

Abstract
Output properties of neurons are greatly shaped by voltage-gated ion channels, whose biophysical properties and localization within axodendritic compartments serve to significantly transform the original input. The hyperpolarization-activated current, Ih, is mediated by hyperpolarization-activated cyclic nucleotide-gated (HCN) channels and plays a fundamental role in influencing neuronal excitability by regulating both membrane potential and input resistance. In neurons such as cortical and hippocampal pyramidal neurons, the subcellular localization of HCN channels plays a critical functional role, yet mechanisms controlling HCN channel trafficking are not fully understood. Because ion channel function and localization are often influenced by interacting proteins, we generated a knock-out mouse lacking the HCN channel auxiliary subunit, tetratricopeptide repeat-containing Rab8b-interacting protein (TRIP8b). Eliminating expression of TRIP8b dramatically reduced Ih expression in hippocampal pyramidal neurons. Loss of Ih-dependent membrane voltage properties was attributable to reduction of HCN channels on the neuronal surface, and there was a striking disruption of the normal expression pattern of HCN channels in pyramidal neuron dendrites. In heterologous cells and neurons, absence of TRIP8b increased HCN subunit targeting to and degradation by lysosomes. Mice lacking TRIP8b demonstrated motor learning deficits and enhanced resistance to multiple tasks of behavioral despair with high predictive validity for antidepressant efficacy. We observed similar resistance to behavioral despair in distinct mutant mice lacking HCN1 or HCN2. These data demonstrate that interaction with the auxiliary subunit TRIP8b is a major mechanism underlying proper expression of HCN channels and Ih in vivo, and suggest that targeting Ih may provide a novel approach to treatment of depression.
Introduction
Neuronal signaling is in part governed by the repertoire of voltage-gated ion channels expressed within a given cell, and biophysical properties and localization within the neuron both determine the physiological role of a specific channel (Lai and Jan, 2006). Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels underlie the hyperpolarization-activated current, Ih, a functionally important current in the nervous system and heart (Biel et al., 2009; Baruscotti et al., 2010). In the brain, subcellular localization of HCN channels is diverse, and this is perhaps most striking in dendrites of hippocampal and cortical pyramidal neurons where HCN channels exist in a gradient of increasing density with increasing distance from the soma (Magee, 1998; Lörincz et al., 2002). Dendritic Ih normalizes the temporal summation of inputs and modulates integrative properties of neurons (Magee, 1999; Williams and Stuart, 2000), and spatially enriched Ih controls local dendritic processes such as Ca2+ spikes and synaptic plasticity (Nolan et al., 2004; Tsay et al., 2007). Although important for normal neuronal function, molecular mechanisms regulating HCN channel biophysical properties and subcellular localization in the mammalian brain are not yet fully understood.
HCN channel localization and function is likely controlled in part by interacting proteins, and although HCN channel binding partners have been reported, functional roles for most of these proteins in vivo remain to be demonstrated (Lewis et al., 2010). Recent work showed that the HCN subunit interacting protein, tetratricopeptide repeat-containing Rab8b-interacting protein (TRIP8b), is an auxiliary subunit of HCN channels in the brain. TRIP8b stoichiometrically coimmunoprecipitates with pore-forming HCN subunits HCN1-4 from native membranes and significantly regulates the voltage gating and kinetics of Ih (Lewis et al., 2009; Santoro et al., 2009; Zolles et al., 2009). Furthermore, alternative splicing of the TRIP8b N terminus generates splice isoforms that differentially influence HCN channel surface trafficking toward or away from the surface plasma membrane (Santoro et al., 2004, 2009; Lewis et al., 2009). We sought to further investigate the importance of TRIP8b in Ih and HCN channel expression in vivo by generating a knock-out mouse lacking all TRIP8b expression (TRIP8b−/−). We primarily focused analysis on pyramidal neurons in hippocampal area CA1, where HCN channels are expressed in a striking and functionally important dendritic gradient.
We found that elimination of TRIP8b decreased Ih and reduced surface HCN channel expression on the surface membrane of CA1 pyramidal neurons. Although HCN channels trafficked to CA1 pyramidal neuron dendrites in TRIP8b−/− mice, the normal gradient of HCN subunits was disrupted. Cell culture and in vivo data suggested increased HCN trafficking to and degradation by lysosomes in the absence of TRIP8b. TRIP8b−/− mice demonstrated enhanced resistance to multiple tasks of behavioral despair with high predictive validity for antidepressant efficacy. We observed similar resistance to behavioral despair in HCN1−/− mice and mice with a spontaneous mutation that eliminates HCN2 expression, apathetic (HCN2ap/ap) mice. Overall, these data characterize a major mechanism for regulation of neuronal Ih in vivo, as well as suggest that Ih plays an important role in regulation of affective behavior.
Materials and Methods
Biochemistry, molecular biology, and imaging
cDNA constructs
cDNA encoding TRIP8b(1a-4), TRIP8b(1c) [previously referred to as “TRIP8b_IsoA4” and “TRIP8b_IsoT,” respectively (Lewis et al., 2009)], and TRIP8b(ΔN) were generated as previously described (Lewis et al., 2009). TRIP8b(1a-4)-GFP and TRIP8b(1c)-GFP were generated by subcloning from pXEGFPC1 into pEGFPN1 (Clontech) at EcoR1/BamHI sites.
Mice
All animal experiments were performed according to protocols approved by the Northwestern University, University of Texas at Austin, University of Texas Southwestern Medical Center, and University of Virginia Institutional Animal Care and Use Committees.
Generation of TRIP8b−/− mice
To generate a mouse with a total body elimination of TRIP8b, recombineering technology was used to generate a targeting vector with a 5′ loxP-Cre recombinase recognition site and a 3′ FRT-neo-FRT cassette and second loxP site flanking exons 6 and 7 of the mouse TRIP8b gene, exons that are common to all TRIP8b isoforms (Pex5l) (see Fig. 1A). Homologous recombination generated four unique neomycin-resistant C57BL/6 ES cell lines (PRX-B6 ES cells derived from Taconic Farms C57BL/6N mice) with the appropriate targeting (see Fig. 1B). Blastocyst injections of two of these lines into pseudopregnant B6(Cg)-Tyrc-2J/J mice produced seven highly chimeric offspring (chimeric Tyrc-2J/Pex5l+/Fneo) that were bred with C57BL/6 albino females [B6(Cg)-Tyrc-2J/J]. Black offspring were genotyped to detect the targeted allele by conventional PCR of the neo cassette contained in the allele. Heterozygous offspring were crossed with heterozygous siblings (Pex5l+/Fneo/Pex5l+/Fneo) to yield animals homozygous for the targeted locus (Pex5lFneo/Fneo) that were crossed with commercially available transgenic mice that express Cre recombinase under the CMV promoter [B6.C-Tg(CMV-cre)1Cgn; The Jackson Laboratory] to produce Pex5l+/− mice. These animals were identified by PCR detection of the cre transgene, as well as discrimination between the wild-type and exon 6–7 deleted alleles (see below, Genotyping TRIP8b−/− mice) (see Fig. 1C). Heterozygous Pex5l+/− animals were bred with each other to generate the conventional knock-out of the Pex5l locus, Pex5l−/−. For clarity and consistency with predominant usage in the literature, mice with the Pex5l−/− genotype are referred to throughout this article as TRIP8b−/− mice. The mice reported herein are on a background that is a mixture of C57BL/6N (from ES cells) and C57BL/6J (from the Cre line).
Figure 1.

Genetic deletion of exons 6–7 of Pex5l eliminates TRIP8b protein expression in mouse brain. A, Schematic illustrating the targeted portion of Pex5l gene (top) and the targeting vector (bottom). Southern blotting probes include 500 bp fragments 5′ and 3′ to the 4.5 kb short arm and 8.0 kb long arm of the targeting vector, and detect an 8.4 kb EcoRI fragment and 11.3 kb NdeI fragment, respectively, in control (wild-type) ES cells. B, Southern blotting of EcoRI- and NdeI-digested DNA from control (lane 1) and neomycin-resistant ES cells reveals 5.8 kb EcoRI and 13.3 kb NdeI fragments reflecting proper targeting in at least four clones. C, Representative PCR genotyping products from TRIP8b+/+, TRIP8b+/−, and TRIP8b−/− DNA separated on ethidium bromide-stained 2% agarose gel. D, Western blot analysis from TRIP8b+/+, TRIP8b+/−, and TRIP8b−/− brain lysates demonstrate no TRIP8b protein or truncated TRIP8b protein using antibodies to the N terminus and C terminus. E, HEK293T cells were either untransfected or transfected with cDNAs encoding full-length TRIP8b [TRIP8b(1a-4)-GFP], TRIP8b C terminus [TRIP8b(1c)-GFP], or GFP, and lysate probed with antibodies to epitopes on the C terminus of TRIP8b (left), N terminus of TRIP8b (middle), or GFP (right). The C-terminal TRIP8b antibody properly recognized both the full-length and truncated TRIP8b isoforms, whereas the N-terminal antibody only recognized the full-length TRIP8b isoform. F, Western blot analysis confirming no expression of TRIP8b splice isoforms using splice isoform-specific antibodies (Lewis et al., 2009).
Genotyping TRIP8b−/− mice
Primers were as follows (written 5′–3′): B6.C-Tg(CMV-cre)1Cgn: primer sequences are available on The Jackson Laboratory website (http://jaxmice.jax.org/strain/006054.html); Pex5l wild-type allele (150 bp): forward, (TSKC5′)-GCCCAATTGATGCATTTACTTTGG, reverse, (1.1b3′)-TGTGCCTATGTCTGCCTTCCCAG; Pex5l knock-out allele (268 bp): forward, TSKC5′, reverse, (TSKB3′)-CTGGACACAAACTAGAGTCACGG.
HCN1−/− mice
Behavioral experiments reported were from HCN1 wild-type (HCN1+/+) and knock-out (HCN1−/−) mouse littermates (Nolan et al., 2003) derived from heterozygous HCN1+/− mice, obtained by crossing animals from the homozygous HCN1 knock-out line (The Jackson Laboratory; stock number 005034) with C57BL/6J mice. Thus, the background of the HCN1−/− mice studied here is the result of a single cross of hybrid C57BL/6J and 129Sv with C57BL/6J.
HCN2ap/ap mutant mice
Behavioral experiments reported were performed on mice with a spontaneous mutation of HCN2, which we have previously characterized and described as apathetic (HCN2ap/ap) (Chung et al., 2009), and their wild-type counterparts. HCN2ap/ap mice lack expression of HCN2, are not fertile, are smaller in size, and display a lack of motor coordination and reduced spontaneous movements. Furthermore, because of poor feeding and reduced viability, they are routinely housed only with other HCN2ap/ap mice. These animals are maintained on a pure C57BL/6J background.
Plasma glucose testing
Peripheral blood was collected from adult male TRIP8b+/+ and TRIP8b−/− littermates and analyzed with a handheld glucometer (Walgreens) at 10:30 A.M. and 4:00 P.M. while mice were given ad libitum access to standard laboratory diet. Food was removed from cages with ad libitum access to water for an overnight 16 h fast, and blood glucose was measured the following morning.
Analysis of plasmalogens, phospholipids, and enzymes of peroxisomal β-oxidation in brain tissue
Analysis of plasmalogens, phospholipids, and enzymes of peroxisomal β-oxidation in brain tissue were performed as previously described (see the following references). Lipid extracts, prepared from one brain hemisphere, were analyzed for phospholipid (Van Veldhoven and Mannaerts, 1987), plasmalogens (Hulshagen et al., 2008), and fatty acid composition (Hellemans et al., 2009) as described (for the latter, a GC column of 60 m was used instead of 30 m). Immunoblots (data not shown) were prepared from brain (one hemisphere) homogenates (40 μg protein/lane), transiently stained with Ponceau S, blocked with defatted milk proteins, and incubated with anti-rat acyl-CoA oxidase 1 (Acox1) (50 kDa subunit) (Van Veldhoven et al., 1994) and anti-rat peroxisomal thiolase (ACAA1) (mature 41 kDa subunit) (Antonenkov et al., 1997), followed by anti-rabbit IgG-alkaline phosphatase conjugates. Immunocomplexes were revealed with NBT/BCIP staining. As control, brain samples of 1-d-old Pex5−/− (Baes et al., 1997) pups were used.
Generation of C-terminal TRIP8b antibody
cDNA encoding amino acids 396–462 of mouse TRIP8b (hinge region between two sets of TPR domains near C terminus of TRIP8b) was generated by PCR using primers (written 5′–3′): forward, CGCGAATTCACCAGCCACCAGCAGGATG, and reverse, CGCCTCGAGCTAGTCAATCATGTCACCATTTTG, and cloned into pET32H (Novagen) at EcoR1/Xho1 sites. pET32H-TRIP8b(396–462) was transformed into BL21 bacteria (Stratagene), and the resulting polyhistidine-tagged fusion protein was purified on a Ni-NTA agarose chromatography column (QIAGEN) according to the manufacturer's instructions. Rabbits were immunized with His-TRIP8b(396–462) (Affinity Bioreagents) to generate immune and preimmune serum, and one rabbit generated sensitive and specific serum used in these studies (see Fig. 1E).
Primary antibodies
The following primary antibodies were used in these studies: guinea pig (gp) anti-HCN1 (Shin and Chetkovich, 2007), rabbit (rb) anti-HCN1 [against amino acids 778–910 (same as gp anti-HCN1)], gp anti-HCN2 (Shin et al., 2006), rb anti-HCN2 (Alomone Labs), mouse (ms) anti-α-tubulin (clone DM1A; Santa Cruz Biotechnology), rb and gp anti-GFP (Lewis et al., 2009), rb and gp anti-TRIP8b N terminus (Shin et al., 2008; Lewis et al., 2009), rb anti-TRIP8b C terminus (see above) (see Fig. 1E), gp anti-TRIP8b exon 1a-5, exon 2, and exon 4 (Lewis et al., 2009) (see Fig. 1F), ms anti-MAP2 (clone HM-2; Sigma-Aldrich), ms anti-KDEL (clone 10C3; Stressgen), ms anti-GM130 (clone 35; BD Biosciences), and rb anti-Lamp1 (Sigma-Aldrich).
Quantitative real-time PCR
Quantitative real-time PCR was performed as previously described (Chung et al., 2009). The amplification program was as follows: 95°C for 2 min followed by 40 cycles of 95°C for 15 s, 56°C for 15 s, and 68°C for 20 s. PCR primer efficiencies differed by no more than 5%, and data were analyzed by the ΔΔCt method. Melting curves were performed to ensure amplification of only one product.
qPCR primer sequences were as follows (all written 5′–3′): HCN1 (231 bp amplicon), forward, AGGTTAATCAGATACATACACC, and reverse, GAGTGCGTAGGAATATTGTTTT; HCN2 (230 bp amplicon), forward, CGGCTCATCCGATATATCCA, and reverse, AGCGCGAACGAGTAGAGCTC; HCN3 (72 bp amplicon), forward, GGCAAGATGTTTGATGAAGAG, and reverse, GAAGTTAATAATCTCCTCCCGA; HCN4 (232 bp amplicon), forward, CCCGCCTCATTCGATACATTCAT, and reverse, AGGGCGTAGGAATACTGCTTC; TRIP8b (196 bp amplicon), forward, GGTCACCGTGAAACTTGACAT, and reverse, CCCCAAACCAAAGCCAAGAAA; Cyclophilin A (91 bp amplicon), forward, GGCTCTTGAAATGGACCCTTC, and reverse, CAGCCAATGCTTGATCATATTCTT.
Preparation of HEK293T cell and brain lysates, pharmacological treatments, Western blotting, and coimmunoprecipitation
Preparation of HEK293T and brain tissue lysates, as well as coimmunoprecipitation, was performed as previously described (Lewis et al., 2009). Primary antibody concentrations used but not previously described were as follows: rb anti-TRIP8b C terminus, 1:2000; rb anti-HCN1, 1:2000; rb anti-HCN2, 1:500; ms anti-KDEL, 1:1000; ms anti-α-tubulin, 1:1000 (HEK293T lysates) or 1:5000 (brain lysates).
(2S,3S)-trans-Epoxysuccinyl-l-leucylamido-3-methylbutane ethyl ester (E-64d) and benzyloxycarbonyl-leucyl-leucyl-leucinal (MG132) (Sigma-Aldrich) were dissolved in DMSO (Sigma-Aldrich) as 100 and 1000× stock concentrations, respectively, and added to fresh 37°C culture media to a final concentration of 10 μm for both inhibitors. Cells were treated with E-64d for 16–20 h or MG132 for 5–6 h before lysis.
Preembedding ultrasmall silver-intensified immunogold electron microscopy
Tissue preparation.
All electron microscopy reagents unless otherwise specified were from Electron Microscopy Sciences. Two sets of adult male littermate TRIP8b+/+ and TRIP8b−/− mice were deeply anesthetized by isoflurane (Isothesia; Butler) and transcardially perfused with 0.9% saline followed by ∼50 ml of ice-cold acidic fixative (2% paraformaldehyde, 1% glutaraldehyde in 0.1 m sodium acetate buffer, pH 6.0) followed by a slow perfusion of ice-cold basic fixative (2% paraformaldehyde, 1% glutaraldehyde in 0.1 m sodium borate buffer, pH 9.0) for 1 h. At the conclusion of the perfusion, brains were removed and placed in ice-cold basic fixative overnight at 4°C on a shaker. The next day, brains were cut into two hemispheres, rinsed three times for 20 min each in 0.12 m Tris-buffered saline (TBS) and cut into 60 μm coronal sections on a microtome, isolating the dorsal hippocampus. Sections were rinsed in TBS five times for 5 min each and treated with 1% NaBH4 in TBS for 30 min. Sections were rinsed in TBS five times for 1 min each and incubated in blocking solution (10% normal goat serum in TBS) for 30 min followed by incubation in primary antibody overnight at 4°C. Primary antibody (gp anti-HCN1; 1:1000) was diluted in 2% NGS plus 0.1 or 0.5% Triton X-100 in TBS. The next day, slices were rinsed in incubation buffer one time for 5 min and in TBS 10 times for 5 min each. Slices were incubated in secondary blocking buffer [2% NGS plus 1% BSA plus 0.3% cold water fish skin gelatin (CWFSG) in TBS] for 1 h followed by incubation in ultrasmall immunogold (Aurion) anti-gp secondary antibody at 1:100 dilution in 2% NGS plus 1% BSA-C plus 0.3% CWFSG at 4°C for ∼40 h. Slices were then rinsed in incubation buffer one time for 5 min, TBS six times for 10 min each, PBS two times for 5 min each, followed by fixation of immunogold with 2% glutaraldehyde in PBS for 1 h. Slices were rinsed two times for 5 min each in PBS, four times for 10 min each in TBS, followed by enhancement conditioning solution (ECS) three times for 10 min each. Slices were then incubated in R-Gent SE-EM enhancement mixture for 90 min in the dark and rinsed in ECS four times for 10 min each, TBS two times for 10 min each, and PBS two times for 10 min each. Slices were osmicated with 0.4% OsO4 in PBS for 8 min at 4°C and rinsed in PBS three times for 10 min each and dH2O two times for 5 min each. Slices were stained in 1% aqueous uranyl acetate for 10 min and rinsed three times for 10 min each in dH2O followed by dehydration in graded ethanols and propylene oxide. Tissue was infiltrated by a 1:1 mixture of Araldite/propylene oxide overnight at room temperature followed by flat embedding between aclar sheets and curing for 48 h at 60°C. Tissue regions of interest were subdissected and reembedded in Araldite and cured overnight at 60°C. The 70 nm ultrathin sections were cut perpendicular to the original plane of section of each hippocampal slice with a diamond knife and placed onto a Formvar-coated slotted grid. Ultrathin sections were then stained with uranyl acetate-lead citrate (for 15 and 10 min, respectively), rinsed in ultrapure dH2O, and allowed to dry at room temperature. This procedure was optimized for immunoreactivity but not ultrastructural preservation. As such, we were unable to perform a reliable quantitative analysis of ultrastructure such as multivesicular bodies, instead limiting our results to observations (see Fig. 7).
Figure 7.

HCN1 is localized to multivesicular bodies in distal dendrites of TRIP8b−/− mice. Representative serial sections taken from TRIP8b−/− mouse area CA1 SLM immunostained for HCN1 demonstrate immunogold localization to multivesicular bodies (MVBs) (arrows), consistent with a lysosomal degradation mechanism. HCN1 localization to MVBs was observed frequently in TRIP8b−/− SLM but rarely in TRIP8b+/+ SLM. Scale bar, 500 nm.
Image acquisition and analysis.
Each serial ultrathin section spanned from stratum pyramidale to the outer molecular layer of the dentate gyrus. Images were obtained with a JEOL 1200EX electron microscope. The distal border of the stratum lacunosum molecular (SLM) was identified by the hippocampal fissure, and SLM was distinguished from stratum radiatum by presence of blood vessels, a band of myelinated fibers at its proximal end, the absence of large-caliber dendrites running perpendicular to the stratum pyramidale, as well as decreased dendritic caliber. The proximal fields of stratum radiatum were within 20–50 μm of the stratum pyramidale, and this region was used for quantification of proximal particles. SLM was used for quantification of distal particles.
Serial sections (range, 16–28) from at least two randomly chosen regions of SLM were taken at 7500× magnification for each animal. For dendritic analysis, all cross-sectioned dendrites displaying membrane immunoreactivity that were completely contained in the image series were identified and analyzed. Analysis included measurements of dendritic surface area, as well as immunogold localization to the dendritic membrane or cytoplasm. Particles were designated as membrane bound if they were within 30 nm of the inner layer of the plasma membrane bilayer (Lörincz et al., 2002). Particle density was determined by dividing the total number of membrane-bound particles by the dendritic surface area over an image series. Dendritic surface area was determined by multiplying the sum of dendritic circumferences (recorded for each image) by 70 nm (the section thickness). Fraction of HCN1 particles localized to the plasma membrane was determined by dividing the number of membrane particles by the total number of particles. Analysis of labeled dendrites was restricted to those that lay between 1 and 5 μm from the tissue surface (i.e., the surface of the tissue that was directly exposed to the reagents during labeling).
Immunohistochemistry
Adult TRIP8b+/+ or TRIP8b−/− mice were deeply anesthetized with isoflurane (Isothesia; Butler) and transcardially perfused with PBS followed by ice-cold 4% paraformaldehyde. Brains were removed and postfixed overnight in 4% paraformaldehyde and transferred to 30% sucrose solution overnight. Brains were then sectioned into 20–50 μm slices on a microtome (Leica). Antigen retrieval was performed by treatment of slices with 10 mm sodium citrate buffer, pH 9.0, for 10 min at 80°C. Slices were blocked in 5% normal goat serum and 5% normal donkey serum in PBS plus 0.03% Triton X-100 for 30 min and incubated in primary antibody diluted in blocking solution overnight. Slices were rinsed and incubated in secondary antibody diluted in blocking buffer for 2 h. Slices were mounted on glass slides with PermaFluor mounting reagent (Thermo Fisher Scientific). Primary antibody dilutions for immunohistochemistry in this study were as follows: gp anti-HCN1, 1:1000; gp anti-HCN2, 1:1000; rb anti-Lamp1, 1:1000; ms anti-GM130, 1:250; ms anti-MAP2, 1:500. For all experiments involving comparison of staining in TRIP8b+/+ and TRIP8b−/−, tissue was always paired and stained at the same time with the same reagents.
Confocal microscopy and image analysis
Images were obtained on a Zeiss LSM 510 META laser-scanning confocal (Plan-Apochromat 63× objective/1.4 numerical aperture oil Ph3). For comparing HCN1- and Lamp1-colocalized puncta in TRIP8b+/+ and TRIP8b−/− hippocampal CA1 regions, images were taken using fixed confocal settings (scaling, X, 0.056 μm, Y, 0.056 μm, Z, 0.5 μm; image size, 512 × 512 pixels, 8 bit; zoom, 5; pixel dwell, 2.51 μs; average, line 8; master gain, 650; digital gain, 1; digital offset, −0.15; lasers, 8% for 488 nm, 15% for 543 nm; pinholes, 86 μm for Ch-488 nm and 96 μm for Ch-543 nm). Lamp1-positive vesicles containing HCN1 were quantified from confocal images of brain slices from TRIP8b+/+ and TRIP8b−/− mice with NIH ImageJ software. All confocal images were obtained from the same range of section depth (the stack containing five slices was taken from a 2 μm range by 0.5 μm interval at the center of 30-μm-thick section). After thresholding with the same parameters for images from both genotypes (20:255), the region of interest (ROI) for Lamp1 was recorded. The ROI was then applied to the HCN1 channel to record HCN1 fluorescence intensity. Lamp1 vesicles containing greater than two times the average HCN1 intensity within the same optical plane were scored as HCN1-positive Lamp1-positive vesicles. Results from this quantification method were verified by manual counting of HCN1- and Lamp1-colocalized puncta. Quantification of CA1 field immunostaining was performed as previously described with some modification (Shin and Chetkovich, 2007). Regions were first identified by drawing a straight line perpendicular from the stratum pyramidale toward the apical [containing stratum radiatum (SR) and SLM] or basal dendritic fields [containing stratum oriens (SO)], and dividing them into equal length sections. SO was divided into two sections, stratum pyramidale (SP) was divided into one section, SR was divided into seven sections, and SLM was divided into three sections. The average line density of staining was then calculated for each region.
Statistics for biochemical, molecular biological, and imaging experiments
All data are expressed as mean ± SEM. Statistical comparisons between two groups were made using unpaired two-tailed Student's t test (one-sample vs 1.0 or two-sample, when appropriate), and comparisons between three or more groups were made using one-way ANOVA followed by Tukey's post hoc test. Calculations were performed using Excel (Microsoft) and Prism (GraphPad) software. Data were considered statistically significant if p < 0.05.
Electrophysiology
Acute hippocampal slices
Hippocampal slices (300 μm) were prepared from 8- to 12-week-old male C57BL/6 TRIP8b+/+ or TRIP8b−/− mice using standard techniques (Routh et al., 2009).
Electrophysiology
Slices were placed individually as needed into a submerged recording chamber continuously perfused with control extracellular saline (see below). Slices were viewed with a Zeiss Axioskop using infrared video microscopy and differential interference contrast optics. For all recordings, the control ACSF solution contained the following (in mm): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, and 12.5 dextrose, and was bubbled continuously with 95% O2/5% CO2 at 31–33°C. Patch pipettes were pulled from borosilicate glass and had a resistance of 4–8 MΩ when filled with the internal recording solution containing the following (in mm): 120 potassium gluconate, 20 KCl, 10 HEPES, 4 NaCl, 4 MgATP, 0.3 Na-GTP, and 7 phosphocreatine, pH 7.3 with KOH. In some cases, Neurobiotin (0.1–0.2%) was included for morphological analysis.
All whole-cell recordings were made from the soma or apical dendrite of CA1 pyramidal neurons using a Dagan BVC-700 or Multiclamp 700A in current-clamp mode using current injection protocols as previously described (Brager and Johnston, 2007). Series resistance was monitored throughout the recording, and experiments in which the series resistance exceeded 30 MΩ were discarded.
Data acquisition and analysis
Data were sampled at 10 kHz, filtered at 3 kHz, and digitized by an ITC-18 interface connected to computer running custom software written in IGOR Pro (Wavemetrics) or using Axograph X. Data analyses were performed with custom-written software in IGOR Pro (Wavemetrics) or Axograph X. Input resistance (RN) was determined by calculating the slope of the linear regression line through the V–I plot (constructed by plotting the amplitude of the steady-state voltage against the corresponding current injection from a family of 500–750 ms current steps). Firing frequency curves were constructed by plotting the numbers of action potentials against the amplitude of the current injections (150 to 300 pA, for 500 ms in 50 pA intervals). Action potential threshold was defined as the voltage where dV/dt = 20 mV/ms. For measurements of temporal summation, simulated EPSPs (αEPSPs) were used to remove any contribution of presynaptic mechanisms. αEPSPs were simulated by the injection of current into the soma using the following function: I = Imax (t/α) e−αt. Imax and α were adjusted to produce EPSP-like waveforms with a peak amplitude of 5 mV and a time-to-peak of 10 ms. Temporal summation ratio was measured as the amplitude of the fifth αEPSP relative to first in a train of five αEPSPs [(αEPSP5 − αEPSP1)/αEPSP1]. Voltage sag was measured as the percentage change between the maximum and steady-state voltage change during hyperpolarizing current injections [(Vmax − Vss)/Vmax] × 100. A chirp stimulus of constant amplitude, with its frequency linearly spanning 0–15 Hz in 15 s, was used to generate an impedance amplitude profile (Narayanan and Johnston, 2007). The magnitude of the ratio of the Fourier transform of the voltage response to the Fourier transform of the chirp stimulus formed the impedance amplitude profile. The frequency at which the impedance amplitude reached its maximum is the resonance frequency. For measurements of resonance at depolarized voltages, 1 μm TTX and 50 μm NiCl2 were added to and the NaH2PO4 was removed from the extracellular saline. Rebound was measured as the slope of the rebound potential amplitude plotted as a function of the steady-state voltage (Brager and Johnston, 2007).
Statistical analyses for electrophysiological studies
All data are expressed as mean ± SEM. Statistical comparisons were made using ANOVA (one-way, two-way, or repeated-measures design as appropriate) followed by Tukey–Kramer multiple-comparisons post hoc test or Student's t test (paired or unpaired as appropriate) with InStat software. Linear fits and correlations were made using either IGOR (Wavemetrics) or InStat. Data were considered statistically significant if p < 0.05.
Drugs and solutions
4-Ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride (ZD7288) (Ascent Scientific) was diluted to the final concentration from stock solutions in DMSO (final concentration of DMSO, ≤0.1%).
Behavior
Overview
All TRIP8b+/+ or TRIP8b−/− mice tested were born within 5 weeks of each other and were tested in a single cohort. Initially, there was a total of 19 littermate pairs of either sex, but after one mouse developed cataracts, that one littermate pair was dropped from subsequent behavioral tasks requiring intact vision (leaving a total of 18 littermate pairs). The HCN1 littermate cohort tested in the forced swim test (FST) included 16 HCN1+/+ mice and 14 HCN1−/− mice of either sex, and in the tail suspension test (TST) included 17 HCN1+/+ mice and 15 HCN1−/− mice of either sex. All mice were born within ∼12 weeks of each other. For the HCN2ap/ap mutants, which must be housed separately from their wild-type littermates, a total of seven age- and gender-matched pairs born within 8 weeks of each other were used in the tail suspension test. Mice were of either sex. All mice were provided with ad libitum food and water (except as noted below). Mice were kept on a 12 h light/dark cycle, and behavioral testing was conducted during the light cycle.
All behavioral testing was conducted by experimenters blind to genotype, and every effort was made to test less stressful behaviors before more stressful behaviors. Behavioral tests were conducted in the following order: elevated plus maze, dark/light box, locomotor activity, marble burying, grooming behavior, social interaction with a juvenile (social learning), social interaction with a caged conspecific, rotarod, fear conditioning, interaction with pheromones, olfactory food finding, nesting behavior, Morris water maze, visible water maze, auditory startle reactivity, tail suspension test, forced swim test, spatial recognition, hot plate, and sucrose preference. Behavioral testing was conducted over 4.5 months, beginning when mice were ∼4–5 months of age. There was always a minimum of 24 h between the different tests. The weight of the mice was measured before the start of behavioral testing. Mice were transported from their housing room and allowed to habituate at the site of behavioral testing for ∼1 h before testing.
Anxiety-like behaviors
The elevated plus maze test was conducted precisely as described previously (Etherton et al., 2009), and the dark/light test was also conducted as described previously (Powell et al., 2004; Blundell et al., 2009).
Locomotor activity
Locomotor activity was measured precisely as described previously (Powell et al., 2004; Tabuchi et al., 2007; Etherton et al., 2009).
Marble burying
Empty home cages were filled with bedding up to 5 cm from the cage floor, and 20 black marbles were placed evenly throughout the cage. Mice were allowed to freely explore the cage (and marbles) for 30 min, and afterward, the number of successfully buried marbles was counted. A marble was defined as “buried” when <25% of the marble was visible.
Grooming behavior
Observation of grooming behavior was conducted as described previously (Etherton et al., 2009).
Social behaviors
The test of social interaction with an adult caged conspecific, modified from the study by Moy et al. (2004), was conducted essentially as described previously (Kwon et al., 2006; Tabuchi et al., 2007; Blundell et al., 2009; Etherton et al., 2009), and TopScan videotracking software (version 2.00; CleverSys) was used to record mouse behavior. Social targets were caged, sex-matched 12- to 14-week-old C57BL/6J mice (The Jackson Laboratory). The test of social interaction with a freely moving juvenile (social learning) was conducted as previously described (Kogan et al., 2000; Kwon et al., 2006; Tabuchi et al., 2007; Blundell et al., 2009; Etherton et al., 2009). The test of interaction with pheromones was conducted as previously described (Etherton et al., 2009). The buried food finding test of olfaction was slightly modified from the study by Moy et al. (2007) and was conducted as previously described (Etherton et al., 2009).
Accelerating rotarod
The accelerating rotarod task was conducted essentially as previously described (Etherton et al., 2009).
Nesting behavior
Nesting behavior was observed and measured as described previously (Etherton et al., 2009).
Fear conditioning
Fear conditioning was conducted as previously described (Powell et al., 2004).
Morris water maze
The Morris water maze was conducted similar to previous descriptions (Powell et al., 2004; Tabuchi et al., 2007; Etherton et al., 2009). Two mice (one littermate pair) were discarded from this task because one member of the littermate pair was unable to swim without the risk of imminent drowning. All data were collected using Ethovision videotracking software (version 2.3.19; Noldus). The visible water maze was conducted during a single day 24 h after the Morris water maze probe trial. A visible cue (black, foam square) was placed on top of the platform, and the platform was placed in a new, random location for each trial. Mice were given a total of six trials, and data were averaged across the six trials to control for the different platform locations. All data were collected using Ethovision videotracking software (version 2.3.19; Noldus).
Auditory startle reactivity
Startle reactivity was conducted as described previously (Kwon et al., 2006; Etherton et al., 2009).
Tail suspension test
The length of each mouse's tail (starting ∼1 cm from the base of the tail) was taped to an aluminum bar, and the aluminum bar was attached to tail suspension chamber (MED Associates) so that the mouse was suspended upside down. Each mouse was suspended by its tail for 6 min. The amount of time that the mouse spent immobile was detected by MED Associates tail suspension software. For tests with HCN1−/− and HCN2ap/ap mutant lines and their respective controls, the mice were also suspended by their tails for 6 min; the sessions were recorded and the video records manually scored for immobility time by an observer blinded to genotype.
Forced swim test
Mice were gently placed in 4 L glass beakers filled with tap water to a depth at which the tail could not contact the bottom of the beaker, and they remained in the beakers for 6 min. Mice were videotaped, and the last 4 min of the test was scored manually by an observer blind to genotype. The total time spent immobile and the latency to the first immobility were measured by the observer.
Spatial recognition
Mice were placed in an open field arena (48 × 48 × 48 cm) with distinct spatial cues on each of the four walls. Mice were habituated to the spatial environment (in the absence of objects) for 5 min on 4 consecutive days. On the fifth day, mice were placed in the open field in the presence of three objects placed at the corners of the open field arena. Mice were given five baseline habituation trials (5 min each) in the presence of the objects. Forty-five minutes after the fifth habituation trial, we conducted a 5 min spatial recognition test in which one object was moved into the previously empty corner. TopScan videotracking software (version 2.00; CleverSys) was used to record mouse behavior, and experimental mice were considered to be interacting with the objects if they were physically interacting with the object or sniffing within a 1.5 cm radius of the object. The difference score was calculated by subtracting the time spent interacting with one of the unmoved objects from the time spent interacting with the moved object.
Hot plate test
The hot plate sensitivity was conducted as described previously (Blundell et al., 2009).
Sucrose preference
The sucrose preference test was conducted over 8 consecutive days, and all mice were single housed for the duration of the test. To habituate mice to drinking from new bottles, two 50 ml conical tubes (BD Biosciences) filled with tap water were placed in each mouse's home cage for 2 d. Next, the two 50 ml conical tubes were filled with 1% sucrose (w/v) in tap water, and they were placed in each mouse's home cage for 2 d to habituate the mice to the sucrose solution. After these 4 habituation days, one conical tube was filled with tap water and the other was filled with 1% sucrose (w/v) in tap water. The two bottles were placed in each mouse's home cage, and they were allowed to drink from them for 4 d. Every day throughout the sucrose preference test, the location of the two bottles was switched to control for possible side bias, and the amount of liquid consumed was measured each day.
Baseline activity
For a measure of baseline activity, mice were placed in a home cage fitted with an infrared mouse motion detector/data logger (Mouse-E-Motion; Infra-E-Motion). The data logging system was set to detect movements of mice placed in this covered chamber on a second-to-second basis, and those values were obtained over a 30 min period; data thus represent the total number of seconds when any movement took place.
Statistical analyses for behavioral studies
Statistical analyses for behavioral studies on TRIP8b−/− mice were conducted using Statistica software (version 5.5; StatSoft). Either two-way ANOVAs or three-way repeated-measures ANOVAs were conducted, as appropriate. Genotype and sex were always included as between-subjects factors, and for the repeated-measures ANOVAs, interaction target (for social interaction with a caged conspecific and three-box social interaction test), trial (for social interaction with a juvenile and rotarod), day (for Morris water maze training and sucrose preference), quadrant (for Morris water maze), platform (for Morris water maze), object (for spatial recognition), time (for nesting behavior), or bin (for locomotor activity), was used as a within-subjects factor. Additional planned comparisons were conducted using Statistica's option for contrast analysis. For HCN1−/− and HCN2ap/ap mutant lines and their respective controls, unpaired t tests were used to compare results. Data were considered statistically significant if p < 0.05.
Results
Generation of TRIP8b knock-out mice
The gene encoding TRIP8b, Pex5l, produces numerous alternatively spliced TRIP8b isoforms, each with different effects on HCN channel trafficking (Lewis et al., 2009; Santoro et al., 2009). To generate a mouse model with elimination of all TRIP8b isoforms, we used homologous recombination to disrupt exons in Pex5l that are present in all TRIP8b splice isoforms (Fig. 1A–C). By breeding chimeric offspring of ES cell-implanted mice, followed by crossing with germline Cre-expressing mice, we generated a total genomic knock-out of TRIP8b mice on a C57BL/6 background. The resulting TRIP8b−/− mouse did not express any full-length or truncated splice isoform protein or mRNA species (Fig. 1D,F) [note that TRIP8b isoform nomenclature is used here consistent with the intuitive scheme previously described by Santoro et al. (2009)]. Previous reports have demonstrated the presence of Ih in pancreatic beta cells (El-Kholy et al., 2007) and a small amount of Pex5l mRNA in pancreas (Amery et al., 2001), although the function of Ih in insulin secretion is unclear (El-Kholy et al., 2007; Zhang et al., 2009). Furthermore, TRIP8b shares significant homology with PEX5, a protein critical for peroxisomal protein import that shares some overlapping affinity for binding partners (Amery et al., 2001; Fransen et al., 2008). However, TRIP8b−/− mice had no abnormalities in weight gain, serum glucose levels, or peroxisomal metabolism (data not shown).
Deletion of TRIP8b significantly reduces Ih in CA1 pyramidal neuron somata and dendrites
To examine how Ih is altered after deletion of TRIP8b, we evaluated Ih-dependent membrane properties in both somata and dendrites of CA1 pyramidal neurons in TRIP8b−/− and TRIP8b+/+ mice. These cells express high levels of Ih, of which the majority is contributed by the HCN1 subunit (Nolan et al., 2004). We have previously demonstrated that short hairpin RNA-mediated knockdown of all TRIP8b isoforms significantly reduces native Ih from cultured hippocampal neurons (Lewis et al., 2009), suggesting that in vivo TRIP8b may also be required for proper expression of Ih. TRIP8b−/− CA1 neurons had a significantly more hyperpolarized somatic resting membrane potential compared with those in TRIP8b+/+ mice (TRIP8b+/+: −66 ± 0.6 mV, n = 19; TRIP8b−/−: −71 ± 0.7 mV, n = 29; p < 0.001, unpaired t test). Therefore, to compare the intrinsic neuronal properties between TRIP8b+/+ and TRIP8b−/− mice, all subsequent measurements were made with the membrane potential held at −70 mV. Recordings from TRIP8b−/− CA1 pyramidal neuron somata had a significantly higher input resistance (RN) and a distinct absence of the characteristic voltage sag (TRIP8b+/+: 14.23 ± 0.96%, n = 23; TRIP8b−/−: 0%, n = 30; p < 0.001, unpaired t test), indicating a significant reduction in Ih compared with TRIP8b+/+ littermate controls (Fig. 2A). The negative slope of the line, when the magnitude of voltage rebound above baseline at the end of a hyperpolarizing step pulse is plotted as a function of the steady-state voltage from which it is released, is another reliable indicator of the presence of Ih (Brager and Johnston, 2007). Consistent with reduced Ih, we found the absence of voltage rebound in TRIP8b−/− but not TRIP8b+/+ neurons (Fig. 2B). Application of the selective Ih blocker ZD7288 (20 μm) significantly increased RN and abolished voltage rebound in TRIP8b+/+ but did not affect TRIP8b−/− neurons (Fig. 2A,B), as would be expected in the absence of Ih.
Figure 2.

Deletion of TRIP8b results in functional loss of Ih in CA1 pyramidal neurons. A, Input resistance (RN) was increased in TRIP8b−/− neurons (128.23 ± 6.03 MΩ; n = 30) compared with TRIP8b+/+ (wild-type) controls (74.97 ± 3.4 MΩ; n = 23). B, Neurons from TRIP8b−/− mice had a significantly lower rebound slope (0.00 ± 0.006 mV/mV; n = 30) compared with wild-type controls (−0.14 ± 0.04 mV/mV; n = 23). The dashed box represents the portion of the traces enlarged. C, TRIP8b−/− mice displayed increased temporal summation (39.61 ± 4.31%; n = 18) compared with wild-type controls (4.75 ± 2.05%; n = 11). A–C, Inset, Average response of three traces for 500 ms current injections from −50 to +50 pA in increments of 10 pA (A), 500 ms current injections from 0 to −200 pA in increments of −20 pA (B), and five αEPSP current injections at 20 Hz (C). D, Neurons from TRIP8b−/− mice (n = 28) show no resonance (resonance frequency, fR = 1.04 ± 0.02 Hz) compared with wild-type controls (n = 23; fR = 3.47 ± 0.17 Hz). A–D, ZD7288 (20 μm) significantly altered RN (A), rebound slope (B), temporal summation (C), and fR (D), in wild-type but not TRIP8b−/− neurons. E, Plot of fR versus VM revealed that TRIP8b−/− neurons (n = 5) have a significantly lower fR at hyperpolarized voltages compared with wild-type mice (n = 5) with no significant difference at depolarized voltages. Inset, Impedance amplitude profiles for wild-type (black) and TRIP8b−/− (red) mice measured at −70 mV (left) and −30 mV (right). F, Depolarizing current injection elicited significantly more action potentials from TRIP8b−/− neurons (n = 10) compared with wild-type neurons (n = 8). In all panels, the open symbols represent individual experiments and the filled symbols represent the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
The high density of Ih in CA1 pyramidal neurons strongly influences integrative properties (Magee, 1999). To assess the effect of TRIP8b deletion on temporal summation, a train of five current pulses (modeled by an α function, αEPSPs) was used to mimic EPSPs. Five αEPSPs were injected into the soma at 20 Hz, a frequency shown to be sensitive to changes in the amount of Ih (Poolos et al., 2002). Consistent with decreased Ih, we found that TRIP8b−/− CA1 pyramidal neurons had significantly more temporal summation compared with TRIP8b+/+ controls, and ZD7288 only increased temporal summation in TRIP8b+/+ but not TRIP8b−/− neurons (Fig. 2C).
Ih can act as a resonating conductance, and changes in Ih are readily observed as changes in the resonance frequency (fR) in CA1 pyramidal neurons (Hutcheon et al., 1996; Brager and Johnston, 2007; Narayanan and Johnston, 2007). CA1 pyramidal neurons from TRIP8b−/− mice did not display any resonance compared with TRIP8b+/+ mice (Fig. 2D), and ZD7288 significantly decreased fR only in TRIP8b+/+ neurons. At −70 mV, HCN channels should be the only conductance that contributes to cell resonance (Hu et al., 2002; Narayanan and Johnston, 2007). To determine whether changes in another resonating conductance may contribute to our observations, we made measurements of fR across a range of voltages (Fig. 2E). We found that resonance in TRIP8b−/− mice was significantly lower compared with controls at all voltages hyperpolarized relative to −50 mV (where Ih dominates) with no significant difference at voltages depolarized relative to −50 mV (where IM dominates). These data demonstrate that deletion of TRIP8b results in a highly significant yet specific reduction of Ih-dependent membrane voltage properties in CA1 pyramidal neurons. Functionally, downregulation of Ih can manifest as increased excitability (Shah et al., 2004; Jung et al., 2007; Shin et al., 2008). Consistent with loss of Ih, CA1 pyramidal neurons from TRIP8b−/− mice showed higher action potential firing frequency compared with TRIP8b+/+ mice (Fig. 2F). This increase was not attributable to a significant change in action potential threshold or half-width (action potential threshold: TRIP8b+/+, −49.2 ± 1.4 mV; TRIP8b−/−, −50.5 ± 1.8 mV; p > 0.05, unpaired t test; and half-width: TRIP8b+/+, 1.03 ± 0.05 ms; TRIP8b−/−, 1.09 ± 0.07 ms; p > 0.05, unpaired t test).
As the majority of Ih is localized within distal dendrites of CA1 pyramidal neurons (Magee, 1998), our somatic recordings cannot exclude the possibility that dendritic HCN channels were affected by deletion of TRIP8b. We therefore performed whole-cell recordings from the apical dendrite of CA1 pyramidal neurons (recording distance from soma: TRIP8b+/+, 161 ± 5 μm, n = 10; TRIP8b−/−, 170 ± 4 μm, n = 11; p > 0.05, unpaired t test). Consistent with our somatic recordings, we found significant differences in all Ih-dependent parameters, specifically, input resistance, rebound slope, resonance frequency, and αEPSP summation, suggesting the absence of any functional Ih in the distal dendrites of TRIP8b−/− mice compared with TRIP8b+/+ mice (Fig. 3). Post hoc morphological analysis of reconstructed neurons from TRIP8b+/+ and TRIP8b−/− mice revealed no significant differences in total dendritic length, volume, or surface area (data not shown). Together with the data obtained from somatic recordings above, the results suggest that deletion of TRIP8b results in a significant functional loss of Ih throughout the entire CA1 pyramidal neuron. It should be noted, however, that application of 20 μm ZD7288 resulted in an increase in input resistance and αEPSP summation at 20 Hz in the TRIP8b−/− dendrites, whereas the absence of resonance and rebound voltage remained unaltered. This finding is suggestive of residual Ih in the dendrites.
Figure 3.

Functional loss of Ih because of knock-out of TRIP8b occurs in the dendrites of CA1 pyramidal neurons. A, Dendritic recordings from CA1 pyramidal neuron dendrites revealed that TRIP8b−/− mice had a significantly higher dendritic RN (102.12 ± 8.12 MΩ; n = 11) compared with TRIP8b+/+ (wild-type) controls (58.03 ± 5.05 MΩ; n = 10). B, Rebound slope measured in the dendrites of CA1 pyramidal neurons from TRIP8b−/− mice was significantly decreased (0.00 ± 0.005 mV/mV; n = 9) compared with wild-type controls (−0.22 ± 0.02 mV/mV; n = 11). The dashed box represents the portion of the traces enlarged. C, Dendritic fR in CA1 pyramidal neurons from TRIP8b−/− mice was significantly lower (1.00 Hz; n = 11) compared with wild-type controls (3.93 ± 0.41 Hz; n = 8). Inset, Impedance amplitude profiles for wild-type and TRIP8b−/− CA1 pyramidal neurons. D, CA1 pyramidal neurons from TRIP8b−/− mice showed significantly more temporal summation in the dendrites (30.56 ± 4.15%; n = 11) compared with wild-type controls (5.85 ± 3.33%; n = 8). A, B, D, Inset, Average response of three traces for 500 ms current injections from −50 to +50 pA in increments of 10 pA (A), 500 ms current injections from 0 to −200 pA in increments of −20 pA (B), and five αEPSP current injections at 20 Hz (D). A–D, ZD7288 (20 μm) significantly altered rebound slope (B) and fR (C) in wild-type but not TRIP8b−/− neurons, whereas it significantly altered RN (A) and temporal summation (D) in TRIP8b−/− as well as TRIP8b+/+ neurons. In all panels, the open symbols represent individual experiments, and the filled symbols represent the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Aberrant trafficking of HCN1 in the absence of TRIP8b
Electrophysiological results demonstrating a large decrease in functional Ih suggested a significant decrease in plasma membrane-localized HCN protein. To evaluate this hypothesis directly, we performed immunogold electron microscopy (immuno-EM) for HCN1, the most abundant and functionally important HCN subunit in hippocampal CA1 neurons (Nolan et al., 2004; Notomi and Shigemoto, 2004; Santoro et al., 2004), in TRIP8b+/+ and TRIP8b−/− mice (see Materials and Methods). We used serial section analysis (Fig. 4A–L) to quantify HCN1 density on the plasma membrane and in the cytoplasm (Fig. 4M,N), which allowed us to assess the proportion of total immunoreactivity that each compartment comprised (Fig. 4O). This approach revealed that, compared with proximal dendrites, distal dendrites of TRIP8b+/+ mice were significantly enriched in plasma membrane HCN1 (Fig. 4M,O), consistent with previous studies (Magee, 1998; Lörincz et al., 2002). Interestingly, cytoplasmic HCN1 was not enriched in the distal dendrites of TRIP8b+/+ mice compared with proximal dendrites (Fig. 4N), suggesting that the increasing Ih density and HCN1 expression gradient observed in distal dendrites of wild-type mice is primarily attributable to a gradient in plasma membrane rather than intracellular HCN1. In contrast, HCN1 membrane density (Fig. 4M) and HCN1 membrane fraction (Fig. 4O) were not significantly enriched in distal dendrites of TRIP8b−/− mice, and the total HCN1 immunoreactivity in distal dendrites of TRIP8b−/− mice was substantially decreased compared with TRIP8b+/+ mice. Together with our electrophysiological studies, this finding suggests that reduction in Ih in TRIP8b−/− mice may result from reduced HCN subunit expression on CA1 neuronal plasma membrane, and that the unique gradient of HCN expression in CA1 pyramidal neuron dendrites is disrupted in the absence of TRIP8b.
Figure 4.
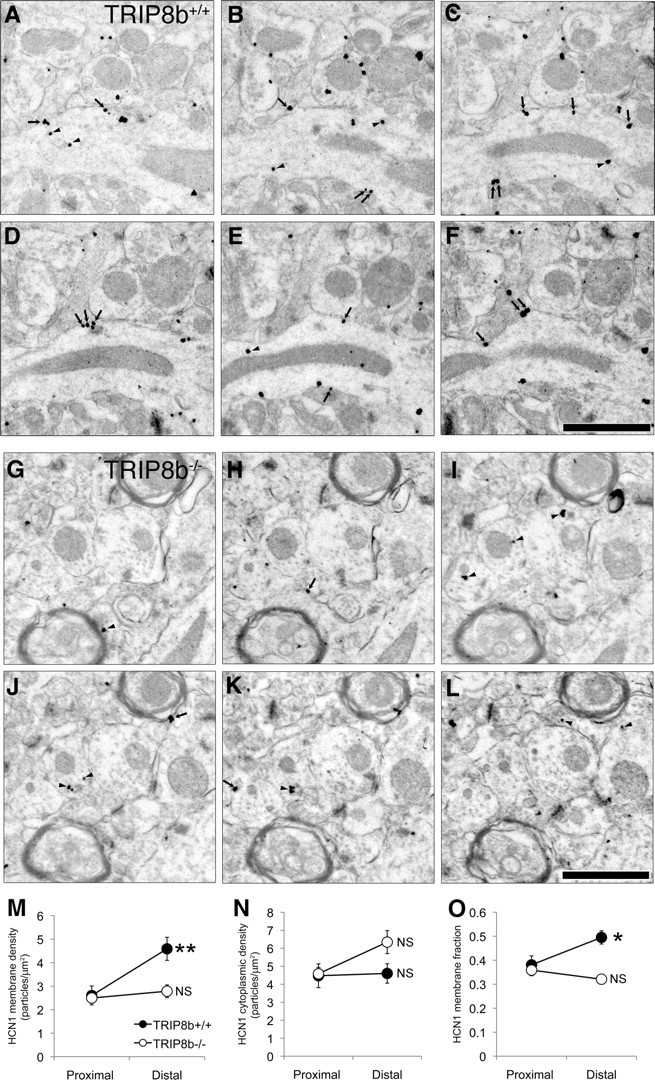
HCN1 protein density is significantly reduced on the plasma membrane of TRIP8b−/− dendrites. A–F, Serial sections through TRIP8b+/+ mouse area CA1 SLM immunostained for HCN1. HCN1 immunogold particles are found on the plasma membrane (arrows) as well as localized in the cytoplasm (arrowheads). Scale bar, 1 μm. G–L, Serial sections through TRIP8b−/− mouse area CA1 SLM immunostained for HCN1 demonstrate both significantly reduced total immunoreactivity, as well as numerous cytoplasm-localized (arrowhead) immunogold particles, although some immunogold particles are observed on the plasma membrane (arrows). Scale bar, 1 μm. Percentages of immunonegative dendrites in TRIP8b−/− mice were higher (proximal, 43%; distal, 38%) compared with TRIP8b+/+ mice (proximal, 17%; distal, 14%). M, Average HCN1 membrane particle density for TRIP8b+/+ and TRIP8b−/− proximal and distal dendrites. **p < 0.01 for TRIP8b+/+, genotype by proximal/distal interaction, F(1,151) = 8.76; multivariate analysis of covariance (MANCOVA), using dendritic diameter as covariate. N, HCN1 cytoplasmic particle density for TRIP8b+/+ and TRIP8b−/− proximal and distal dendrites. Although there was no statistically significant effect of genotype on cytoplasmic particle density, a planned comparison of TRIP8b+/+ distal and TRIP8b−/− distal cytoplasmic particle density yielded a p = 0.0586, which indicates a trend toward higher cytoplasmic immunoreactivity in the TRIP8b−/− mice. O, Quantification of fraction of HCN1 particles localized to the plasma membrane. *p < 0.05 for TRIP8b+/+, genotype by proximal/distal interaction, F(1,151) = 6.19; MANCOVA with dendritic diameter as covariate. For M–O, NS signifies p > 0.05, genotype by proximal/distal interaction, MANCOVA with dendritic diameter as covariate. For all calculations, total n = 156; TRIP8b+/+ proximal, 23; TRIP8b+/+ distal, 34; TRIP8b−/− proximal, 67; TRIP8b−/− distal, 32.
Reduction of HCN subunit proteins in TRIP8b−/− mice
To further characterize the mechanism by which Ih and plasma membrane HCN subunit expression is reduced in TRIP8b−/− mice, we next examined whether deletion of TRIP8b resulted in the loss of HCN channel subunits. Quantitative RT-PCR did not reveal any change in mRNA of Hcn1, Hcn2, Hcn3, or Hcn4, suggesting that, as expected, TRIP8b does not regulate transcription of HCN channel genes (Fig. 5A). However, unlike mRNA, HCN1 and HCN2 protein levels were both reduced by ∼40% in lysates prepared from subdissected TRIP8b−/− hippocampal area CA1 (Fig. 5B). These findings suggest a posttranslational reduction in HCN subunit levels that could result from reduced synthesis or increased degradation. Evaluating coimmunoprecipitation of HCN1 with HCN2 revealed that heteromerization of HCN1 with HCN2 was unaffected by TRIP8b deletion (Fig. 5C), suggesting that reduction of functional Ih does not result from defective channel assembly. We also found no evidence that decreased HCN1 levels resulted from impaired early trafficking through the endoplasmic reticulum or Golgi apparatus, as there was no upregulation of the unfolded protein response protein, BiP (relative BiP protein levels: TRIP8b+/+, 1.00 ± 0.27; TRIP8b−/−, 0.64 ± 0.05; n = 4; p > 0.58 unpaired t test) (Fig. 5D) (Vandenberghe et al., 2005), no change in HCN1 protein glycosylation as manifested by altered gel mobility (Fig. 5B), and HCN1 was not sequestered in Golgi (Fig. 5E). These findings suggest that HCN1 is not degraded as a result of retention within secretory pathway structures.
Figure 5.

TRIP8b deletion reduces HCN1 and HCN2 protein levels without affecting HCN1/HCN2 heteromerization, upregulation of the unfolded protein response protein BiP (Grp78), or sequestration of HCN1 protein in Golgi apparatus. A, qPCR from hippocampal mRNA shows absence of Pex5l mRNA in TRIP8b−/− mice but no significant differences in mRNA coding for HCN channel subunits (***p < 0.001, n = 3, unpaired t test). B, Immunoblots of lysates from subdissected hippocampal area CA1 show decreased HCN1 and HCN2 subunit protein in TRIP8b−/− mice (*p < 0.05, ***p < 0.001, n = 4–6, unpaired t test). C, Immunoprecipitation (IP) of HCN1 followed by immunoblotting (IB) for HCN2 (left, top panels) and IP of HCN2 followed by IB for HCN1 (left, bottom panels) from hippocampal lysates was not significantly different in TRIP8b−/− mice compared with TRIP8b+/+ mice (n = 3 mice/genotype; p > 0.10, unpaired t test). Input is 5% of total lysate immunoprecipitated. The bottom blot shows shorter exposure of top blot, used to quantify the IP fraction compared with the input quantified from top blot. NS, Not significant. D, Immunoblotting of hippocampal lysates from TRIP8b+/+ and TRIP8b−/− mice with antibodies against BiP (Grp78), TRIP8b, and α-tubulin. E, Immunohistochemistry for HCN1 (green) and GM130 (marker of Golgi apparatus; red) in CA1 stratum pyramidale from TRIP8b+/+ and TRIP8b−/− mice demonstrates no change in colocalization, suggesting TRIP8b is not required for HCN1 transit from Golgi apparatus. Scale bar, 10 μm.
HCN subunit degradation is increased in the absence of TRIP8b
HCN channel plasma membrane steady-state protein levels are regulated by dynamic trafficking to and from recycling endosomes in cardiac or heterologous cells that lack TRIP8b expression (Hardel et al., 2008). We wondered whether TRIP8b might maintain HCN channels in a cycling vesicular pool, thereby limiting trafficking of channels to late endosomes and ultimate lysosomal degradation. In this case, reduction of HCN protein in hippocampal pyramidal neurons could result from increased lysosomal degradation in the absence of TRIP8b. To explore whether lysosomal degradation contributes to reduced HCN1 protein levels in cells lacking TRIP8b, we transfected HEK293T cells with HCN1 and an isoform of TRIP8b known to enhance HCN1 surface expression, TRIP8b(1a-4), or with a TRIP8b construct that binds TRIP8b but lacks the alternatively spliced N terminus that controls surface expression (TRIP8bΔN) (Lewis et al., 2009; Santoro et al., 2009). We reasoned that if TRIP8b N-terminal interaction with endocytic sorting molecules keeps HCN1 cycling rather being degraded, then HCN1 coexpression with TRIP8b(1a-4) but not TRIP8bΔN should enhance HCN1 protein levels. However, if interaction of HCN1 with TRIP8b alone enhances HCN1 protein expression or stability, then coexpression with either TRIP8b(1a-4) or TRIP8bΔN should enhance HCN1 protein levels. We found that HCN1 protein levels are significantly increased over controls by coexpression with TRIP8b(1a-4), but not when coexpressed with TRIP8bΔN (Fig. 6A). This result suggests that binding to TRIP8b alone does not increase HCN1 stability, but rather the alternatively spliced N terminus is required for enhanced protein levels.
Figure 6.

HCN1 is targeted to lysosomes in neurons lacking TRIP8b. A, Immunoblots from HEK293T cells cotransfected with HCN1 and TRIP8b(1a-4) or TRIP8bΔN demonstrates the TRIP8b N terminus is required for increased HCN1 protein level (*p < 0.05 vs HCN1 only, one-sample t test; #p < 0.05 vs TRIP8bΔN, unpaired t test; n = 5 sets of transfections). NS, Not significant versus HCN1 only. B, HEK293T cells expressing HCN1 and GFP were treated with E-64d (10 μm) or MG132 (10 μm) and HCN1 levels analyzed by Western blot, demonstrating treatment with E-64d enhances HCN1 protein levels similarly to TRIP8b(1a-4) (*p < 0.05 vs HCN1 control, one-sample t test; #p < 0.05 vs MG132-treatment, one-way ANOVA with Tukey's post hoc test; n = 5 sets of transfections). NS, Not significant versus control. C, HEK293T cells expressing HCN1 and TRIP8b(1a-4) were treated with vehicle or E-64d, lysed, and immunoblotted, demonstrating that lysosomal blockade does not further enhance HCN1 protein levels in TRIP8b(1a-4)-expressing cells. D, Confocal images of MAP2 (blue), HCN1 (green), and Lamp1 (red) immunostaining in SLM dendrites reveals increased colocalization of HCN1 and Lamp1 in dendrites of TRIP8b−/− mice versus controls. Scale bar, 5 μm.
If HCN channel lysosomal degradation is increased in the absence of TRIP8b, then inhibitors of lysosomal proteases should increase HCN subunit protein levels. Treatment of HCN1-transiently transfected HEK293T cells with E-64d, an inhibitor of lysosomal cysteine proteases, significantly increased the level of HCN1 protein over control to a similar extent as HCN1 coexpressed with TRIP8b(1a-4) (Fig. 6B). Increased HCN1 levels from either E-64d treatment or coexpression with TRIP8b(1a-4) is likely via a similar mechanism, as E-64d treatment of cells expressing HCN1 plus TRIP8b(1a-4) did not further increase HCN1 protein levels (relative HCN1 protein level: vehicle, 1.00 ± 0.10; E-64d, 0.79 ± 0.06; n = 5; p > 0.05, unpaired t test) (Fig. 6C). Although E-64d can also inhibit nonlysosomal cysteine proteases such as calpain, that E-64d treatment enhanced HCN1 levels when expressed alone [and not when coexpressed TRIP8b(1a-4)] suggests that E-64d effects result from inhibition of lysosomal proteases rather than more ubiquitous proteases. Furthermore, treatment with an inhibitor of proteosomal degradation, MG132, did not similarly enhance HCN1 levels (Fig. 6B), suggesting that proteosomal HCN1 degradation may be less important than lysosomal degradation in heterologous cells. Thus, in vitro studies suggest that TRIP8b limits lysosomal degradation of HCN1.
Increased HCN1 trafficking to lysosomes in TRIP8b−/− mice
We next explored in vivo whether reduction of HCN subunit levels in TRIP8b−/− mice results from enhanced lysosomal degradation. We performed colocalization experiments using antibodies to HCN1, Lamp1, a lysosomal marker, and MAP2 to confirm dendritic localization. In the SLM of TRIP8b−/− mouse CA1 pyramidal neuron dendrites, we again observed decreased HCN1 staining consistent with our electron microscopy results and also found increased colocalization of HCN1 with Lamp1-positive vesicles [TRIP8b+/+, 28.54 ± 2.94%; TRIP8b−/−, 51.16 ± 4.24%; n = 37 (TRIP8b+/+) and 35 (TRIP8b−/−) from four animals of each genotype; p < 0.001, unpaired t test] (Fig. 6D). There was no significant change in density of Lamp1-positive vesicles between genotypes (TRIP8b+/+, 18.78 ± 3.43 particles/100 mm2; TRIP8b−/−, 23.50 ± 2.79/100 mm2; n = 4 animals of each genotype; p > 0.28, unpaired t test), arguing against a general increase in lysosomes in TRIP8b−/− mice. These findings suggest that the absence of TRIP8b in vivo leads to increased trafficking of the HCN1 protein to lysosomes. Next, we explored subcellular HCN1 localization by immuno-EM. Although optimization for immunolocalization leads to ultrastructural changes that preclude an unequivocal analysis of lysosomal localization of HCN immunoreactivity, tissue from TRIP8b−/− mice (but not TRIP8b+/+ mice) showed many clear instances of labeled multivesicular bodies, which represent late endosomes involved in lysosomal degradation (Fig. 7) (Von Bartheld and Altick, 2011). This observation combined with increased colocalization of HCN1 with Lamp1 together suggest that reduced HCN subunit levels in TRIP8b−/− mice result from increased targeting to and degradation by lysosomes.
Disruption of the dendritic gradient of HCN subunits in the TRIP8b−/− mice
Electrophysiological studies showed that some Ih persists in dendrites in the absence of TRIP8b, a finding supported by immuno-EM studies. Thus, it is clear that TRIP8b is not required for HCN channels to reach dendritic membrane. However, another important aspect of HCN trafficking is the dramatic enrichment of HCN channels in a proximal to distal gradient along CA1 and cortical pyramidal neurons. Immuno-EM suggested a loss of enrichment of HCN1 in distal versus proximal TRIP8b−/− CA1 pyramidal neuron dendrites (Fig. 4M,O). We sought to further evaluate the importance of TRIP8b for the expression of the HCN channel gradients. Immunofluorescence staining for HCN1 in neocortical pyramidal neurons of TRIP8b−/− mice revealed loss of characteristic distal dendritic enrichment (Fig. 8A). Furthermore, immunostaining for HCN1 and HCN2 in hippocampal area CA1 similarly demonstrated disruption of the normal HCN channel gradient (Fig. 8B) with no concurrent change in MAP2 distribution (data not shown). Together, these findings illustrate that TRIP8b is required for the distance-dependent enrichment of HCN channels in hippocampal and cortical pyramidal neuron dendrites.
Figure 8.

TRIP8b is required for expression of dendritic HCN channel gradients. A, Immunostaining reveals loss of HCN1 enrichment in TRIP8b−/− layer V neocortical pyramidal neuron dendrites. Scale bar, 50 μm. B, Hippocampal immunostaining for HCN1 or HCN2 (green) demonstrates loss of distal enrichment and decreased protein levels in TRIP8b−/− mice (**p < 0.01, ***p < 0.001, n = 3, unpaired t test). Regions are denoted by drawing a straight line perpendicular from the stratum pyramidale toward the apical (containing SR and SLM) or basal dendritic fields (containing SO), and dividing them into equal length sections. SO was divided into two sections, SP was divided into one section, SR was divided into seven sections, and SLM was divided into three sections. This method was adapted from the study by Shin and Chetkovich (2007). Scale bar, 400 μm.
TRIP8b knock-out mice show impaired motor learning, and TRIP8b, HCN1 knock-out, and apathetic mice demonstrate enhanced antidepressant behavior
Mice with complete deletion of HCN1 demonstrate altered motor learning (Nolan et al., 2003), whereas mice with a forebrain-restricted deletion of HCN1 demonstrate significantly reduced hippocampal Ih as well as improved performance in a subset of short- and long-term spatial memory tests, implying that Ih constrains some specific forms of memory (Nolan et al., 2004). TRIP8b−/− mice show an even more pronounced reduction in hippocampal Ih-dependent membrane voltage properties than do the HCN1 forebrain deletion mice, and thus provide a novel tool for understanding the general role of Ih in spatial learning and memory.
Similar to HCN1−/− mice, TRIP8b−/− mice do not exhibit widespread behavioral abnormalities and were comparable with controls in anxiety-like behavior [time in open arms (in seconds)/time in both arms (in seconds) of elevated plus maze: TRIP8b+/+, 0.16 ± 0.04; TRIP8b−/−, 0.17 ± 0.05; n = 19 littermate pairs; p > 0.05, two-way ANOVA, main effect of genotype; time in dark/light boxes (in seconds): TRIP8b+/+, 76.64 ± 9.92 s; TRIP8b−/−, 69.86 ± 12.69 s; n = 19 littermate pairs; p > 0.05, two-way ANOVA, main effect of genotype], locomotor activity (Fig. 9A, for 5 min binned beam brakes) (total beam breaks in 2 h: TRIP8b+/+, 3236.16 ± 436.00 breaks; TRIP8b−/−, 3129.00 ± 264.74 breaks; n = 19 littermate pairs; p > 0.05, two-way ANOVA, main effect of genotype), repetitive behavior (Fig. 9B for grooming behavior) (number of marbles buried during 30 min period: TRIP8b+/+, 7.21 ± 1.10 marbles; TRIP8b−/−, 6.89 ± 1.37 marbles; n = 19 littermate pairs; p > 0.05, two-way ANOVA, main effect of genotype), social behavior (Fig. 9C,D), tests of olfaction [time spent interacting with a pheromone-coated slide (in seconds): TRIP8b+/+, 11.81 ± 1.26 s; TRIP8b−/−, 11.71 ± 2.17 s; n = 19 littermate pairs; p > 0.05, two-way ANOVA, main effect of genotype; time to find a buried peanut butter cookie: TRIP8b+/+, 236.11 ± 25.02 s; TRIP8b−/−, 283.05 ± 27.59 s; n = 19 littermate pairs; p > 0.05, two-way ANOVA, main effect of genotype], a test of nociception (time to lick/shake hindpaw on hot plate test: TRIP8b+/+, 13.80 ± 1.14 s; TRIP8b−/−, 15.06 ± 1.09 s; n = 19 littermate pairs; p > 0.05, two-way ANOVA, main effect of genotype), and sucrose preference behavior (Fig. 9E). We also did not find that TRIP8b−/− mice differed from controls in behavioral tests of hippocampal-dependent spatial memory, including the Morris water maze (Fig. 10A, for water maze training trials; B, for water maze probe trial) [distance traveled over time to reach visible platform in visible water maze (in centimeters): TRIP8b+/+, 78.99 ± 7.60 cm; TRIP8b−/−, 83.98 ± 12.95 cm; n = 18 littermate pairs; p > 0.05, two-way ANOVA, main effect of genotype], contextual fear conditioning (Fig. 10C), and spatial object recognition memory [time spent interacting with object in new spatial location minus time interacting with unmoved object (difference score): TRIP8b+/+, 4.05 ± 1.34; TRIP8b−/−, 4.80 ± 1.87; n = 18 littermate pairs; p > 0.05, two-way ANOVA, main effect of genotype]. These results are consistent with those of Nolan et al. (2003) who found no difference on classical versions of the Morris water maze for mice with complete knock-out of HCN1. Rather, HCN1−/− showed short-term learning and memory changes in the Morris water maze dependent on a priming procedure (Nolan et al., 2004). However, we did observe that TRIP8b−/− mice, like HCN1−/− mice, demonstrated impaired motor learning on the accelerating rotarod task, strongly suggesting a role for Ih in motor learning (Fig. 10D) (Nolan et al., 2003). Additionally, TRIP8b−/− mice exhibited impaired nest-building behavior and decreased auditory startle responses (Fig. 11).
Figure 9.

TRIP8b−/− mice do not exhibit gross behavioral abnormalities. A, Locomotor activity. Data represent the number of photobeam breaks during 5 min bins. B, Grooming behavior. Data represent the total time spent grooming (left bars and y-axis) and the total number of individual grooming bouts (right bars and y-axis) during a 10 min period of observation. C, Social interaction with a juvenile (social learning). Experimental mice were placed in a novel cage with a juvenile mouse for 2 min (initial), and the test was repeated 3 d later (recognition) (###p < 0.001 compared with “initial,” planned comparisons, contrast analysis). D, Social interaction with a caged conspecific. Data represent the time spent interacting with an empty cage (inanimate target) or with a caged sex-matched conspecific (social target). #p < 0.05, ###p < 0.001 compared with the inanimate target, planned comparisons, contrast analysis. E, Sucrose preference test. Data represent the percentage of the total daily volume of liquid (sucrose or water) consumed that was consumed from the sucrose bottle (volume of sucrose solution consumed/total volume consumed). The dotted line represents chance performance. All data are from 19 littermate pairs.
Figure 10.

TRIP8b−/−, HCN2ap/ap, and HCN1−/− mice exhibit reduced depression-like behavior, and TRIP8b−/− mice exhibit impaired motor learning and normal hippocampal-dependent learning and memory. A, Water maze training trials. Distance traveled to reach the hidden platform over time (n = 18 littermate pairs). B, Water maze probe trial. Percentage time spent in each quadrant of the maze (n = 18 littermate pairs; #p < 0.05, ###p < 0.001 vs target quadrant, planned comparisons, contrast analysis). C, Contextual fear conditioning. Percentage time spent freezing during initial habituation period (pretrain) and during contextual memory test (context) 24 h later (n = 19 littermate pairs). D, Rotarod. Time to fall off accelerating rotating rod (n = 19 littermate pairs; *p < 0.05, ***p < 0.001 vs TRIP8b+/+, planned comparisons, contrast analysis). E, FST on TRIP8b−/− and HCN1−/− mice along with respective controls. Total time immobile [*p < 0.05, TRIP8b−/− vs TRIP8b+/+, n = 19 littermate pairs, two-way ANOVA, main effect of genotype; ***p < 0.001, HCN1−/− vs HCN1+/+, n = 14 (HCN1−/−); n = 16 (HCN1+/+), unpaired t test]. F, TST on TRIP8b−/−, HCN1−/−, and HCN2ap/ap mice along with respective controls. Total time immobile [*p < 0.05, TRIP8b−/− vs TRIP8b+/+, n = 19 littermate pairs, two-way ANOVA, main effect of genotype; *p < 0.05, HCN1−/− vs HCN1+/+, n = 15 (HCN1−/−), n = 17 (HCN1+/+), unpaired t test; ***p < 0.001, HCN2ap/ap vs HCN2+/+, n = 7 each, unpaired t test].
Figure 11.
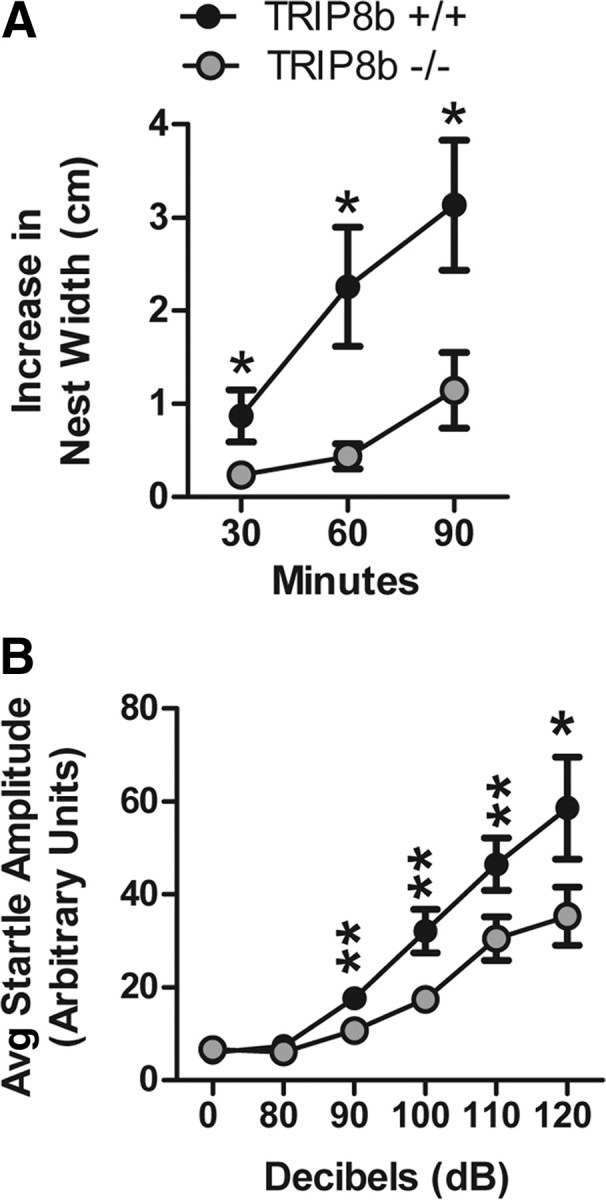
TRIP8b−/− mice exhibit impaired nest-building behavior and decreased auditory startle response. A, Nest-building behavior. Data represent the increase in nest width, measured at 30, 60, and 90 min after placing a new nestlet in the cage (n = 19 littermate pairs; *p < 0.05 compared with TRIP8b+/+, planned comparisons, contrast analysis). The key in A also applies to B. B, Auditory startle response. Data represent the mean startle amplitude of mice when presented with auditory tones of different decibel levels (n = 19 littermate pairs; *p < 0.05, **p < 0.01 compared with TRIP8b+/+, planned comparisons, contrast analysis).
We also examined TRIP8b−/− mice in the FST and the TST, two behavioral models of despair with high predictive validity for antidepressant drugs. Interestingly, in both tasks, we found that TRIP8b−/− mice spent significantly less time immobile compared with TRIP8b+/+ mice (Fig. 10E,F). Furthermore, in the FST, TRIP8b−/− mice also exhibited an increased latency to first immobility (TRIP8b+/+, 5.37 ± 2.32 s; TRIP8b−/−, 10.84 ± 2.99 s; n = 19; p < 0.05, Mann–Whitney U test). These data suggest that deletion of TRIP8b leads to a decrease in behavioral despair consistent with an antidepressant effect.
To determine whether antidepressant-like effects of TRIP8b deletion could be explained by loss of Ih, we evaluated behavioral despair in mice with reduction in Ih resulting from deletion or mutation of genes encoding either HCN1 or HCN2. Interestingly, HCN1−/− mice were significantly less immobile in both the FST and TST than wild-type littermate counterparts (Fig. 10E,F). The reduced immobility of HCN1−/− mice in both of these tests was not attributable to a difference in baseline activity (percentage of time active: HCN1+/+, 29.6 ± 1.7%, n = 8; HCN1−/−, 32.1 ± 6.1%, n = 7; p > 0.65, unpaired t test). Although HCN2ap/ap mice cannot swim and thus could not undergo the FST, in the TST HCN2ap/ap mice were significantly less immobile than their wild-type counterparts (Fig. 10F). Together, these results strongly suggest that Ih plays an important role in controlling affective behavior in mice.
Discussion
In this study, we have generated a mouse with a genetic deletion of Pex5l, the gene encoding TRIP8b, to determine whether TRIP8b is required for proper expression of Ih and HCN channels in the mouse brain. Although many distinct types of neurons express Ih, we focused our analysis on CA1 pyramidal neurons, in which Ih and HCN channel proteins have been well characterized, as well as where TRIP8b is highly expressed. The main findings of our study are that (1) deletion of TRIP8b significantly reduces evidence of functional Ih expression in CA1 pyramidal neurons, (2) loss of TRIP8b reduces HCN channels on the plasma membrane and disrupts the gradient of HCN subunit protein expression in cortical and CA1 pyramidal neuron dendrites, (3) in the absence of TRIP8b there is increased HCN channel targeting to and degradation by lysosomes, and (4) mice with reduced Ih resulting from absence of TRIP8b, HCN1, or HCN2 all demonstrate enhanced resistance in tests of behavioral despair. The importance of our study is thus in characterizing a key mechanism for expression of Ih in the hippocampus, as well as illuminating a novel potential target for development of antidepressant therapy.
Importance of TRIP8b for hippocampal Ih
Electrophysiological recordings found little evidence of functional Ih in TRIP8b−/− CA1 somata. However, in distal CA1 dendrites where Ih current densities in rats have previously been shown to be over sixfold higher than in somata (Magee, 1998), surrogates for functional Ih were dramatically reduced but not completely absent in TRIP8b−/− neurons, as the Ih blocker ZD7288 increased the input resistance and αEPSP summation. Consistent with electrophysiological studies, immuno-EM analysis for HCN1 indicated reduced but residual presence of surface-expressed HCN1 in TRIP8b−/− dendrites. Together, these findings reveal an important but not obligate role for TRIP8b in the expression of Ih in CA1 pyramidal neurons, a finding not unexpected when considering that HCN channels can be trafficked to plasma membrane in the absence of TRIP8b in heart.
HCN channels, TRIP8b, and endocytosis
Previous studies in cells lacking TRIP8b revealed that overexpressed HCN channels are endocytosed and trafficked to a Rab11-positive/Rme1-positive endocytic recycling compartment (ERC) that serves as a dynamic reservoir for HCN channel proteins (Hardel et al., 2008). We speculate that the role of TRIP8b in neurons is to maintain HCN channels in an analogous cycling vesicular pool, with the balance of surface versus intracellular trafficking in a cell influenced by expression of distinct upregulating versus downregulating TRIP8b isoforms (see model) (Fig. 12). Consistent with this model, overexpression of a downregulating isoform of TRIP8b enhances transport of surface HCN channels to EEA1-positive early endosomes (Santoro et al., 2004, 2009). We found in vitro evidence suggesting lysosomal degradation of HCN1 is reduced by coexpression with the surface upregulating TRIP8b(1a-4) (but not a mutant TRIP8b construct lacking the ability to influence HCN1 trafficking). Furthermore, our observation of reduced HCN channel protein combined with increased HCN1 colocalization with Lamp1 and presence in multivesicular bodies in distal CA1 dendrites of TRIP8b−/− mice also supports a role for TRIP8b in preventing lysosomal degradation. In addition to increased lysosomal targeting, selective loss of plasma membrane HCN channel expression in TRIP8b−/− CA1 neurons is consistent with the most abundant hippocampal TRIP8b isoforms being upregulating (Lewis et al., 2009). Additional studies are yet needed to confirm this potential role for TRIP8b as an adapter for HCN channels in a cycling compartment in neurons.
Figure 12.

Schematic model of TRIP8b function in hippocampal CA1 neurons. A, Sample CA1 pyramidal neuron. B, Schematic drawing of CA1 pyramidal neuron soma (top) and distal dendrite (bottom). HCN channels (green) are expressed in endoplasmic reticulum (ER), assembled as tetramers, sorted into Golgi (1), and then trafficked into dendrites (2) without requiring TRIP8b (red). In distal dendrites, TRIP8b facilitates targeting of HCN channels to plasma membranes by unknown mechanisms (3). C, Schematic of CA1 pyramidal neuron distal dendrite showing TRIP8b (red) colocalized with HCN channels (green) and favoring trafficking from an endocytic recycling compartment (ERC) to plasma membrane (thick arrow) (PM), and limiting trafficking to multivesicular bodies/lysosomes. D, In the TRIP8b−/− mice, HCN channels are not efficiently targeted to plasma membrane resulting in increased targeting to multivesicular bodies and degradation by lysosomes (thick arrow).
TRIP8b and the dendritic HCN channel gradient
A central question of HCN channel biology concerns the mechanism underlying the striking gradient of enrichment of HCN channels along dendrites of CA1 and neocortical pyramidal neurons. Although electrophysiological studies revealed that some HCN channel trafficking into dendrites occurred in the absence of TRIP8b, fluorescence microscopy studies revealed elimination of the HCN1 and HCN2 gradients in TRIP8b−/− CA1 pyramidal neuron dendrites (and HCN1 in cortical pyramidal neurons). Furthermore, serial immuno-EM showed that the gradient of HCN1 particles on the plasma membrane of proximal to distal CA1 pyramidal neuron dendrites is eliminated in TRIP8b−/− mice. Thus, our data showed that enrichment of HCN1 and HCN2 along dendrites, but not presence in dendrites, requires TRIP8b. Because of evidence that TRIP8b might favor trafficking of HCN channels in a cycling vesicular pool, it would be interesting to determine whether there is unique pool of vesicles (or vesicles imbued with specific proteins) found in a gradient similar to that of TRIP8b/HCN in dendrites. Regardless, studies to resolve the TRIP8b/HCN channel interactome could also help identify mechanisms by which TRIP8b controls dendritic HCN channel localization.
Behavioral similarities between TRIP8b and HCN1 knock-out mice
Despite a large battery of behavioral experiments, only a small subset of tests demonstrated behavioral alterations in TRIP8b−/− mice, arguing these changes are specifically attributable to the mutation. Mice lacking HCN1 either in the whole brain or in the forebrain also have limited behavioral changes (Nolan et al., 2003, 2004). TRIP8b−/− mice, like HCN1 forebrain knock-out mice (Nolan et al., 2004), did not differ from TRIP8b+/+ mice when tested in the Morris water maze without a priming procedure, demonstrating that deletion of TRIP8b does not profoundly alter swimming or locomotion. However, like mice containing a total body deletion of HCN1 (Nolan et al., 2003), the TRIP8b−/− mice displayed motor learning deficits on an accelerating rotarod. We have observed expression of TRIP8b mRNA in rat Purkinje cells (data not shown) and TRIP8b protein in whole cerebellar lysates (Lewis et al., 2009), but additional experimentation is required to determine whether the deficit in TRIP8b−/− mice is attributable to abnormal Ih in cerebellar Purkinje neurons (Nolan et al., 2003). Regardless, our findings overall suggest that changes in Ih because of either loss of HCN1 or loss of TRIP8 result in a limited and mostly overlapping set of behavioral consequences and are likely not attributable to the effects of battery testing.
Antidepressant-like behavior in mice lacking TRIP8b, HCN1, or HCN2
Although deletion of TRIP8b did not alter many behaviors, we surprisingly found significant resistance to multiple tests of behavioral despair in not only the TRIP8b−/− mice but also in mice that lack expression of either HCN1 or HCN2. Other than reduced Ih, there are no shared factors that easily explain the shared antidepressant-like effects in the three genetically distinct mutant mice. HCN2ap/ap mutant mice have profound deficits in locomotion (Chung et al., 2009), but TRIP8b−/− and HCN1−/− do not. Furthermore, impairment in locomotion would be expected to increase immobility on the TST or FST tests, whereas the opposite is seen in all three mutants. HCN2ap/ap mice (Chung et al., 2009) and TRIP8b−/− mice (Jaramillo et al., 2009) both demonstrate electroencephalographic findings suggestive of absence epilepsy. However, absence seizures in these mice are manifest by 1–2 s behavioral arrest episodes, events that must be correlated with EEG to be identified as abnormal. Because they are so brief (and in the case of the TRIP8b−/− mice occur less than once per 5 min), these events would not be scored in the TST or FST. Furthermore, HCN1−/− mice do not exhibit spontaneous seizures (Huang et al., 2009; Santoro et al., 2010). Overall, we reason that the shared resistance to behavioral despair phenotype observed in these three distinct mutant mice is not attributable to locomotor defects or seizures.
Despite qualitatively similar results in behavioral despair tests in the three mutants, the magnitude of the effect in the HCN1−/− mice was similar to that in the HCN2ap/ap mice, and both were greater than in the TRIP8b−/− mice. Differences in effect magnitude is not easily attributable to seizures (seen in TRIP8b−/− and HCN2ap/ap, but not HCN1−/− mice) or locomotor defects (profound in HCN2ap/ap, but not HCN1−/− or TRIP8b−/− mice). Genetic background differences are one potential explanation for differences in effect magnitude, as all three mice were on different genetic backgrounds. Another potential explanation for differences in effect magnitude in these lines is that TRIP8b is less important than HCN1 or HCN2 for expression of Ih in whatever cells are involved in affective behavior. Future experiments with all mutations on the same genetic background and with floxed HCN or TRIP8b lines and promoter-specific Cre expression will be helpful in determining how and where distinct HCN subunits and TRIP8b individually contribute to controlling affective behavior. Regardless of these and other unresolved issues, the shared phenotype in these three distinct lines of mutant mice strongly implicates Ih (as opposed to other effects ascribed to TRIP8b, HCN1, or HCN2 individually) as playing a role in some aspects of depression.
Depression and related psychiatric disorders represent extraordinary challenges for the development of therapeutics because research has been unable to pinpoint a specific and common mechanism for such illnesses. Existing drugs effective for treating depression primarily target monoamine neurotransmitters (Berton and Nestler, 2006), yet more than one-half of patients with depression do not enter full remission after treatment. Our data suggest that targeting TRIP8b/HCN channel function could lead to new agents for treatment of depression that are distinct from currently available therapies. Because TRIP8b is primarily a brain-specific protein, manipulating the interaction of TRIP8b with HCN channels may represent a method to differentially regulate Ih without side effects from disrupting cardiac Ih (Barbuti and DiFrancesco, 2008). Future research is needed to determine the most appropriate strategies for pharmacological manipulation of this interaction.
Footnotes
This work was supported by National Institutes of Health Grants NS064757 (A.S.L.), NS05595 and NS059934 (D.M.C.), AG031574 and AG017139 (D.A.N.), MH081164 and HD065290 (C.M.P.), and MH048432 (D.J.), by a Brain Research Foundation Seed Grant (D.M.C.), by a Young Investigator Award from National Alliance for Research on Schizophrenia and Depression (X.C.), and by Deutsche Forschungsgemeinschaft Grant BE4107/2-1 (R.A.B.). We thank Quratul-Ain Ismail, Robert J. Heuermann, Pujan Patel, Sara Kee, and Elena Ramos for technical support, Alfred Rademaker for statistical assistance, and Danish Jaffer for assistance with neuron reconstructions. Additionally, we are grateful to Nelson Spruston for a camera lucida drawing of a CA1 pyramidal neuron we used for illustration purposes.
References
- Amery L, Sano H, Mannaerts GP, Snider J, Van Looy J, Fransen M, Van Veldhoven PP. Identification of PEX5p-related novel peroxisome-targeting signal 1 (PTS1)-binding proteins in mammals. Biochem J. 2001;357:635–646. doi: 10.1042/0264-6021:3570635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonenkov VD, Van Veldhoven PP, Waelkens E, Mannaerts GP. Substrate specificities of 3-oxoacyl-CoA thiolase A and sterol carrier protein 2/3-oxoacyl-CoA thiolase purified from normal rat liver peroxisomes. Sterol carrier protein 2/3-oxoacyl-CoA thiolase is involved in the metabolism of 2-methyl-branched fatty acids and bile acid intermediates. J Biol Chem. 1997;272:26023–26031. doi: 10.1074/jbc.272.41.26023. [DOI] [PubMed] [Google Scholar]
- Baes M, Gressens P, Baumgart E, Carmeliet P, Casteels M, Fransen M, Evrard P, Fahimi D, Declercq PE, Collen D, van Veldhoven PP, Mannaerts GP. A mouse model for Zellweger syndrome. Nat Genet. 1997;17:49–57. doi: 10.1038/ng0997-49. [DOI] [PubMed] [Google Scholar]
- Barbuti A, DiFrancesco D. Control of cardiac rate by “funny” channels in health and disease. Ann N Y Acad Sci. 2008;1123:213–223. doi: 10.1196/annals.1420.024. [DOI] [PubMed] [Google Scholar]
- Baruscotti M, Barbuti A, Bucchi A. The cardiac pacemaker current. J Mol Cell Cardiol. 2010;48:55–64. doi: 10.1016/j.yjmcc.2009.06.019. [DOI] [PubMed] [Google Scholar]
- Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci. 2006;7:137–151. doi: 10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- Blundell J, Tabuchi K, Bolliger MF, Blaiss CA, Brose N, Liu X, Südhof TC, Powell CM. Increased anxiety-like behavior in mice lacking the inhibitory synapse cell adhesion molecule neuroligin 2. Genes Brain Behav. 2009;8:114–126. doi: 10.1111/j.1601-183X.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH, Johnston D. Plasticity of intrinsic excitability during long-term depression is mediated through mGluR-dependent changes in Ih in hippocampal CA1 pyramidal neurons. J Neurosci. 2007;27:13926–13937. doi: 10.1523/JNEUROSCI.3520-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WK, Shin M, Jaramillo TC, Leibel RL, LeDuc CA, Fischer SG, Tzilianos E, Gheith AA, Lewis AS, Chetkovich DM. Absence epilepsy in apathetic, a spontaneous mutant mouse lacking the h channel subunit, HCN2. Neurobiol Dis. 2009;33:499–508. doi: 10.1016/j.nbd.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Kholy W, MacDonald PE, Fox JM, Bhattacharjee A, Xue T, Gao X, Zhang Y, Stieber J, Li RA, Tsushima RG, Wheeler MB. Hyperpolarization-activated cyclic nucleotide-gated channels in pancreatic beta-cells. Mol Endocrinol. 2007;21:753–764. doi: 10.1210/me.2006-0258. [DOI] [PubMed] [Google Scholar]
- Etherton MR, Blaiss CA, Powell CM, Südhof TC. Mouse neurexin-1α deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci U S A. 2009;106:17998–18003. doi: 10.1073/pnas.0910297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen M, Amery L, Hartig A, Brees C, Rabijns A, Mannaerts GP, Van Veldhoven PP. Comparison of the PTS1- and Rab8b-binding properties of Pex5p and Pex5Rp/TRIP8b. Biochim Biophys Acta. 2008;1783:864–873. doi: 10.1016/j.bbamcr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- Hardel N, Harmel N, Zolles G, Fakler B, Klöcker N. Recycling endosomes supply cardiac pacemaker channels for regulated surface expression. Cardiovasc Res. 2008;79:52–60. doi: 10.1093/cvr/cvn062. [DOI] [PubMed] [Google Scholar]
- Hellemans KH, Hannaert JC, Denys B, Steffensen KR, Raemdonck C, Martens GA, Van Veldhoven PP, Gustafsson JA, Pipeleers D. Susceptibility of pancreatic beta cells to fatty acids is regulated by LXR/PPARalpha-dependent stearoyl-coenzyme A desaturase. PLoS One. 2009;4:e7266. doi: 10.1371/journal.pone.0007266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J Physiol. 2002;545:783–805. doi: 10.1113/jphysiol.2002.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Walker MC, Shah MM. Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J Neurosci. 2009;29:10979–10988. doi: 10.1523/JNEUROSCI.1531-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulshagen L, Krysko O, Bottelbergs A, Huyghe S, Klein R, Van Veldhoven PP, De Deyn PP, D'Hooge R, Hartmann D, Baes M. Absence of functional peroxisomes from mouse CNS causes dysmyelination and axon degeneration. J Neurosci. 2008;28:4015–4027. doi: 10.1523/JNEUROSCI.4968-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheon B, Miura RM, Puil E. Subthreshold membrane resonance in neocortical neurons. J Neurophysiol. 1996;76:683–697. doi: 10.1152/jn.1996.76.2.683. [DOI] [PubMed] [Google Scholar]
- Jaramillo TC, Ying SW, Lewis AS, Popov A, Goldstein PA, Chetkovich DM. TRIP8b knockout mice exhibit reduced thalamocortical Ih and pure absence epilepsy. Soc Neurosci Abstr. 2009;35:716.7. [Google Scholar]
- Jung S, Jones TD, Lugo JN, Jr, Sheerin AH, Miller JW, D'Ambrosio R, Anderson AE, Poolos NP. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan JH, Frankland PW, Silva AJ. Long-term memory underlying hippocampus-dependent social recognition in mice. Hippocampus. 2000;10:47–56. doi: 10.1002/(SICI)1098-1063(2000)10:1<47::AID-HIPO5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci. 2006;7:548–562. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- Lewis AS, Schwartz E, Chan CS, Noam Y, Shin M, Wadman WJ, Surmeier DJ, Baram TZ, Macdonald RL, Chetkovich DM. Alternatively spliced isoforms of TRIP8b differentially control h channel trafficking and function. J Neurosci. 2009;29:6250–6265. doi: 10.1523/JNEUROSCI.0856-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AS, Estep CM, Chetkovich DM. The fast and slow ups and downs of HCN channel regulation. Channels (Austin) 2010;4:215–231. doi: 10.4161/chan.4.3.11630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lörincz A, Notomi T, Tamás G, Shigemoto R, Nusser Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci. 2002;5:1185–1193. doi: 10.1038/nn962. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:508–514. doi: 10.1038/12229. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP, Barbaro JR, Wilson LM, Threadgill DW, Lauder JM, Magnuson TR, Crawley JN. Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav Brain Res. 2007;176:4–20. doi: 10.1016/j.bbr.2006.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Johnston D. Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron. 2007;56:1061–1075. doi: 10.1016/j.neuron.2007.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, Morozov A. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell. 2003;115:551–564. doi: 10.1016/s0092-8674(03)00884-5. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- Powell CM, Schoch S, Monteggia L, Barrot M, Matos MF, Feldmann N, Südhof TC, Nestler EJ. The presynaptic active zone protein RIM1alpha is critical for normal learning and memory. Neuron. 2004;42:143–153. doi: 10.1016/s0896-6273(04)00146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routh BN, Johnston D, Harris K, Chitwood RA. Anatomical and electrophysiological comparison of CA1 pyramidal neurons of the rat and mouse. J Neurophysiol. 2009;102:2288–2302. doi: 10.1152/jn.00082.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Wainger BJ, Siegelbaum SA. Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. J Neurosci. 2004;24:10750–10762. doi: 10.1523/JNEUROSCI.3300-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Piskorowski RA, Pian P, Hu L, Liu H, Siegelbaum SA. TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channels in the brain. Neuron. 2009;62:802–813. doi: 10.1016/j.neuron.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Lee JY, Englot DJ, Gildersleeve S, Piskorowski RA, Siegelbaum SA, Winawer MR, Blumenfeld H. Increased seizure severity and seizure-related death in mice lacking HCN1 channels. Epilepsia. 2010;51:1624–1627. doi: 10.1111/j.1528-1167.2010.02554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Chetkovich DM. Activity-dependent regulation of h channel distribution in hippocampal CA1 pyramidal neurons. J Biol Chem. 2007;282:33168–33180. doi: 10.1074/jbc.M703736200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Simkin D, Suyeoka GM, Chetkovich DM. Evaluation of HCN2 abnormalities as a cause of juvenile audiogenic seizures in Black Swiss mice. Brain Res. 2006;1083:14–20. doi: 10.1016/j.brainres.2006.01.102. [DOI] [PubMed] [Google Scholar]
- Shin M, Brager D, Jaramillo TC, Johnston D, Chetkovich DM. Mislocalization of h channel subunits underlies h channelopathy in temporal lobe epilepsy. Neurobiol Dis. 2008;32:26–36. doi: 10.1016/j.nbd.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsay D, Dudman JT, Siegelbaum SA. HCN1 channels constrain synaptically evoked Ca2+ spikes in distal dendrites of CA1 pyramidal neurons. Neuron. 2007;56:1076–1089. doi: 10.1016/j.neuron.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe W, Nicoll RA, Bredt DS. Interaction with the unfolded protein response reveals a role for stargazin in biosynthetic AMPA receptor transport. J Neurosci. 2005;25:1095–1102. doi: 10.1523/JNEUROSCI.3568-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Veldhoven PP, Mannaerts GP. Inorganic and organic phosphate measurements in the nanomolar range. Anal Biochem. 1987;161:45–48. doi: 10.1016/0003-2697(87)90649-x. [DOI] [PubMed] [Google Scholar]
- Van Veldhoven PP, Van Rompuy P, Fransen M, De Béthune B, Mannaerts GP. Large-scale purification and further characterization of rat pristanoyl-CoA oxidase. Eur J Biochem. 1994;222:795–801. doi: 10.1111/j.1432-1033.1994.tb18926.x. [DOI] [PubMed] [Google Scholar]
- Von Bartheld CS, Altick AL. Multivesicular bodies in neurons: distribution; protein content; and trafficking functions. Prog Neurobiol. 2011;93:313–340. doi: 10.1016/j.pneurobio.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Site independence of EPSP time course is mediated by dendritic Ih in neocortical pyramidal neurons. J Neurophysiol. 2000;83:3177–3182. doi: 10.1152/jn.2000.83.5.3177. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu Y, Qu J, Hardy A, Zhang N, Diao J, Strijbos PJ, Tsushima R, Robinson RB, Gaisano HY, Wang Q, Wheeler MB. Functional characterization of hyperpolarization-activated cyclic nucleotide-gated channels in rat pancreatic beta cells. J Endocrinol. 2009;203:45–53. doi: 10.1677/JOE-09-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolles G, Wenzel D, Bildl W, Schulte U, Hofmann A, Müller CS, Thumfart JO, Vlachos A, Deller T, Pfeifer A, Fleischmann BK, Roeper J, Fakler B, Klöcker N. Association with the auxiliary subunit PEX5R/Trip8b controls responsiveness of HCN channels to cAMP and adrenergic stimulation. Neuron. 2009;62:814–825. doi: 10.1016/j.neuron.2009.05.008. [DOI] [PubMed] [Google Scholar]