

To the Editor
When the road map for sequencing the human genome was laid out the study of genetic variation was deemed a critical component1; with the mapping of Single Nucleotide Polymorphisms (SNPs) initially being a priority. The availability of a high quality human reference assembly2 facilitated the discovery and characterization of structural variation (SV) of DNA with copy number variation (CNV) being its most abundant form3,4,5. As other multi-cellular organisms were sequenced, SV was observed to be a ubiquitous feature of genomes6,7. dbSNP8 was created early in 1998 to manage SNP and small scale variation but it was not designed for larger and more complex SV data. The explosion of data from diverse SV studies now necessitates that a public data archive be developed. Here we describe two official companion databases dbVar and DGVa serving this community role.
Public data archives (PDA) play a major role in supporting the scientific community. PDAs provide stable and traceable identifiers and allow for a single point of access for data collections, facilitating download and meta-analysis across studies. The Database of Genomic Variants (DGV) was developed in 2004 to support public access to human genomic SV data for biomedical studies. DGV has served a very important role in collecting and analyzing SV studies but is not able to provide a comprehensive and perpetual archive and will discontinue accepting direct submissions. Instead, DGV will work in partnership with the new archives and use these as the primary source of SV information, ensuring that data in DGV is synchronized with accessioned SVs in dbVar and DGVa. The main role of DGV going forward will be to curate and visualize selected studies to facilitate interpretation of SV data, including implementing the highest-level quality standards required by the clinical and diagnostic communities. The complex nature of SV often makes it less amenable than other genomic data (e.g. SNPs) to single-step electronic mapping to reference genomes necessitating significant manual curation efforts; a critical contribution which DGV will continue to perform (figure 1).
Figure 1.
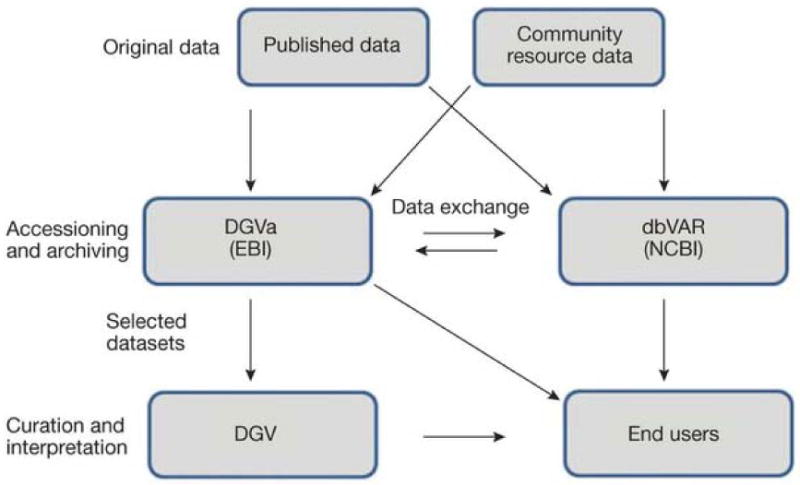
This diagram shows the workflow for data submission and curation. Data from published works or community resources will be submitted to either dbVar or DGVa. The groups use a common tracking system for data loading and currently coordinate monthly releases. Once data has been synchronized, DGV will collect data from DGVa in order to curate selected human data sets. End users will be able to obtain data from all three sources.
Efforts are ongoing to populate dbVar/DGVa with the historical studies from DGV and 14 of these studies will be public with the June data release with the remaining studies to follow shortly. As part of this transition, the scope of data represented will be expanded. Studies looking at the association of SVs with phenotypes and the SV characterization of cancer genomes will be represented. Studies with participants that have not been consented for their genetic information to be released in an open, public database can submit sample level data to the European Genome-phenome Archive (EGA) or the Database of Genotype and Phenotype (dbGaP). Sample level data will be held in the controlled access archives and only summary level data will be released to the DGVa and dbVar archives. One such study, nstd11 (Walter et al)9, has already been submitted via this pathway. Additionally, studies from diverse species will be represented in an effort to support comparative analysis of SV.
dbVar and DGVa use a common data model (Supplementary Figure 1), share identifiers and exchange data on a regular basis; the sites expect to be synchronized with respect to data content by the end of 2010. Identifiers are assigned at 3 levels, at the study level (std) and the variants region level (sv) and at the supporting variant level (ssv). Identifiers assigned at EBI will be prefixed with an ‘e’ and those assigned at NCBI will be prefixed with an ‘n’. At this time, DGV will also be updated to be synchronized with the data in the new archives. The majority of data loaded to date has been formatted by internal staff, in consultation with the authors, although we are moving toward direct submissions and a limited number of studies have been submitted this way, nstd19 (Quinlan et al)10, nstd31 (Alkan et al)11, nstd35 (Kidd et al)12. Submission instructions and example submissions for dbVar can be found on the dbVar website. Submission information for DGVa will be available through the EBI submission page. Most of the studies currently available do not have base level precision, and the technologies for identifying SV are rapidly changing. In fact, few variants are defined at the breakpoint level and both archives encourage submitters to provide the breakpoint range and level of resolution for each analysis. Both archives are attempting to develop more robust ways to graphically represent the lack of precision. We collect all supporting information for variant assertions, including descriptions of methods, analysis and the raw data used in the study (via Array Express/GEO or the SRA/ERA). Additionally, variant regions are only considered validated if a separate experimental analysis has been performed and this data is also collected and presented to users. We can accept population based data, such as genotype or copy number analysis, although this is not required. We will accept data for all studies that have been submitted to a peer review journal and we will also accept unpublished data from large-scale community based projects prior to publication to facilitate community access. We are working directly with several large-scale projects such as the Human Structural Variation Database, 1000 Genomes and the ISCA Consortium13 to ensure their data are updated in the database regularly.
While dbVar and DGVa plan to store the same data, they have different and complementary plans for providing data access and providing value added tools. DGVa data will be integrated with other EBI resources including the comprehensive EBI search and the Ensembl genome browser14. DGVa has been designed to facilitate the curatorial work of DGV. In addition to coordinating with public browsers, dbVar has an independent site where users can use the Entrez8 query system to search for individual variants or studies. This site is integrated with other NCBI resources and links to other databases are readily available. dbVar is currently working to map all data onto common coordinate systems and provide some minimal curation by cross referencing this data with information from the Genome Reference Consortium in order to identify called variants that are likely artifacts due to errors in previous version of the genome reference assembly for GRC supported assemblies. Both sites provide XML and tab delimited files on their FTP site and dbVar provides data in GFF format and DGVa will provide data in this format shortly so that users can readily load data to their browser of choice. Ultimately, through this project, the SV archives promise to greatly enable all studies of genetic variation.
Footnotes
URL List:
dbVar: http://www.ncbi.nlm.nih.gov/dbvar
DGVa: http://www.ebi.ac.uk/dgva
DGV: http://projects.tcag.ca/variation
EGA: http://www.ebi.ac.uk/ega/
dbGaP: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap
dbVar Submissions: http://www.ncbi.nlm.nih.gov/dbvar/content/submission/
DGVa Submissions: http://www.ebi.ac.uk/Submissions/
Human Structural Variation Database: http://hgsv.washington.edu/
1000 Genomes: http://1000genomes.org/page.php
Genome Reference Consortium: http://genomereference.org
Author contributions:
DMC, SWS and PF conceived of the databases and oversaw their development. IL, TPS, JH, MM, JL, JG,JP and LF developed the databases and processed data to populate the databases.
MD and EY developed schemas and tools for the data model at NCBI
Contributor Information
Deanna M. Church, Email: church@ncbi.nlm.nih.gov.
Paul Flicek, Email: flicek@ebi.ac.uk.
References
- 1.Collins FS, et al. New goals for the U.S. Human Genome Project: 1998-2003. Science. 1998;282:682–689. doi: 10.1126/science.282.5389.682. [DOI] [PubMed] [Google Scholar]
- 2.Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 3.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 5.Eichler EE, et al. Completing the map of human genetic variation. Nature. 2007;447:161–165. doi: 10.1038/447161a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.She X, Cheng Z, Zöllner S, Church DM, Eichler EE. Mouse segmental duplication and copy number variation. Nat Genet. 2008;40:909–14. doi: 10.1038/ng.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emerson JJ, Cardoso-Moreira M, Borevitz JO, Long M. Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science. 2008;320:1629–1631. doi: 10.1126/science.1158078. [DOI] [PubMed] [Google Scholar]
- 8.Wheeler DL, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walter MJ, et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci USA. 2009;106:12950–12955. doi: 10.1073/pnas.0903091106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quinlan AR, et al. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 2010;20:623–635. doi: 10.1101/gr.102970.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alkan C, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kidd JM, et al. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat Methods. 2010;7:365–371. doi: 10.1038/nmeth.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller DT, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flicek P, et al. Ensembl's 10th year. Nucleic Acids Res. 2010;38:D557–562. doi: 10.1093/nar/gkp972. [DOI] [PMC free article] [PubMed] [Google Scholar]