. Cells were γ-irradiated at 2.5 krads and released into 23°C media containing α factor. Time points were taken every 60 min to determine the amount of Clb2 and Cdc28 protein and Clb2-associated histone H1 kinase activity (HH1). The APC was immunoprecipitated with anti-Cdc26 antibodies and its Hct1-dependent cyclin-ubiquitin ligase activity was measured as described in Materials and Methods. A ∼8-kD ladder of ubiquitin conjugates appears above the unconjugated cyclin B1 fragment at the bottom of the gel. The asterisk denotes a background band present in the nonubiquitinated substrate. Deletion of RAD52 was not required in these experiments as anaphase cells cannot undergo homologous double-stranded break repair. (C) cdc15-2 and cdc15-2 rad9Δ strains, both carrying an integrated GAL–PDS1 fusion, were arrested in anaphase by shifting to 37°C. Cells were γ-irradiated at 2.5 krad and GAL–PDS1 was induced for 1 hr by the addition of 2% galactose. Dextrose was then added to shut off PDS1 expression and cells remained arrested at 37°C. Samples were taken every 60 min and analyzed by western blotting with antibodies against Pds1 or Cdc28. (D) A cdc15-2 strain was either arrested in metaphase with nocodazole at 23°C or in anaphase by shifting to 37°C. After 3.5 hr, anaphase-arrested cells were divided and one portion was γ-irradiated, whereas the other half remained undamaged. All strains remained arrested for an additional 3 hr. Samples were taken at the indicated times and analyzed by Western blotting with antibodies against Pds1 or Cdc28.
It has been proposed that the DNA damage checkpoint acts simply by inhibiting Cdc20 function (Lim and Surana 1996; Hwang et al. 1998). If this were the case, then damaged cells lacking Pds1 would be expected to arrest in late anaphase with high Clb2 levels, as Cdc20 is required for cyclin destruction as well as Pds1 destruction (Lim et al. 1998). However, we found that CDC20 is required for mitotic exit in pds1Δ cells after DNA damage (Fig. 1A), implying that Cdc20 is active in these cells. These results suggest that DNA damage does not cause a complete inhibition of Cdc20 activity (at least in pds1Δ cells). A simpler interpretation is that the DNA damage checkpoint acts through Pds1, not Cdc20, to inhibit both anaphase and cytokinesis.
The majority of Pds1 is degraded at the metaphase-to-anaphase transition, and only a low level of the protein persists in mutants arrested in anaphase (Cohen-Fix et al. 1996; Jaspersen et al. 1998). Therefore, we reasoned that if Pds1 is required for the DNA damage checkpoint, then anaphase-arrested cells might be only partially responsive to DNA damage. To test this possibility, cdc15-2 cells were arrested in late anaphase, irradiated, and then returned to the permissive temperature in medium containing α factor. Interestingly, irradiated anaphase cells exhibited a complete RAD9-dependent cell cycle arrest; Clb2 levels and activity remained high, whereas Hct1–APC activity was undetectable (Fig. 1B). We could not test directly the role of Pds1 in these experiments because pds1Δ cdc15-2 cells do not exhibit a uniform, reversible anaphase arrest at the restrictive temperature. However, our observation that pds1Δ cells exit mitosis after DNA damage in metaphase (Fig. 1A) indicates that Pds1 is required not only for anaphase inhibition after damage but also for the inhibition of mitotic exit.
Pds1 is stabilized in response to DNA damage
If Pds1 is essential for the damage-induced cell cycle arrest, then it may be protected from degradation in response to irradiation. To test this possibility, we analyzed Pds1 stability in cdc15-2 cells arrested in late anaphase, where Pds1 is normally unstable (Jaspersen et al. 1998). cdc15-2 and cdc15-2 rad9Δ strains carrying PDS1 under the control of the regulatable GAL promoter were arrested in anaphase and irradiated. A transient pulse of PDS1 expression was then induced by addition of galactose, after which glucose was added to repress PDS1 expression. Irradiation resulted in the complete stabilization of Pds1 for >3 hr, whereas Pds1 was degraded within 1 hr in rad9Δ cells (Fig. 1C). Consistent with these results, DNA damage induced an increase in the levels of endogenous Pds1 in anaphase-arrested cells (Fig. 1D). The amount of Pds1 increased in these cells to levels approaching those found in undamaged, metaphase-arrested cells (Fig. 1D).
Pds1 blocks Clb2 proteolysis and inactivation in metaphase and late anaphase but not in G1
The requirement for Pds1 in the damage response implies that Pds1 is able to inhibit cyclin destruction as well as sister separation. We tested this possibility by analyzing cyclin levels in cells overproducing Pds1Δdb, a mutant version of Pds1 that is resistant to APC-dependent destruction. A wild-type strain and a strain carrying GAL–pds1Δdb were arrested in metaphase with nocodazole, incubated in galactose medium to induce expression of pds1Δdb, and released from nocodazole into galactose medium containing α factor. Overexpression of pds1Δdb prevented Clb2 proteolysis and significantly delayed Clb2-associated kinase inactivation (Fig. 2A). We observed consistently that kinase activity decreased more rapidly than Clb2 levels, presumably because of the increase in Sic1 protein levels at later time points. After 4–5 hr, Cdk1 inactivation was complete and 80%–87% of cells underwent cytokinesis without properly segregating their sister chromatids (data not shown).
Figure 2.



Overexpression of pds1Δdb inhibits cyclin destruction and Sic1 accumulation in metaphase and anaphase. (A) Wild-type cells with or without an integrated copy of GAL–pds1Δdb were arrested in metaphase with nocodazole at 23°C, followed by galactose addition for 30 min. Cells were then released from nocodazole into media containing α factor and galactose, and cell lysates were prepared from samples taken every 60 min. Western blotting of Clb2, Sic1, Pds1Δdb, and Cdc28 was performed, and Clb2-associated histone H1 kinase activity (HH1) was measured as described in Materials and Methods. (B) cdc15-2 strains with or without GAL–pds1Δdb were arrested in late anaphase by shifting to 37°C. Galactose was added for 30 min, followed by the addition of α factor, and cells were released back to the permissive temperature of 23°C. At the times indicated, samples were withdrawn to analyze Clb2, Sic1, Pds1Δdb, and Cdc28 protein levels, as well as Clb2-associated histone H1 kinase activity. (C) cdc15-2, cdc15-2 rad9Δ, and cdc15-2 mad1Δ strains containing GAL–pds1Δdb were arrested in late anaphase by shifting to 37°C. Dextrose or galactose was added for 30 min as indicated, followed by the addition of α factor, and cells were returned to the permissive temperature of 23°C. At 75-min intervals, samples were analyzed by Western blotting with antibodies against Mad1, Clb2, Pds1Δdb, and Cdc28.
We also analyzed the effects of Pds1Δdb in a late anaphase arrest. cdc15-2 strains, with or without GAL–pds1Δdb, were arrested in late anaphase by growth at the restrictive temperature. Pds1Δdb production was induced and cells were returned to the permissive temperature in the presence of α factor. Again, the pds1Δdb mutant blocked the destruction of Clb2 and delayed kinase inactivation (Fig. 2B); after 4 hr, Cdk1 inactivation was complete and mitotic exit occurred in 88% of cells (data not shown). This result clearly demonstrates that Pds1 blocks cyclin destruction by a mechanism that is independent of sister chromatid separation. It is also consistent with our observation that DNA damage can inhibit cyclin destruction in late anaphase (Fig. 1B).
Although the Pds1Δdb protein lacks the amino acid sequence required for APC recognition, it is still possible that its overexpression results simply in competitive inhibition of APC activity. However, we found that pds1Δdb overexpression did not inhibit the activity of the APC in G1 cells and did not cause stabilization of Clb2 (data not shown). It is also known that overexpression of pds1Δdb does not inhibit endogenous Pds1 destruction (Cohen-Fix et al. 1996; see Fig. 3 below). Therefore, we conclude that Pds1Δdb overexpression does not act simply by competitive inhibition of the APC and cannot inhibit the APC when it is already active. Instead, our results suggest that Pds1 specifically inhibits the processes that trigger APC activation toward cyclins in late mitosis.
Figure 3.

Overexpression of pds1Δdb and mutation of ESP1 delay cyclin destruction and Cdk1 inactivation. Wild-type or esp1-1 strains with or without GAL–pds1Δdb were arrested in G1 with α factor, and then released from α factor into 37°C media containing galactose. After 75 min, when >90% of cells had formed buds, α factor was added. Cell lysates were prepared from samples taken every 60 min, and Western blotting was performed to detect endogenous Pds1, Clb2, and Pds1Δdb proteins. Histone H1 kinase activity (HH1) was measured in anti-Clb2 immunoprecipitates. At t = 240 min, aliquots were fixed and processed for DNA staining with DAPI. The percent of large-budded cells with a single DNA mass (metaphase), large-budded cells with a divided nucleus (anaphase), unbudded cells with a single DNA mass (G1), and unbudded cells with little or no DNA mass (aneuploid) were scored as follows: wild-type, 99% G1, 1% anaphase; pds1Δdb, 33% G1, 35% aneuploid, 32% metaphase; esp1-1, 42% G1, 42% aneuploid, 16% metaphase, esp1-1 pds1Δdb, 35% G1, 35% aneuploid, 30% metaphase.
It is possible that overproduction of Pds1Δdb blocks cyclin destruction by stimulating the DNA damage response pathway. However, we found that the cyclin-stabilizing effects of Pds1Δdb were unaffected by mutations in RAD9 or RAD53 (Fig. 2C; data not shown). We also tested whether the inhibition of Clb2 proteolysis is dependent on the spindle assembly checkpoint, as overexpression of pds1Δdb in metaphase causes spindle abnormalities. Inhibition of Clb degradation by Pds1Δdb did not depend on the function of Mad1 (Fig. 2C).
Esp1 is required for normal cyclin destruction
Pds1 is thought to inhibit anaphase by forming a complex with Esp1, whose release after Pds1 destruction initiates sister chromatid separation (Ciosk et al. 1998). Therefore, we tested the possibility that Esp1 promotes cyclin destruction as well as anaphase, providing a simple mechanism whereby Pds1 (and DNA damage) might block cyclin destruction and mitotic exit. We first tested this possibility by analyzing cyclin levels in the absence of Esp1 function. Although previous evidence suggests that Cdk1 inactivation can occur in esp1-1 mutants (Surana et al. 1993), the effect of this mutant on the timing of cyclin destruction has not been determined. Wild-type and esp1-1 strains, with or without GAL–pds1Δdb, were arrested in G1 with α factor and then released at the nonpermissive temperature into media containing galactose. After the initiation of S phase, α factor was added again to arrest cells in the next G1. As before, we found that overexpression of pds1Δdb severely delayed both Clb2 proteolysis and inactivation (Fig. 3). Interestingly, Clb2 degradation and kinase inactivation were also defective in the esp1-1 mutant; in addition, mutation of ESP1 (like overexpression of pds1Δdb) appeared to cause a greater delay in cyclin destruction than in kinase inactivation. Cells carrying both the esp1-1 mutation and overexpressed pds1Δdb displayed defects identical to those in cells carrying pds1Δdb alone (Fig. 3), indicating that the effects of the two mutations are not additive. Pds1 destruction occurred normally in esp1-1 cells and in cells expressing pds1Δdb, as shown previously (Cohen-Fix et al. 1996; Ciosk et al. 1998); thus, the delay in cyclin destruction in these cells is not attributable to a general slowing of cell cycle progression or to activation of the spindle assembly checkpoint. These results are consistent with our hypothesis that Pds1 inhibits cyclin destruction (but not its own destruction) by inhibiting Esp1 function. Because the effects of the esp1-1 mutation are less pronounced than those of pds1Δdb expression, we cannot eliminate the possibility that Pds1 also acts independently of Esp1 to inhibit Clb2 proteolysis. However, we believe that this is unlikely (see Discussion).
ESP1 overexpression triggers cyclin destruction in metaphase-arrested cells
To test further whether Esp1 is a positive regulator of Cdk1 inactivation, we analyzed the effects of ESP1 overexpression on Clb2 levels and activity. Overexpression of ESP1 from the GAL promoter had no effect in cells arrested in S phase with hydroxyurea; however, induction of ESP1 expression in metaphase-arrested cells resulted in a decrease in Clb2 levels and Clb2-associated kinase activity (Fig. 4). Interestingly, Esp1 also triggered a dramatic decrease in Pds1 levels.
Figure 4.

Overexpression of ESP1 triggers Pds1 and Clb2 degradation in metaphase-arrested cells. Wild-type and pds1Δ cells containing an integrated triple-copy of GAL–ESP1 were either arrested in S phase with HU or in metaphase with nocodazole. α Factor was added, followed by the addition of 2% dextrose or 2% galactose to repress (−) or induce (+) ESP1 expression, and cells were incubated at 23°C for an additional 3 hr. Pds1 and Clb2 were detected by Western blotting and Clb2-associated histone H1 kinase assays (HHI) were performed. Note that more extensive degradation of Clb2 in wild-type cells can be achieved when ESP1 expression is induced with 4% galactose (see Figs. 5 and 6).
The effect of ESP1 overexpression on Clb2 levels was not due to the sequestration or inhibition of Pds1, as Clb2 degradation was still stimulated by Esp1 in pds1Δ cells (Fig. 4). Overexpression of ESP1 was consistently more effective in pds1Δ cells, presumably because its antagonist was removed. It is unlikely that Esp1 is triggering Clb2 proteolysis indirectly by promoting sister chromatid separation, as separation has presumably occurred in nocodazole-arrested pds1Δ cells (Ciosk et al. 1998). Thus, ESP1 overexpression overrides the proteolysis defect imposed by the spindle assembly checkpoint, by a mechanism that is independent of sister chromatid separation.
ESP1 overexpression bypasses the DNA damage checkpoint
Our results clearly lead to the hypothesis that Pds1 mediates the DNA damage-induced cell cycle arrest by blocking the ability of Esp1 to promote cyclin destruction. To test this hypothesis, a cdc15-2 GAL–ESP1 strain was arrested in metaphase with nocodazole or in anaphase by shifting to the nonpermissive temperature. Cells were then irradiated and released from their respective arrests into media containing α factor with or without galactose. ESP1 overexpression bypassed the mitotic arrest seen in cells irradiated in metaphase and anaphase (Fig. 5). Tubulin staining revealed the presence of G1 asters in the cells that bypassed the arrest (data not shown). The ability of ESP1 overexpression to bypass the damage response is clearly consistent with the possibility that damage inhibits Esp1 by stabilizing its inhibitor Pds1. In addition, the ability of Esp1 to bypass the damage response in late anaphase indicates that Esp1, like Pds1Δdb, can control cyclin destruction independently of its effects on sister separation.
Figure 5.

Overexpression of ESP1 bypasses the DNA damage checkpoint in metaphase and late anaphase. A cdc15-2 rad52Δ strain carrying triple-integrated GAL–ESP1 was arrested either in metaphase with nocodazole at 23°C or in anaphase by shifting to 37°C. Cells were γ-irradiated with 2.5 krad, and each culture was divided as follows: One-half was released into 23°C media containing dextrose (ESP1 off) and α factor; the other half was released into 23°C media containing galactose (ESP1 on) and α factor. Samples taken at the indicated times were analyzed by Western blotting of Clb2, Sic1, and Cdc28 protein, as well as Clb2-associated histone H1 kinase activity (HH1).
Esp1-induced Cdk1 inactivation requires Cdc20 and the mitotic exit network
Our observation that Esp1 triggers Pds1 destruction (see Fig. 4) raised the intriguing possibility that Esp1 acts primarily through the activation of Cdc20. Consistent with this possibility, we found that Cdc20 is required for Esp1 to induce cyclin degradation and accumulation of Sic1 in nocodazole-arrested cells (Fig. 6A). Cdc16 function was also required, further suggesting that Esp1 is acting through activation of APC-dependent proteolysis (Fig. 6A). Thus, we conclude that overexpression of ESP1 promotes cyclin destruction and Cdk1 inactivation through a Cdc20-dependent pathway.
Figure 6.

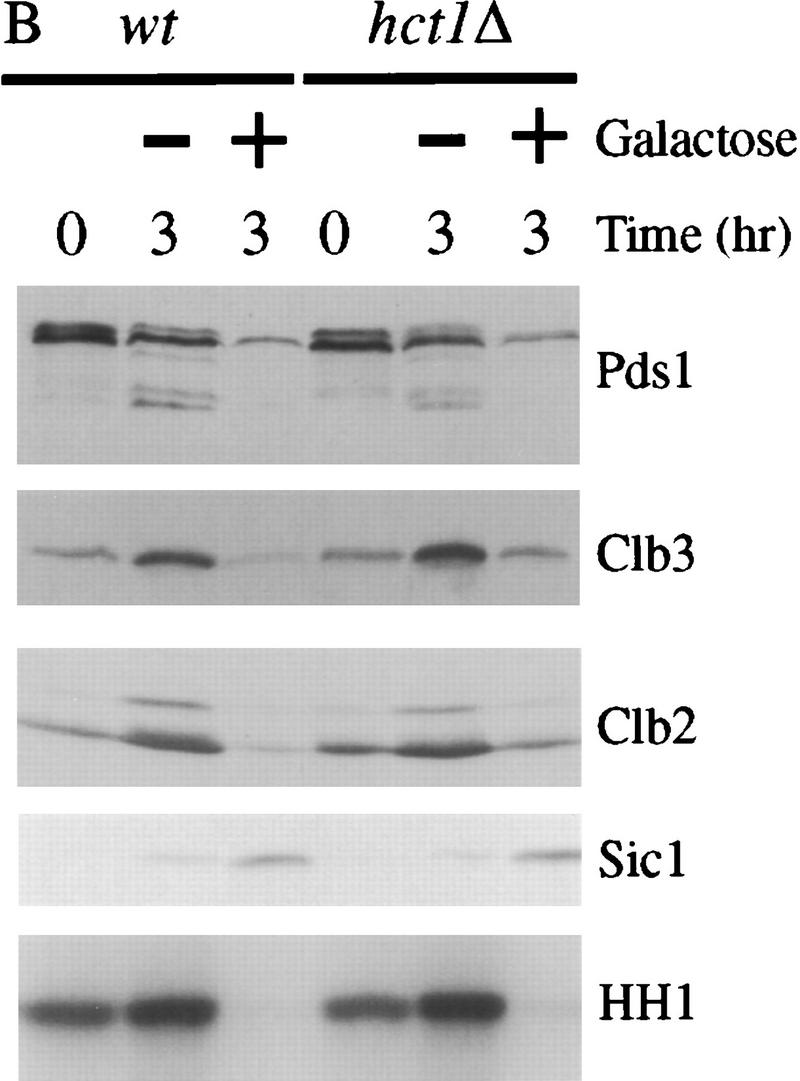


Esp1-induced cyclin destruction and Sic1 accumulation are dependent on Cdc20 and the mitotic exit network. (A) Wild-type, cdc20-1, and cdc16-1 strains containing GAL–ESP1 were arrested in metaphase with nocodazole for 3 hr. Cells were then shifted to 35°C for 30 min. Dextrose or galactose was added, along with α factor, and cells were incubated at the nonpermissive temperature for an additional 3 hr. Cell lysates were analyzed by Western blotting of Clb3, Clb2, Sic1, and Cdc28, and Clb2-associated histone H1 kinase (HH1) activity was measured. Although GAL–ESP1 had no effect on Clb levels in the cdc16 mutant, it did trigger spindle elongation and nuclear division (data not shown; see also Ciosk et al. 1998). (B) Wild-type and hct1Δ strains containing GAL–ESP1 were arrested in metaphase with nocodazole at 23°C. α Factor was then added, followed by the addition of dextrose or galactose, and incubation continued for 3 hr. Cell lysates were analyzed by Western blotting of Pds1, Clb3, Clb2, and Sic1, and Clb2-associated histone H1 kinase activity was measured. (C) Wild-type and hct1Δ cells containing GAL–CDC20 on a CEN/ARS plasmid were arrested in metaphase with nocodazole at 23°C. Dextrose or galactose was added, in addition to α factor, and cells were incubated for an additional 3 hr. Clb2, Clb3, Cdc20, and Cdc28 levels were analyzed by Western blotting, and Clb2-associated histone H1 kinase activity was measured. In control experiments (data not shown), addition of galactose caused cyclin destruction only in cells carrying GAL–ESP1 or GAL–CDC20 and not in cells lacking these plasmids. (D) Wild-type, cdc14-1, and tem1-3 strains containing GAL–ESP1 were treated and analyzed as described in A. Cell lysates were analyzed by Western blotting of Pds1, Clb3, Clb2, and Sic1, and Clb2-associated histone H1 kinase activity was measured.
Deletion of HCT1 did not affect the ability of ESP1 overexpression to trigger Pds1 destruction and Sic1 accumulation in nocodazole-arrested cells (Fig. 6B), further demonstrating that Esp1 acts primarily through stimulation of Cdc20–APC activity. Deletion of HCT1 did reduce partially the effects of ESP1 on the levels of Clb2 and Clb3 (Fig. 6B). We also found that ESP1 expression in wild-type cells leads to activation of the Hct1–APC (data not shown). Therefore, we suspect that the effects of overexpressed Esp1 on Clb2 and Clb3 levels are due in part to the direct action of the Cdc20–APC and in part to Cdc20-dependent destruction of an inhibitor of Hct1–APC (see Discussion).
We also analyzed the effects of CDC20 overexpression on Clb2 and Clb3 levels in nocodazole-arrested cells. As in our experiments with GAL–ESP1, GAL–CDC20 expression led to a decrease in Clb2 and Clb3 levels that was partially dependent on the presence of Hct1 (Fig. 6C). The ability of Esp1 and Cdc20 to induce partially Hct1-independent cyclin destruction and Cdk1 inactivation further supports the notion that Esp1 acts by stimulating Cdc20.
Because the mitotic exit network is known to be required for Hct1–APC activation and Sic1 accumulation, we tested its role in the effects of Esp1 in nocodazole-arrested cells. We found that Esp1-induced Clb2 destruction and Sic1 accumulation were blocked in cdc14 and tem1 mutants (Fig. 6D). The degradation of Pds1 and Clb3 was also reduced partially in these mutants, raising the possibility that Cdc14 and Tem1 are required for complete activation of the Cdc20–APC. We also found that the effects of ESP1 overexpression on Clb2 levels were blocked in cdc5 and cdc15 mutants arrested in late anaphase (data not shown). Thus, components of the mitotic exit network are required for Esp1 to exert its inhibitory effects on Clb2 levels and activity.
Cdc20 activity is required for exit from a late anaphase cdc15 arrest
In our earlier experiments, we showed that DNA damage and production of the Pds1Δdb protein blocked exit from cdc15-arrested cells. Considering our hypothesis that Pds1 acts by inhibiting Esp1-dependent Cdc20 activation, these results imply that Cdc20 function is required for exit from the cdc15 arrest. To confirm this possibility, a cdc15-2 cdc20Δ GAL–CDC20 strain was grown in galactose and arrested in late anaphase by shifting to the nonpermissive temperature. Cells were then released to the permissive temperature in either dextrose (to repress CDC20 expression) or galactose (to maintain CDC20 expression). Cells lacking Cdc20 were unable to degrade Clb2 or promote Sic1 accumulation (Fig. 7); the absence of Cdc20 activity allowed Clb2 protein levels and kinase activity to increase, further suggesting that Cdc20 contributes to Clb2 degradation. These results demonstrate that Cdc20 function is required for exit from the cdc15 arrest, consistent with our hypothesis that Pds1Δdb blocks exit from this arrest by inhibiting Esp1-dependent Cdc20 activation. These results are also consistent with previous evidence that Cdc20 function is required not only for sister separation (i.e., Pds1 destruction), but also for cyclin destruction and mitotic exit (Yamamoto et al. 1996b; Lim et al. 1998).
Figure 7.

Cdc20 is required for late anaphase cells to exit mitosis. A cdc20Δ GAL–CDC20 cdc15-2 strain growing in galactose-containing media was arrested in anaphase by shifting to the nonpermissive temperature. The culture was then divided and pelleted by centrifugation. One-half was resuspended in media containing galactose; the other half was resuspended in media containing dextrose to repress CDC20 expression. α Factor was added, and cells were allowed to resume cell division at 23°C. Lysates were analyzed by Western blotting of Clb2 and Sic1, as well as Clb2-associated kinase activity.
Discussion
Pds1 inhibits cyclin destruction and mitotic exit by inhibiting Esp1
To ensure the correct order of late mitotic events in yeast, Cdc20–APC activation occurs before Hct1–APC activation; as a result, Clb2 degradation (and mitotic exit) occurs after the degradation of the anaphase inhibitor Pds1. Our results suggest that this sequence of events is due, at least in part, to the ability of Pds1 to inhibit cyclin destruction and Cdk1 inactivation. Thus, cyclin destruction is initiated only after Pds1 destruction occurs and anaphase is presumably under way.
Our evidence suggests that Pds1 inhibits mitotic exit by inhibiting Esp1. We were originally uncertain about this possibility because previously reported phenotypes of pds1Δdb and the esp1-1 mutant appeared quite distinct: pds1Δdb overexpression was thought to cause a near-permanent inhibition of mitotic exit, whereas esp1-1 mutants appeared to inactivate Cdk1 and progress through mitotic exit as effectively as wild-type cells (cyclin levels were not analyzed) (Surana et al. 1993; Cohen-Fix et al. 1996). However, we found that cells expressing Pds1Δdb eventually inactivate Cdk1 and exit mitosis; in addition, we found that esp1-1 mutants exhibit a delay in mitotic exit that is accompanied by a significant defect in cyclin destruction (the defect in Cdk1 inactivation is less severe). Thus, the mitotic exit defects in pds1Δdb and esp1-1 cells are more similar than previously realized, consistent with the hypothesis that Pds1 inhibits mitotic exit by inhibiting Esp1.
It is possible that Pds1 also acts by mechanisms other than the inhibition of Esp1, as the effects of the esp1-1 mutant are less striking than those of GAL–pds1Δdb. However, the timing of Clb2 destruction and inactivation in pds1Δdb-expressing cells is not affected by the esp1-1 mutation, indicating that the effects of pds1Δdb overexpression and the esp1-1 mutation are not additive (Fig. 3). The simplest interpretation of these results is that pds1Δdb and the esp1-1 mutation cause cyclin stabilization by the same mechanism. We also suspect that the esp1-1 mutant is not completely defective. Although this mutant undergoes extensive chromosome nondisjunction (whereby the daughter cell receives the bulk of the DNA and both spindle poles) (McGrew et al. 1992), 15%–20% of sister chromatids eventually separate (Ciosk et al. 1998).
We also found that overexpression of ESP1 induces premature disappearance of Clb2 in metaphase and anaphase cells (Figs. 4–6). These effects are dependent on APC function, suggesting that Esp1 is acting through stimulation of APC-dependent proteolysis of Clb2. ESP1 overexpression is not acting by blocking some other inhibitory action of Pds1, because Esp1 still triggers cyclin destruction in the absence of Pds1. Therefore, cyclin destruction is increased at high levels of ESP1 expression and decreased at low levels (esp1-1 mutants), supporting the possibility that Esp1 is a limiting regulator of cyclin degradation.
Several lines of evidence indicate that Pds1 and Esp1 do not regulate cyclin destruction and mitotic exit indirectly through their actions on sister chromatid separation. First, Pds1Δdb expression blocks cyclin destruction in cdc15 cells arrested in late anaphase (Fig. 2B). Second, ESP1 overexpression triggers cyclin destruction in nocodazole-arrested pds1Δ cells (where sisters have separated) (Fig. 4) and in cells subjected to DNA damage in late anaphase (Fig. 5).
Esp1 triggers mitotic exit by a Cdc20- dependent mechanism
Our results indicate that the effects of Esp1 on cyclin destruction are dependent on CDC20. We found that Esp1 promotes Pds1 destruction as well as that of Clb2, and that these effects were blocked in the cdc20-1 mutant. We conclude that Esp1 somehow promotes Cdc20-dependent APC activity, which then leads to Hct1–APC activation and Sic1 accumulation.
Overexpression of CDC20 in cells arrested in S phase with hydroxyurea is sufficient to induce Pds1 degradation (Visintin et al. 1997), whereas we did not see any effect of ESP1 overexpression in S phase (Fig. 4). Because the actions of Esp1 require Cdc20, these results are consistent with previous evidence that Cdc20 protein is absent in S phase (Prinz et al. 1998). In nocodazole-arrested cells, we found that Esp1 triggers the destruction of Pds1, Clb3, and Clb2; here again, our results are consistent with previous experiments showing that CDC20 overexpression in these cells causes degradation of both Pds1 and Clb2 (Visintin et al. 1997). Interestingly, we also found that the effects of overexpressed ESP1 and CDC20 on Clb3, Clb2, and Sic1 levels were affected similarly by the deletion of HCT1, further suggesting that Esp1 acts through Cdc20 (Fig. 6).
We do not believe that Esp1 is normally required for the initial activation of the Cdc20-dependent APC, as Pds1 destruction occurs normally in the esp1-1 mutant and in cells expressing pds1Δdb (Fig. 3). Nor do we believe that Esp1-dependent stimulation of Cdc20 normally causes Hct1-independent Clb2 destruction, as the majority of Clb2 destruction in vivo is dependent on Hct1-APC activity (Schwab et al. 1997; Visintin et al. 1997). Instead, we suspect that Cdc20 has a low level of Clb2-targeting activity that can cause a reduction in Clb2 levels when CDC20 or ESP1 are overexpressed. In a normal cell cycle, it seems likely that Cdc20, with a push from Esp1, promotes the destruction of some cyclins and causes a slight decrease in Cdk1 activity, which might be sufficient to trigger the Hct1–Sic1 activation switch (Fig. 8). In the absence of Esp1 (or in the presence of Pds1Δdb), this activation may be delayed until the accumulation of Cdc20 activity overcomes the requirement for Esp1.
Figure 8.

A speculative model of the regulatory relationships governing exit from mitosis. As described in the text, our evidence suggests that the activation of Esp1 after Pds1 destruction leads not only to sister chromatid separation but also enhances Cdc20-dependent APC activation, leading to destruction of an unidentified inhibitor (‘X’) of cyclin proteolysis and Cdk1 inactivation. DNA damage blocks both anaphase and mitotic exit by stabilizing Pds1 and preventing Esp1 action.
Esp1-induced cyclin destruction and Sic1 accumulation are dependent on the mitotic exit network
We also found that the ability of Esp1 to trigger Clb2 proteolysis and Sic1 accumulation is dependent on components of the mitotic exit network, including Tem1, Cdc14, Cdc5, and Cdc15 (Fig. 6; data not shown). ESP1-induced degradation of Pds1 was also slightly reduced in late mitotic mutants. Combined with our evidence that the effects of Esp1 require Cdc20, these results point to the possibility that the mitotic exit network is also required for complete activation of the Cdc20–APC (Fig. 8).
Therefore, our results are consistent with several previous lines of evidence that the mitotic exit network does not act simply by promoting Hct1–APC activity but has more general actions that include activation of the Cdc20–APC as well. First, the late mitotic mutants all arrest with low but still significant levels of Pds1 protein; complete destruction of Pds1 does not occur until mitotic exit (Jaspersen et al. 1998). Second, the ability of overexpressed CDC5 and CDC14 to trigger cyclin destruction is dependent on CDC20 (Charles et al. 1998; Visintin et al. 1998). There is also biochemical evidence that the vertebrate Cdc5 homolog phosphorylates the APC core and promotes its activity, and in yeast there is evidence that Cdc5 and Cdc15 are required for some modification of the APC that increases its responsiveness to Hct1 in vitro (Kotani et al. 1998; Jaspersen et al. 1999). One explanation for these results (but not the only one) is that some components of the mitotic exit network act by promoting the activity of the Cdc20–APC as well as that of the Hct1–APC. Clearly, they are not required for the initial burst of Cdc20 activity that triggers most Pds1 destruction and causes anaphase, but they may be required for complete Cdc20-dependent destruction of Pds1 and other inhibitors of cyclin destruction.
The role of Pds1 in the DNA damage response
In most eukaryotic cells, DNA damage blocks entry into mitosis by inhibiting mitotic Cdk activation (Elledge 1996; Weinert 1998). However, in S. cerevisiae, inhibition of Cdk activity does not occur in the damage response, and instead cells arrest with high Cdk activity (Amon et al. 1992; Sorger and Murray 1992). Budding yeast are also unique in that they assemble their mitotic spindle during S phase even if DNA damage occurs. These and other results suggest that the DNA damage checkpoint in budding yeast must be able to prevent exit from, rather than entry into, metaphase. We have confirmed this possibility and shown that the key target in the damage response is Pds1, whose stabilization after DNA damage blocks the initiation of both anaphase and cytokinesis.
Irradiated pds1Δ cells undergo anaphase and exit mitosis. Therefore, we do not believe that Cdc20 is the major target for inhibition by the DNA damage checkpoint because irradiated pds1Δ cells would then be expected to arrest after anaphase like cdc20 pds1 double mutants (Fig. 1). It also seems unlikely that Pds1 is the only target of the DNA damage checkpoint because stabilization of Pds1 alone (i.e., overexpression of pds1Δdb) does not delay cytokinesis as effectively as DNA damage (cf. Figs. 1 and 2). In addition, because pds1Δ rad9Δ cells bypass the damage response more completely than pds1Δ cells (Fig. 1), it appears that deletion of PDS1 alone does not cause a complete bypass of the damage checkpoint. It should also be mentioned that rad9Δ, rad17Δ, and rad24Δ cells are each less sensitive to DNA damage than double or triple mutant combinations, suggesting that multiple pathways control the DNA damage response (Weinert and Hartwell 1990; Paulovich et al. 1997). Nevertheless, Pds1 is clearly a key component in the DNA damage response.
The DNA damage checkpoint works in late anaphase and requires Pds1
Previous studies have shown that cells containing a dicentric chromosome pause transiently in mid-anaphase in a RAD9-dependent fashion, presumably attributable to chromosome breakage (Yang et al. 1997). We now show that late anaphase cells are also responsive to DNA damage, and that the exit from anaphase is blocked in these cells by Pds1. PDS1 mRNA and protein are present at low levels in anaphase-arrested cells (Yamamoto et al. 1996a; Jaspersen et al. 1998), and our results suggest that Pds1 protein is stabilized and accumulates to near-peak levels in anaphase cells subjected to DNA damage (Fig. 1). Pds1 probably blocks mitotic exit after DNA damage by inhibiting Esp1, as ESP1 overexpression bypasses the cell cycle arrest that occurs when metaphase or anaphase cells are subjected to DNA damage (Fig. 5).
What purpose, if any, does a DNA damage checkpoint serve in late anaphase? Because repair of double-stranded breaks requires DNA homology, damage induced by γ-irradiation cannot be repaired after sister chromatids separate in anaphase; therefore, a mitotic arrest in such cells may not serve to allow repair, although it would prevent damaged cells from further propagation. However, it is conceivable that single-stranded DNA breaks that require DNA mismatch-repair mechanisms are reparable in late anaphase.
DNA damage cannot inhibit APC activity in G1 (R. Tinker-Kulberg, unpubl.). In fact, such a mechanism would be deleterious because inactivation of the APC in G1 would lead to cyclin accumulation and would drive cells into S phase. It is therefore more appropriate that DNA damage operates through Pds1 to inhibit the activation of the APC and not the maintenance of its activity in G1. Interestingly, the components of the mitotic exit network are also required for the activation of the APC but not for its maintenance in G1 (Jaspersen et al. 1998). These results are consistent with the possibility that Pds1 inhibits the activation of the mitotic exit network.
Esp1 links sister chromatid separation and cytokinesis
On the basis of our findings, we speculate that the ability of Pds1 to inhibit Cdk1 inactivation provides a mechanism by which cytokinesis is restrained until the separation of sister chromatids begins (Fig. 8). Mitotic exit begins with the completion of spindle assembly, which helps initiate Cdc20–APC activation and the destruction of Pds1 (and perhaps some cyclins as well). Degradation of the majority of Pds1 liberates Esp1, which leads directly to sister chromatid separation. Esp1 also enhances Cdc20-dependent APC activity; its mechanism of action remains obscure, and may involve either a direct action on the Cdc20–APC or the activation of regulators in the mitotic exit network that stimulate Cdc20 activity. Esp1-dependent activation of the Cdc20–APC then leads to further destruction of Cdc20 targets, including the cyclin destruction inhibitor (X in Fig. 8) whose existence is suggested by evidence that Cdc20 is required for mitotic exit in pds1Δ cells and in cells released from a cdc15 arrest. Destruction of X then allows activation of the Hct1–APC and accumulation of Sic1, leading to Cdk1 inactivation and mitotic exit.
Materials and methods
Plasmids and strains
Yeast strains and genotypes are shown in Table 1. All strains are derivatives of W303 (MATa ade2-1 trp1-1 leu2-3, 112 his3-11,15 ura3-1 can1-100). Yeast transformations and genetic manipulations were performed according to published methods (Guthrie and Fink 1991).
Table 1.
Yeast strains
| Strain
|
Relevant genotype
|
Source
|
|---|---|---|
| AFS92a | MATa ade2-1 can1-100 ura3-1 leu2-3, 112, his-11,15, trp-1 bar1 | A. Straight (UCSF) |
| RTK164 | MATa bar1 trp1∷GAL–pds1ΔdbHA–TRP1 | this study |
| SLJ127 | MATa bar1 cdc15-2 | S. Jaspersen (UCSF) |
| RTK44 | MATa bar1 cdc15-2 trp1∷GAL–pds1ΔdbHA–TRP1 | this study |
| RTK310 | MATa bar1 cdc15-2 rad9Δ∷LEU2 trp1∷GAL–pds1ΔdbHA–TRP1 | this study |
| RTK306 | MATa bar1 cdc15-2 mad1Δ∷URA3 trp1∷GAL–pds1ΔdbHA–TRP1 | this study |
| RTK313 | MATa bar1 pds1∷PDS1 myc18–LEU2 | this study |
| RTK335 | MATa bar1 pds1::PDS1 myc18–LEU2 trpl::GAL-pdslΔdbHA–TRPl | this study |
| RTK337 | MATa bar1 pds1∷PDS1 myc18–LEU2 esp1-1 | this study |
| RTK336 | MATa bar1 pds1∷PDS1 myc18–LEU2 esp1-1 trp1∷GAL–pdsΔdbHA–TRP1 | this study |
| K7346 | MATα BAR1 trp∷3X(GAL–ESP1-TRP1) ura3∷URA3 tetOs leu2∷LEU2 tetR–GFP | F. Uhlmann (IMP, Vienna, Austria) |
| RTK314 | MATa bar1 trp∷3X(GAL–ESP1-TRP1) pds1∷PDS1 myc18–Leu2 | this study |
| RTK312 | MATa bar1 pds1Δ∷LEU2 trp∷3X(GAL–ESP1–TRP1) | this study |
| RTK338 | MATa bar1 hct1-Δ1∷HIS3 trp∷3X(GAL–ESP1–TRP1)pds1∷PDS1 myc18–LEU2 | this study |
| RTK327 | MATa bar1 cdc16-1 trp∷3X(GAL–ESP1–TRP1) | this study |
| RTK339 | MATa bar1 cdc14-1 trp∷3X(GAL–ESP1–TRP1) pds1∷PDS1 myc18–LEU2 | this study |
| RTK344 | MATa bar1 tem1-3 trp∷3X(GAL–ESP1–TRP1) pds1∷PDS1 myc18–LEU2 | this study |
| RTK223 | MATa bar1 (YCGAL–CDC20∷URA) | this study |
| RTK211 | MATa bar1 hct1Δ∷LEU2 (YCGAL–CDC20∷URA) | this study |
| RTK260 | MATa bar1 cdc15-2 cdc20∷LEU2 trp1∷GAL–CDC20–TRP1 | this study |
| RTK238 | MATa bar1 rad52Δ∷URA3 | this study |
| RTK239 | MATa bar1 rad52Δ∷URA3 pds1Δ∷LEU2 | this study |
| RTK210 | MATa bar1 rad52Δ∷URA3 pds1Δ∷LEU2 rad9Δ∷HIS3 | this study |
| RTK303 | MATa bar1 rad52Δ∷URA3 cdc15-2 trp∷3X(GAL–ESP1–TRP) | this study |
| RTK12 | MATa bar1 cdc15-2 rad9Δ∷LEU2 | this study |
| RTK43 | MATa bar1 cdc15-2 trp1∷GAL–PDS1HA–TRP1 | this study |
| RTK35 | MATa bar1 cdc15-2 trp1∷GAL–PDS1HA–TRP1 rad9Δ∷HIS3 | this study |
| RTK330 | MATa bar1 cdc15-2 pds1∷PDS1 myc18–LEU2 | this study |
All strains are in a W303 background.
To construct pRTK-C2 (GAL–pds1ΔdbHA), the FspI–NdeI fragment of pRTK-C1 (GAL–PDS1HA) (Jaspersen et al. 1998) was replaced by a PCR fragment from which the Pds1 destruction box (RLPLAAKDN) was deleted and replaced with amino acids LE, creating an XhoI restriction site (Cohen-Fix et al. 1996). To make strain RTK44, RTK–C2 was digested with Bsu36I and integrated at the TRP1 locus. All strains containing GAL–pdsΔdb HA were obtained by crossing to RTK44. Strains containing a triple integrant of the GAL–ESP1 construct were obtained from crossing to K7346 (Ciosk et al. 1998). RTK313, RTK314, RTK330, RTK335–339, and RTK344 were obtained by replacing PDS1 with a version of the gene that encoded a carboxy-terminally myc18-epitope-tagged Pds1 protein as described (Shirayama et al. 1998). Deletion of RAD9 was performed as described (Weinert and Hartwell 1990) and confirmed by Southern blot hybridization. PDS1 was deleted as described (Yamamoto et al. 1996a) and deletions were identified by screening for temperature sensitivity (i.e., at 30°C), sensitivity to the microtubule poison benomyl, and the inability to complement a pds7A-1F temperature-sensitive mutant (gift from Sue Biggins, UCSF). Deletion of RAD52 was carried out as described (Kaytor and Livingston 1994) and confirmed by screening for hydroxyurea and radiation sensitivity. Deletion of MAD1 was performed as described (Hardwick and Murray 1995) and confirmed by PCR and Western blot analysis. All other strains were derived from crosses using standard methods (Guthrie and Fink 1991). RTK223 and RTK211 were derived by transformation with the URA3-based centromeric plasmid pLH68, which contains the CDC20 gene fused to a triple hemagglutinin (HA) tag and a six-histidine tag at its carboxyl terminus (Hwang et al. 1998).
Cell-cycle synchronization
Standard protocols were used for cell propagation (Guthrie and Fink 1991). To arrest at G1, S phase, or metaphase, cells were grown at 23°C (or at 19°C in case of pds1Δ strains) to mid-log phase (OD600 of 0.35) and arrested with 1.5 μg/ml α-factor, 0.1 m hydroxyurea (HU), or 15 μg/ml nocodazole at 23°C for 3.5 hr, respectively (unless otherwise indicated). To release from a G1, S phase, or metaphase arrest, cells were pelleted by centrifugation and washed with the appropriate media three times, and resuspended in 23°C or 37°C media as indicated. To arrest temperature-sensitive strains, cells were grown to mid-log phase at 23°C and arrested by shifting cells to 37°C for 3.5 hr. Cells were released from the arrest by returning them to 23°C. In all cases, 90%–95% of cells displayed the correct arrest morphology. All strains were either grown in YEP media (Rose et al. 1990) supplemented with 2% dextrose (YEPD) or 2% raffinose (YEPraf) if galactose induction was going to take place. To express from the GAL promoter, cells were grown in YEPraf and expression was induced by the addition of galactose to 4% (unless otherwise indicated). Transcription was repressed by the addition of 4% dextrose (unless otherwise indicated). Strains containing the centromeric plasmid with CDC20 were grown to mid-log phase in minimal medium lacking uracil and containing 2% raffinose. Strains were then pelleted and resuspended in YEPraf to perform the experiment described.
Irradiation
After the indicated cell cycle arrest, 20 OD600 units were pelleted by centrifugation and resuspended in 1 ml of media. Cells were placed in one well of a 3043-microtiter dish (Fisher) and γ-irradiated at 2.5 krads using a cesium source at 360 rads/min (1 rad = 0.1 Gy). Cells were resuspended in the appropriate media as described.
Lysate preparation and Western blot analysis
Protein extracts were prepared by resuspending cells in 3 to 4-pellet volumes of ice-cold LLB [50 mm HEPES-NaOH (pH 7.4), 75 mm KCl, 50 mm NaF, 50 mm β-glycerophosphate, 1 mm EGTA, 0.1% NP40, 1 mm DTT, 1 mm phenylmethylsulfonylfluoride, 2 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin] and an equal volume of glass beads. Samples were lysed by mechanical disruption in a Beadbeater (Biospec) for 2 min. Lysates were clarified by centrifugation at 14,000g for 15 min at 4°C and protein concentrations were determined using the Bio-Rad protein assay.
For Western blotting, 25 μg of lysate was used for detection of endogenous Clb2, Cdc28, and Pds1-myc18, whereas 60 μg of lysate was used for detection of endogenous Sic1 and Clb3 and galactose-overexpressed Pds1HA and Pds1ΔdbHA proteins. Clb2, Cdc28, Sic1, and Clb3 proteins were detected with affinity-purified polyclonal antibodies as previously described (Gerber et al. 1995; Charles et al. 1998; Jaspersen et al. 1998). For detection of HA-tagged proteins, the mouse monoclonal antibody 16B12 was used as described (Gerber et al. 1995). Pds1myc18 immunoblots were performed with a 1:1000 dilution of α-myc polyclonal antibodies (Santa Cruz).
Kinase assays
To measure Clb2-associated histone H1 kinase activity, 75 μg of cell lysate was incubated with 0.3 μg of affinity-purified anti-Clb2 antibody and 20 μl of a 1:1 slurry of protein A–sepharose (Sigma) for 1.5 hr at 4°C. Immune complexes were washed two times in LLB and once in Buffer A [50 mm HEPES-NaOH (pH 7.4) and 1 μm DTT], and incubated for 15 min at 23°C in a 20-μl reaction mixture containing 100 μm ATP, 1 mm MgCl2, 5 μg of histone H1 protein, and 2.5 μCi [γ-32P] ATP (3000 mCi/mmole) in buffer A. Reaction products were resolved on 12% SDS-PAGE gels and detected by autoradiography.
Cyclin ubiquitination assay
Cyclin ubiquitin–ligase activity of the APC was measured as described (Charles et al. 1998). Briefly, the APC was immunoprecipitated by incubating 400 μg of yeast lysate with 0.5 μg of anti-Cdc26 polyclonal antibodies and 20 μl of protein A–Sepharose for 1.5 hr at 4°C. Immune complexes were washed three times in LLB, twice in High Salt QA [20 mm Tris-HCl (pH 7.6), 250 mm KCl, 1 mm MgCl2, 1 mm DTT], and twice in buffer QA [20 mm Tris-HCl (pH 7.6), 100 mm KCl, 1 mm MgCl2, 1 mm DTT]. A mix (15 μl) containing 3.5 pmoles of Uba1, 47 pmoles of Ubc4, 1 mm ATP, 20 μg of bovine ubiquitin (Sigma), and 0.25 μl 125I-labeled sea urchin (13-91) cyclin B1 in buffer QA was added and the reaction was allowed to proceed for 20 min at 23°C. Reaction products were resolved on 12.5% SDS–polyacrylamide gels and ubiquitin conjugates were detected by autoradiography with the BioMaxMS System (Kodak). It should be noted that this in vitro assay detects only Hct1-dependent APC activity and does not detect Cdc20–APC activity (Charles et al. 1998).
Acknowledgments
We thank J. Charles, S. Jaspersen, A. Rudner, H. Funabiki, and A. Murray for advice and helpful discussions during the course of this work, T. Weinert, L. Hu, and D. Dean for instruction and advice on γ-irradiation, and U. Surana, D. Toczyski, F. Uhlmann, T. Weinert, and various members of the Morgan and Murray laboratories for plasmids, strains, and antibodies. R.L.T-K. thanks the Cold Spring Harbor Yeast Genetic Course instructors A. Adams, D. Gottschling, and C. Kaiser for their valuable teachings and her family for their endless support. We are also grateful to A. Rudner, J. Charles, H. Funabiki, A. Szidon, A. Murray, J. Nourse, S. Jaspersen, and S. Biggins for comments on the manuscript. R. L. T-K. was supported by the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation Fellowship, DRG-1375. This work was supported by funding from the National Institute of General Medical Sciences (to D.O.M.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL dmorgan@cgl.ucsf.edu; FAX (415) 476-4929.
References
- Alexandru G, Zachariae W, Schleiffer A, Nasmyth K. Sister chromatid separation and chromosome re-duplication are regulated by different mechanisms in response to spindle damage. EMBO J. 1999;18:2707–2721. doi: 10.1093/emboj/18.10.2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon A, Surana U, Muroff I, Nasmyth K. Regulation of p34CDC28 tyrosine phosphorylation is not required for entry into mitosis in S. cerevisiae. Nature. 1992;355:368–371. doi: 10.1038/355368a0. [DOI] [PubMed] [Google Scholar]
- Charles JF, Jaspersen SL, Tinker-Kulberg RL, Hwang L, Szidon A, Morgan DO. The Polo-related kinase Cdc5 activates and is destroyed by the mitotic cyclin destruction machinery in S. cerevisiae. Curr Biol. 1998;8:497–507. doi: 10.1016/s0960-9822(98)70201-5. [DOI] [PubMed] [Google Scholar]
- Ciosk R, Zachariae W, Michaelis C, Shevchenko A, Mann M, Nasmyth K. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell. 1998;93:1067–1076. doi: 10.1016/s0092-8674(00)81211-8. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Koshland D. The anaphase inhibitor of Saccharomyces cerevisiae Pds1p is a target of the DNA damage checkpoint pathway. Proc Natl Acad Sci. 1997;94:14361–14366. doi: 10.1073/pnas.94.26.14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Fix O, Peters J-M, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes & Dev. 1996;10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- Donovan JD, Toyn JH, Johnson AL, Johnston LH. P40SDB25, a putative CDK inhibitor, has a role in the M/G1 transition in Saccharomyces cerevisiae. Genes & Dev. 1994;8:1640–1653. doi: 10.1101/gad.8.14.1640. [DOI] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: Preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW. Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol Cell. 1998;2:163–171. doi: 10.1016/s1097-2765(00)80126-4. [DOI] [PubMed] [Google Scholar]
- Fesquet D, Fitzpatrick PJ, Johnson AL, Kramer KM, Toyn JH, Johnston LH. A Bub2p-dependent spindle checkpoint pathway regulates the Dbf2 kinase in budding yeast. EMBO J. 1999;18:2424–2434. doi: 10.1093/emboj/18.9.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funabiki H, Kumada K, Yanagida M. Fission yeast Cut1 and Cut2 are essential for sister chromatid separation, concentrate along the metaphase spindle and form large complexes. EMBO J. 1996;15:6617–6628. [PMC free article] [PubMed] [Google Scholar]
- Gerber MR, Farrell A, Deshaies R, Herskowitz I, Morgan DO. Cdc37 is required for association of the protein kinase Cdc28 with G1 and mitotic cyclins. Proc Natl Acad Sci. 1995;92:4651–4655. doi: 10.1073/pnas.92.10.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR, editors. Guide to yeast genetics and molecular biology. Methods in enzymology. San Diego, CA: Academic Press; 1991. [PubMed] [Google Scholar]
- Hardwick KG, Murray AW. Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J Cell Biol. 1995;131:709–720. doi: 10.1083/jcb.131.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: Controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Hershko A. Roles of ubiquitin-mediated proteolysis in cell cycle control. Curr Opin Cell Biol. 1997;9:788–799. doi: 10.1016/s0955-0674(97)80079-8. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ganoth D, Sudakin V, Dahan A, Cohen LH, Luca FC, Ruderman JV, Eytan E. Components of a system that ligates cyclin to ubiquitin and their regulation by the protein kinase cdc2. J Biol Chem. 1994;269:4940–4946. [PubMed] [Google Scholar]
- Holloway SL, Glotzer M, King RW, Murray AW. Anaphase is initiated by proteolysis rather than by the inactivation of maturation-promoting factor. Cell. 1993;73:1393–1402. doi: 10.1016/0092-8674(93)90364-v. [DOI] [PubMed] [Google Scholar]
- Hwang LH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW. Budding yeast Cdc20: A target of the spindle checkpoint. Science. 1998;279:1041–1044. doi: 10.1126/science.279.5353.1041. [DOI] [PubMed] [Google Scholar]
- Irniger S, Piatti S, Michaelis C, Nasmyth K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 1995;81:269–277. doi: 10.1016/0092-8674(95)90337-2. [DOI] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Tinker-Kulberg RL, Morgan DO. A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. Mol Biol Cell. 1998;9:2803–2817. doi: 10.1091/mbc.9.10.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Morgan DO. Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol. 1999;9:227–236. doi: 10.1016/s0960-9822(99)80111-0. [DOI] [PubMed] [Google Scholar]
- Kaytor MD, Livingston DM. Saccharomyces cerevisiae RAD52 alleles temperature-sensitive for the repair of DNA double-strand breaks. Genetics. 1994;137:933–944. doi: 10.1093/genetics/137.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Lin DP, Matsumoto S, Kitazono A, Matsumoto T. Fission yeast Slp1: An effector of the Mad2-dependent spindle checkpoint. Science. 1998;279:1045–1047. doi: 10.1126/science.279.5353.1045. [DOI] [PubMed] [Google Scholar]
- King RW, Peters J-M, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Kitada K, Johnson AL, Johnston LH, Sugino A. A multicopy suppressor gene of the Saccharomyces cerevisiae G1 cell cycle mutant gene dbf4 encodes a protein kinase and is identified as CDC5. Mol Cell Biol. 1993;13:4445–4457. doi: 10.1128/mcb.13.7.4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani S, Tugendreich S, Fujii M, Jorgensen P, Watanabe N, Hoog C, Hieter P, Todokoro K. PKA and MPF-activated Polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol Cell. 1998;1:371–380. doi: 10.1016/s1097-2765(00)80037-4. [DOI] [PubMed] [Google Scholar]
- Lahav-Baratz S, Sudakin V, Ruderman JV, Hershko A. Reversible phosphorylation controls the activity of cyclosome-associated cyclin-ubiquitin ligase. Proc Natl Acad Sci. 1995;92:9303–9307. doi: 10.1073/pnas.92.20.9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. Bifurcation of the mitotic checkpoint pathway in budding yeast. Proc Natl Acad Sci. 1999;96:4989–4994. doi: 10.1073/pnas.96.9.4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gorbea C, Mahaffey D, Rechsteiner M, Benezra R. MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. Proc Natl Acad Sci. 1997;94:12431–12436. doi: 10.1073/pnas.94.23.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HH, Surana U. Cdc20, a β-transducin homologue, links RAD9-mediated G2/M checkpoint control to mitosis in Saccharomyces cerevisiae. Mol & Gen Genet. 1996;253:138–148. doi: 10.1007/s004380050306. [DOI] [PubMed] [Google Scholar]
- Lim HH, Goh P, Surana U. Cdc20 is essential for the cyclosome-mediated proteolysis of both Pds1 and Clb2 during M phase in budding yeast. Curr Biol. 1998;8:231–234. doi: 10.1016/s0960-9822(98)70088-0. [DOI] [PubMed] [Google Scholar]
- McGrew JT, Goetsch L, Byers B, Baum P. Requirement for ESP1 in the nuclear division of Saccharomyces cerevisiae. Mol Biol Cell. 1992;3:1443–1454. doi: 10.1091/mbc.3.12.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll T, Tebb G, Surana U, Robitsch H, Nasmyth K. The role of phosphorylation and the CDC28 protein kinase in the cell cycle-regulated nuclear import of the S. cerevisiae transcription factor SWI5. Cell. 1991;66:743–758. doi: 10.1016/0092-8674(91)90118-i. [DOI] [PubMed] [Google Scholar]
- Morgan DO. Regulation of the APC and the exit from mitosis. Nature Cell Biol. 1999;1:E47–E53. doi: 10.1038/10039. [DOI] [PubMed] [Google Scholar]
- Murray AW. The genetics of cell cycle checkpoints. Curr Opin Genet Dev. 1994;5:5–11. doi: 10.1016/s0959-437x(95)90046-2. [DOI] [PubMed] [Google Scholar]
- ————— Cyclin ubiquitination: The destructive end of mitosis. Cell. 1995;81:149–152. doi: 10.1016/0092-8674(95)90322-4. [DOI] [PubMed] [Google Scholar]
- New JH, Sugiyama T, Zaitseva E, Kowalczykowski SC. Rad52 protein stimulates DNA strand exchange by Rad51 and rereplication protein A. Nature. 1998;391:401–410. doi: 10.1038/34950. [DOI] [PubMed] [Google Scholar]
- Paulovich AG, Margulies RU, Garvik BM, Hartwell LH. RAD9, RAD17, and RAD24 are required for S phase regulation in Saccharomyces cerevisiae in response to DNA damage. Genetics. 1997;145:45–62. doi: 10.1093/genetics/145.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J-M. SCF and APC: The Yin and Yang of cell cycle regulated proteolysis. Curr Opin Cell Biol. 1998;10:759–768. doi: 10.1016/s0955-0674(98)80119-1. [DOI] [PubMed] [Google Scholar]
- Prinz S, Hwang ES, Visintin R, Amon A. The regulation of Cdc20 proteolysis reveals a role for the APC components Cdc23 and Cdc27 during S phase and early mitosis. Curr Biol. 1998;8:750–760. doi: 10.1016/s0960-9822(98)70298-2. [DOI] [PubMed] [Google Scholar]
- Rose MD, Winston F, Hieter P. Methods in yeast genetics—A laboratory course manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- Rudner AD, Murray AW. The spindle assembly checkpoint. Curr Opin Cell Biol. 1996;8:773–780. doi: 10.1016/s0955-0674(96)80077-9. [DOI] [PubMed] [Google Scholar]
- Schwab M, Lutum AS, Seufert W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 1997;90:683–693. doi: 10.1016/s0092-8674(00)80529-2. [DOI] [PubMed] [Google Scholar]
- Schweitzer B, Philippsen P. CDC15, an essential cell cycle gene in Saccharomyces cerevisiae, encodes a protein kinase domain. Yeast. 1991;7:265–273. doi: 10.1002/yea.320070308. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Matsui Y, Toh-e A. The yeast TEM1 gene, which encodes a GTP-binding protein, is involved in termination of M phase. Mol Cell Biol. 1994;14:7476–7482. doi: 10.1128/mcb.14.11.7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Dominant mutant alleles of yeast protein kinase gene CDC15 suppress the lte1 defect in termination of M phase and genetically interact with CDC14. Mol Gen & Genet. 1996;251:176–185. doi: 10.1007/BF02172916. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Zachariae W, Ciosk R, Nasmyth K. The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J. 1998;17:1336–1349. doi: 10.1093/emboj/17.5.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Sorger PK, Murray AW. S-phase feedback control in budding yeast independent of tyrosine phosphorylation of p34cdc28. Nature. 1992;355:365–368. doi: 10.1038/355365a0. [DOI] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A. The cyclosome, a large complex containing cyclin-selective ubiquitin-ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–198. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surana U, Amon A, Dowzer C, McGrew J, Byers B, Nasmyth K. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase-to-anaphase transition in budding yeast. EMBO J. 1993;12:1969–1978. doi: 10.1002/j.1460-2075.1993.tb05846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor GS, Liu Y, Baskerville C, Charbonneau H. The activity of Cdc14p, an oligomeric dual specificity protein phosphatase from Saccharomyces cerevisiae, is required for cell cycle progression. J Biol Chem. 1997;272:24054–24063. doi: 10.1074/jbc.272.38.24054. [DOI] [PubMed] [Google Scholar]
- Toyn JH, Johnson AL, Donovan JD, Toone WM, Johnston LH. The Swi5 transcription factor of Saccharomyces cerevisiae has a role in exit from mitosis through induction of the Cdk-inhibitor Sic1 in telophase. Genetics. 1996;145:85–96. doi: 10.1093/genetics/145.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science. 1997;278:455–460. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A. CDC20 and CDH1: A family of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell. 1998;2:709–718. doi: 10.1016/s1097-2765(00)80286-5. [DOI] [PubMed] [Google Scholar]
- Wan J, Xu H, Grunstein M. CDC14 of Saccharomyces cerevisiae. J Biol Chem. 1992;267:11274–11280. [PubMed] [Google Scholar]
- Weinert T. DNA damage checkpoints update: Getting molecular. Curr Opin Genet Dev. 1998;8:185–193. doi: 10.1016/s0959-437x(98)80140-8. [DOI] [PubMed] [Google Scholar]
- Weinert TA, Hartwell LH. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- ————— Characterization of RAD9 of Saccharomyces cerevisiae and evidence that its function acts posttranslationally in cell cycle arrest after DNA damage. Mol Cell Biol. 1990;10:6554–6564. doi: 10.1128/mcb.10.12.6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Guacci V, Koshland D. Pds1p is required for the faithful execution of anaphase in yeast. J Cell Biol. 1996a;133:85–97. doi: 10.1083/jcb.133.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Pds1, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s) J Cell Biol. 1996b;133:99–110. doi: 10.1083/jcb.133.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SS, Yeh E, Salmon ED, Bloom K. Identification of a mid-anaphase checkpoint in budding yeast. J Cell Biol. 1997;136:345–354. doi: 10.1083/jcb.136.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae W, Nasmyth K. TPR proteins required for anaphase progression mediate ubiquitination of mitotic B-type cyclins in yeast. Mol Biol Cell. 1996;7:791–801. doi: 10.1091/mbc.7.5.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 1998;282:1721–1724. doi: 10.1126/science.282.5394.1721. [DOI] [PubMed] [Google Scholar]