

Abstract
Nicotine stimulates the activity of mesolimbic dopamine neurons, which is believed to mediate the rewarding and addictive properties of tobacco use. Accumulating evidence suggests that the endocannabinoid system might play a major role in neuronal mechanisms underlying the rewarding properties of drugs of abuse, including nicotine. Here, we investigated the modulation of nicotine effects by the endocannabinoid system on dopamine neurons in the ventral tegmental area with electrophysiological techniques in vivo and in vitro. We discovered that pharmacological inhibition of fatty acid amide hydrolase (FAAH), the enzyme that catabolizes fatty acid ethanolamides, among which the endocannabinoid anandamide (AEA) is the best known, suppressed nicotine-induced excitation of dopamine cells. Importantly, this effect was mimicked by the administration of the FAAH substrates oleoylethanolamide (OEA) and palmitoylethanolamide (PEA), but not methanandamide, the hydrolysis resistant analog of AEA. OEA and PEA are naturally occurring lipid signaling molecules structurally related to AEA, but devoid of affinity for cannabinoid receptors. They blocked the effects of nicotine by activation of the peroxisome proliferator-activated receptor-α (PPAR-α), a nuclear receptor transcription factor involved in several aspects of lipid metabolism and energy balance. Activation of PPAR-α triggered a nongenomic stimulation of tyrosine kinases, which might lead to phosphorylation and negative regulation of neuronal nicotinic acetylcholine receptors. These data indicate for the first time that the anorexic lipids OEA and PEA possess neuromodulatory properties as endogenous ligands of PPAR-α in the brain and provide a potential new target for the treatment of nicotine addiction.
Keywords: dopamine neurons, nicotine, electrophysiology, endocannabinoids, fatty acid amide hydrolase, patch clamp, peroxisome proliferator-activated receptor
Introduction
Nicotine is the main active component in tobacco smoke, which initiates and sustains tobacco addiction. Hence, nicotine induces drug-seeking behavior in animals and many additional effects commonly seen with addictive drugs (Stolerman and Shoaib, 1991). Among these effects, stimulation of mesolimbic dopamine (DA) transmission is considered to be one of the hallmarks to define the addicting potential of nicotine, being one of the common features of all abused drugs (Di Chiara and Imperato, 1988; Wise, 2004). By acting at neuronal nicotinic acetylcholine receptors (nAChRs), nicotine activates ventral tegmental area (VTA) DA neurons (Mereu et al., 1987; Pidoplichko et al., 1997) and induces DA release in the nucleus accumbens (Di Chiara and Imperato, 1988).
Among medications aimed at achieving smoking cessation, antagonists at the cannabinoid type-1 (CB1) receptors show promise, based on preclinical data indicating that these compounds, such as rimonabant (SR141716A) or AM251, reduce nicotine self-administration or conditioned place preference (CPP) (Cohen et al., 2002; Le Foll and Goldberg, 2004; Forget et al., 2005; Shoaib, 2008), nicotine-induced DA release in the nucleus accumbens (Cohen et al., 2002; Cheer et al., 2007), or smoking cessation in humans (Cahill and Ussher, 2007). These data strongly point to a facilitatory effect of the endocannabinoid system in the motivational and DA-releasing properties of nicotine. Indeed, endogenous cannabinoids, such as arachidonoylethanolamide (anandamide, AEA) and 2-arachidonoylglycerol, and CB1 receptors are involved in the neuronal mechanisms underlying the rewarding effects of most drugs of abuse, including nicotine (Castañé et al., 2005; Le Foll and Goldberg, 2005; Le Foll et al., 2008; Solinas et al., 2008).
The present study was designed to investigate modulation of nicotine effects by the endocannabinoid system, and to clarify the role of DA neurons in the mediation of the antiaddicting properties of CB1 antagonists. To this aim, the electrophysiological responses of DA neurons to nicotine administration were studied following either blockade of CB1 receptors or, conversely, enhancement of brain endocannabinoid levels by inhibiting fatty acid amide hydrolase (FAAH) (Kathuria et al., 2003; Fegley et al., 2005), the major hydrolyzing enzyme for AEA and other endogenous fatty acid ethanolamides (FAEs), such as the N-acylamines oleoylethanolamide (OEA) and palmitoylethanolamide (PEA). Unlike AEA, both OEA and PEA have no affinity for cannabinoid receptors, but bind to the peroxisome proliferator-activated receptor (PPAR), a family of nuclear receptor transcription factors (Fu et al., 2003; Lo Verme et al., 2005). Three subtypes of PPARs (α, β/δ and γ) play important roles in lipid metabolism, insulin sensitivity, glucose homeostasis and inflammation (Berger and Moller, 2002). Through PPAR-α, OEA and PEA are peripherally acting satiety signals that modulate feeding, body weight and lipid metabolism (Rodriguez de Fonseca et al., 2001; Fu et al., 2003) and have antinociceptive effects in visceral and inflammatory pain models (Lo Verme et al., 2005; D'Agostino et al., 2007; Suardiaz et al., 2007).
We discovered that, contrary to our expectations, enhancement of brain FAE levels, rather than blockade of CB1 receptors, inhibited the responses of DA neurons to nicotine. More importantly, the noncannabinoid FAEs OEA and PEA play a novel and unsuspected role, as PPAR-α agonists, in the negative regulation of neuronal responses to nicotine.
Materials and Methods
Electrophysiology: single unit recordings.
We performed the experiments in strict accordance with the Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research (National Research Council 2004) and EEC Council Directive of 24 November 1986 (86/609). We made all efforts to minimize pain and suffering and to reduce the number of animals used.
Male Sprague Dawley rats (250–350 g) (Harlan) were housed in groups of three to six in standard conditions of temperature and humidity under a 12 h light/dark cycle (with lights on at 7:00 A.M.) with food and water available ad libitum.
Animals were anesthetized with urethane (1300 mg/kg, i.p.), their femoral vein was cannulated for intravenous administration of pharmacological agents, and they were placed in the stereotaxic apparatus (Kopf) with their body temperature maintained at 37 ± 1°C by a heating pad. Thereafter, the scalp was retracted and one burr hole was drilled above the VTA (−6.0 mm anteroposterior from bregma, 0.3–0.6 mm lateral from midline) for the placement of a recording electrode. For intracerebroventricular drug administration, a guide cannula (23 gauge stainless steel) was placed into the ventricle ipsilateral to the recording side (1.0 mm posterior, 1.4 mm lateral to bregma and 4.0 mm ventral to the cortical surface). Structures were localized according to the stereotaxic atlas of Paxinos and Watson (1997). Intracerebroventricular injections were made through a prefilled inner cannula (30 gauge stainless steel tubing) connected to a 50 μl Hamilton microsyringe and extending 1.0 mm below the tip of the guide into the ventricle. Infusion rate was set at 2.5 μl/min by an electrically driven mini-pump.
Single unit activity of neurons located in the VTA (V 7.0–8.0 mm from the cortical surface) was recorded extracellularly with glass micropipettes filled with 2% pontamine sky blue dissolved in 0.5 m sodium acetate (impedance 2–5 MΩ). Single unit activity was filtered (bandpass 500–5000 Hz) and individual spikes were isolated by means of a window discriminator (Digitimer), displayed on a digital storage oscilloscope (TDS 3012, Tektronics) and digitally recorded. Experiments were sampled on line and off line with Spike2 software (Cambridge Electronic Design) by a computer connected to CED 1401 interface (Cambridge Electronic Design).
Single units were isolated and identified according to already published criteria (Grace and Bunney, 1983, 1984; Ungless et al., 2004). Since only one cell was recorded per rat, VTA DA neurons were selected when all criteria for identification were fulfilled: firing rate <10 Hz, duration of action potential >2.5 ms, inhibitory responses to hindpaw pinching. Bursts were defined as the occurrence of two spikes at an interspike interval <80 ms, and terminated when the interspike interval exceeded 160 ms (Grace and Bunney, 1983).
At the end of each recording section, direct current (10 μA for 15 min) was passed through the recording electrode to eject Pontamine sky blue, which allowed the identification of the recorded cells. Brains were removed and fixed in 8% formalin solution. The position of the electrodes was microscopically identified on serial sections (60 μm) stained with cresyl violet.
Electrophysiology: patch-clamp recordings.
The preparation of VTA slices was as described previously (Johnson and North, 1992). Briefly, male Sprague Dawley rats (10–28 d) were anesthetized with halothane in a vapor chamber and killed by decapitation. A block of tissue containing the midbrain was rapidly dissected and sliced in the horizontal plane (300 μm) with a vibratome (Leica VT1000S) in ice-cold low-Ca2+ solution containing (in mm): 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 0.625 CaCl2, 18 NaHCO3, and 11 glucose. Slices were transferred to a holding chamber with artificial CSF (ACSF, 37°C) saturated with 95% O2 and 5% CO2 containing (in mm): 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 18 NaHCO3, and 11 glucose. Slices were allowed to recover for at least 1 h before being placed in the recording chamber and superfused with the ACSF (37°C) saturated with 95% O2 and 5% CO2. Cells were visualized with an upright microscope with infrared illumination (Axioskop FS 2 plus, Zeiss), and whole-cell patch-clamp recordings were made by using an Axopatch 200B amplifier (Axon Instruments). Both voltage- and current-clamp experiments were made with electrodes filled with a solution containing the following (in mm): 117 KCl 144, 10 HEPES, BAPTA 3.45, CaCl 1, 2.5 Mg2ATP, and 0.25 Mg2GTP (pH 7.2–7.4, 275–285 mOsm). Experiments were begun only after series resistance had stabilized (typically 15–40 MΩ). Series and input resistance were monitored continuously on-line with a 5 mV depolarizing step (25 ms). Data were filtered at 2 kHz, digitized at 10 kHz, and collected on-line with acquisition software (pClamp 8.2, Axon Instruments). DA neurons from the posterior VTA were identified by the presence of a large Ih current (Johnson and North, 1992) that was assayed immediately after break-in, using a series of incremental 10 mV hyperpolarizing steps from a holding potential of −70 mV. Each slice received only a single drug exposure.
Drugs.
Nicotine ((−)-nicotine hydrogen tartrate) was purchased from Sigma. OEA, PEA, methanandamide (mAEA), AM281, AM251, capsazepine, WY14643, MK886, genistein, PP2 were purchased from Tocris. Rimonabant (SR141716A) was a generous gift of Sanofi-Aventis Recherche (Montpellier). Nicotine was diluted in saline (pH = 7). For i.c.v. administration, OEA or mAEA were dissolved in 40% w/v 2-hydroxypropyl-β-cyclodextrin. mAEA for i.v. injections was dissolved in 2% Tween 80 and 2% ethanol and then diluted in saline. Rimonabant was emulsified in 1% Tween 80, then diluted in saline solution and sonicated. URB597 and MK886 were dissolved in DMSO (100 μg/μl) and diluted to the final concentration in saline. All drugs for patch-clamp experiments were dissolved in DMSO as stock solutions and then dilute to the final volume in ACSF (final concentration <0.01%).
Statistical analysis.
Drug-induced changes in firing rate and pattern were calculated by averaging the effects after drug administration (2 min or 30 s bins for in vivo and in vitro electrophysiology, respectively) and normalizing to the predrug baseline. All the numerical data are given as mean ± SEM. Data were compared and analyzed by using two-way ANOVA for repeated measures (treatment × time), or one-way ANOVA or Student's t test for repeated measures, when appropriate. Post hoc multiple comparisons were made using the Dunnett's test. Statistical analysis was performed by means of the NCSS program. The significance level was established at p < 0.05.
Results
Effects of rimonabant and URB597 on activation of VTA dopamine neurons by nicotine in vivo
We recorded the activity of VTA DA neurons in urethane anesthetized rats. Cells were recorded only when they fulfilled all established criteria for VTA DA neuron identification (see Materials and Methods). A typical DA neuron broad waveform is shown in Figure 1A. Consistent with previous in vivo studies (Mereu et al., 1987; Erhardt et al., 2002), nicotine (0.2 mg/kg, i.v.) enhanced firing rate of VTA DA neurons to 144.2 ± 24.2% of baseline and burst firing to + 10.6 ± 3.8% (F(5,71) = 4.06, n = 23, p < 0.05; F(5,71) = 2.89, n = 23, p < 0.05, respectively, one-way ANOVA for repeated measure) (Fig. 1B–D). As previously reported, nicotine-induced excitation was short lasting, being significant 2 and 4 min following administration (Dunnett's post hoc). Injections of all the different vehicles (either intraperitoneal, intravenous, or intracerebroventricular) that were used in all subsequent experiments neither significantly changed the discharge activity of DA neurons nor modulated the effect of the subsequent nicotine administration. Therefore, all control experiments with nicotine were pooled.
Figure 1.
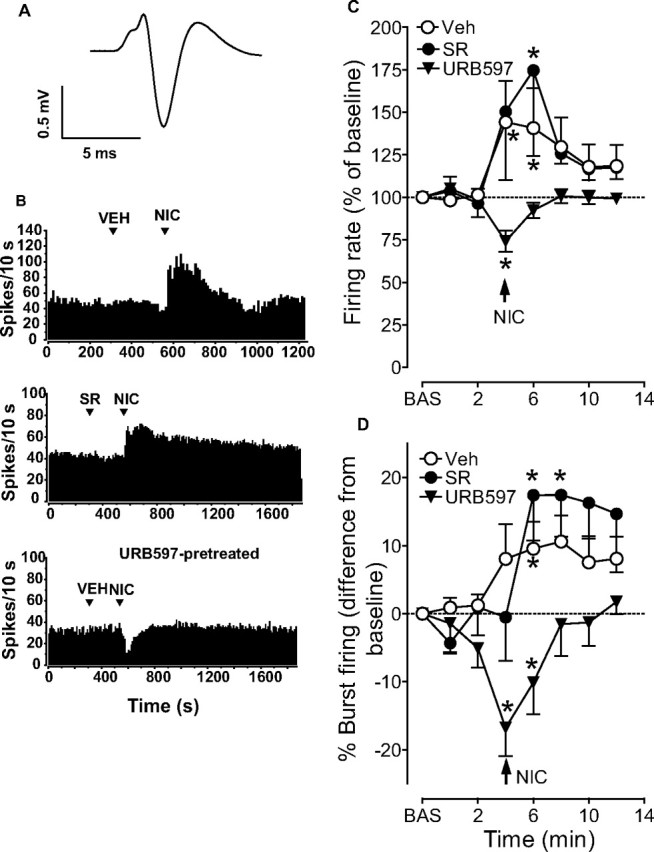
Effects of rimonabant and URB597 on activation of VTA dopamine neurons by nicotine. A, Average trace, acquired from a digital storage oscilloscope, showing the typical, broad, notched waveform of a VTA dopamine neuron recorded from an anesthetized rat. B, Representative firing rate histograms showing effects of intravenous nicotine (NIC, injected at arrowheads) on discharge activity of individual VTA dopamine neurons recorded from anesthetized rats. The top panel shows the typical response to 0.2 mg/kg nicotine in control conditions following intravenous injection of vehicle (VEH). The middle panel shows the lack of effect of rimonabant (SR, 0.5 mg/kg, i.v.) on spontaneous firing rate of dopamine neurons and on the subsequent effects of nicotine. The bottom panel shows the effect of nicotine in a URB597 pretreated animal, where nicotine induced a transient inhibition of firing activity. C, D, Graphs illustrating the time course of nicotine's effects on firing rate and burst firing of VTA dopamine neurons. Pretreatment with URB597 (0.1 mg/kg, i.v.), but not rimonabant (0.5 mg/kg, i.v.), prevented the nicotine-induced increases in firing rate (C) and burst firing (D) of VTA dopamine neurons. Results are means, with vertical bars representing the SEM of firing rates and burst firing, expressed as a percentage of or difference from baseline (BAS) values. Arrows represent time of nicotine injection. *p < 0.05 versus baseline (one-way ANOVA for repeated measures and Dunnett's test).
We first studied the effect of CB1 receptor blockade on nicotine-induced excitation of DA neurons. Rimonabant (SR141716A, 0.5 mg/kg, i.v. 4 min before nicotine) did not modify the excitatory response of DA neurons to nicotine compared with vehicle (F(1,103) = 1.10, n = 11, p = 0.3, two-way ANOVA for repeated measures) (Fig. 1B–D). Next, we investigated the effect of enhancement of endogenous endocannabinoid tone. Rats were pretreated between 60 and 120 min (average 72.4 min) before electrophysiological recordings with URB597 (0.1 mg/kg, i.v.), an irreversible FAAH inhibitor. The interval between URB597 administration and recordings varied among experiments, nevertheless, this dose of URB597 is within the range of doses shown to enhance persistently (>6 h) brain AEA levels (Kathuria et al., 2003; Fegley et al., 2005), with maximal effects 1–2 h following administration (Fegley et al., 2005). URB597 did not change spontaneous baseline firing rate (3.7 ± 0.26 Hz, n = 48, in control animals; 3.9 ± 0.14, n = 14, in URB-pretreated animals, p = 0.36, Student's t test) or burst firing (22.6 ± 3.64% of spikes in bursts, n = 48, in control animals; 23.0 ± 5.15%, n = 14; in URB-pretreated animals, p = 0.96, Student's t test) of VTA DA neurons assessed before nicotine administration, but, unexpectedly, it completely prevented nicotine's effects on DA neuronal activity (74.1 ± 6.2% and −16.8 ± 4.1% of baseline, for firing rate and burst firing, respectively) (Fig. 1B,C,D). Two-way ANOVA showed a highly significant effect of URB597 treatment on the effects of nicotine on firing rate (F(1,95) = 4.95, n = 6, p < 0.01) and burst firing (F(1,107) = 7.98, n = 6, p < 0.01). Hence, following URB597 pretreatment, nicotine transiently inhibited, rather than excited, firing rate and burst firing of DA neurons (F(3,23) = 3.73, p < 0.001, n = 6; F(3,23) = 9.44, p < 0.01, one-way ANOVA for repeated measures) (Fig. 1B,C,D).
Contribution of CB1 receptors and PPAR-α to URB597 antagonism of nicotine effects in vivo
URB597 has been shown to increase not only AEA, but also OEA and PEA levels (Kathuria et al., 2003). We first investigated whether AEA mediates URB597's antagonism of nicotine effects by acting at CB1 receptors. For this purpose, we administered the CB1 antagonist rimonabant together with URB597. Rimonabant (0.5 mg/kg, i.v. 1 min before URB597) significantly reduced the ability of URB597 to block nicotine-induced stimulation of DA neuron discharge rate (123.7 ± 11.5% of baseline), but not of burst firing (−18.4 ± 8.0% of baseline) (F(1,23) = 11.04, n = 9, p < 0.01; F(1,23) = 2.30, n = 9, p = 0.2, respectively, two-way ANOVA for repeated measures) (Fig. 2A,B). The CB1 receptor antagonist AM251 (1.0 mg/kg, i.v. 1 min before URB597, n = 5) fully replicated the effects of rimonabant (Fig. 2A,B) (firing rate: F(1,61) = 10.96, n = 6, p < 0.01; burst firing: F(1,75) = 0.00, n = 6, p = 0.9, two-way ANOVA for repeated measures). This dissociation between URB597 effects on nicotine-induced increases in firing rate and burst firing was further analyzed by assessing the contribution of the noncannabinoid FAEs (OEA and PEA), which are ligands at PPAR-α (Fu et al., 2003). We asked whether the effect of URB597 could be antagonized by the synthetic selective PPAR-α antagonist MK886 (3 mg/kg, i.p.). MK886 significantly prevented URB597 from altering nicotine-induced stimulation of bursting (+10.5 ± 3.5% of baseline) (F(1,72) = 4.90, n = 13, p < 0.05, two-way ANOVA for repeated measures) (Fig. 2B), but not firing rate (F(1,75) = 0.0, n = 13, p = 0.95, two-way ANOVA for repeated measures) (Fig. 2A). These results suggest that diverse FAEs may modulate nicotine effects on DA neurons through different mechanisms.
Figure 2.

Contribution of CB1 receptors and PPAR-α to URB597's reversal of nicotine's effects in vivo. A, The CB1 receptor antagonists rimonabant (SR, 0.5 mg/kg, i.v.) or AM251 (AM, 1.0 mg/kg, i.v.) reversed URB597's blockade of nicotine-induced increases in firing rate of VTA dopaminergic neurons produced by nicotine (NIC, 0.2 mg/kg), whereas the PPAR-α antagonist MK886 (MK, 3 mg/kg, i.p.) was ineffective. B, In contrast, MK886 administration reversed URB597's blockade of nicotine-induced increase in bursting activity of VTA dopamine neurons, whereas rimonabant and AM251 were ineffective. Results are means, with vertical bars representing the SEM of firing rates and burst firing, expressed as a percentage of or difference from baseline (BAS) values. Arrows represent time of nicotine injection. *p < 0.05 versus baseline (one-way ANOVA for repeated measures and Dunnett's test).
Oleoylethanolamide blocks nicotine effects in vivo via PPAR-α
To determine the precise contribution of either CB1 or PPAR-α receptors in the observed effects, we assessed whether mAEA, the metabolically stable analog of AEA, and OEA modulated the response to nicotine of VTA DA neurons. mAEA was administered intravenously at doses of 1 and 5 mg/kg (n = 6 each group) (Fig. 3A,B), or i.c.v. at a dose of 5 μg/5 μl (n = 6) (Fig. 3B) 4 min before nicotine administration. These doses, which exert CB1 receptor-mediated behavioral effects in vivo (Solinas et al., 2006, 2007), did not affect either baseline firing rate or burst firing of DA neurons or modulate the excitatory response to nicotine administration, compared with vehicle (F(3,234) = 0.68, n = 6, p = 0.57, two-way ANOVA for repeated measures). Due to the poor metabolic stability of OEA, we chose to administer it (20 μg/5 μl), or a corresponding volume of vehicle (40% w/v 2-hydroxypropyl-β-cyclodextrin), into the lateral ventricle 4 min before nicotine. In contrast to mAEA, OEA completely prevented the activation of DA neurons induced by nicotine (92.7 ± 13.5% of baseline at 2 min postnicotine) (Fig. 3C,D), whereas vehicle injection was inactive (n = 6, data not shown). Two-way ANOVA showed a significant effect of OEA treatment on nicotine-induced stimulation of firing rate and burst firing (F(1,99) = 5.61, n = 6, p < 0.05; F(1,107) = 4.28, n = 6, p < 0.05, respectively, two-way ANOVA for repeated measures). Neither OEA nor vehicle produced significant changes in the spontaneous firing rate or burst firing of DA neurons (Fig. 3C,D). Next, MK886 pretreatment (3 mg/kg, i.p., 30 min before recordings) prevented the blockade by OEA of nicotine's excitatory effects (122.8 ± 7.2% of baseline at 4 min postnicotine), when compared with OEA alone (F(1,55) = 6.06, n = 8, p < 0.05, two-way ANOVA for repeated measures) (Fig. 3C,D), thus highlighting the role of PPAR-α in the effects of OEA.
Figure 3.
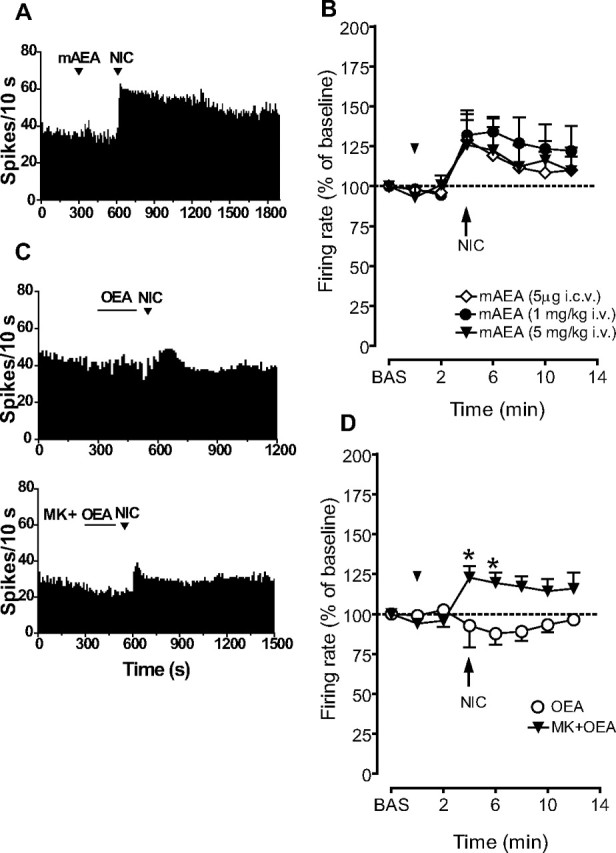
OEA, but not mAEA, prevented increases in firing rate of VTA dopaminergic neurons produced by nicotine. A, Representative firing rate histograms showing the effects of nicotine (NIC, 0.2 mg/kg i.v., injected at arrowheads) on the discharge activity of individual VTA dopamine neurons recorded following injection of mAEA (1 mg/kg, i.v.). B, Graph showing that nicotine-induced excitation of VTA dopamine neurons was not changed following the administration of mAEA, either intravenously (1 and 5 mg/kg) or intracerebroventricularly (5 μg/5 μl). C, Representative firing rate histograms showing the effects of NIC (0.2 mg/kg i.v., injected at arrowheads) on the discharge activity of individual VTA dopamine neurons recorded following injection of OEA (20 μg/5 μl, i.c.v.; top). MK886 (MK, 3 mg/kg, i.p.) reversed the OEA-induced blockade of nicotine's effects (bottom). Neither OEA nor vehicle (40% w/v 2-hydroxypropyl-β-cyclodextrin) produced significant changes in spontaneous firing rate or burst firing. D, Graph showing that nicotine-induced excitation of VTA dopamine neurons was abolished by OEA. MK886 (MK, 3 mg/kg, i.p.) reversed the OEA-induced blockade of nicotine's effects. Results are means, with vertical bars representing SEM of firing rates, expressed as a percentage of baseline (BAS) values. Arrows represent the time of intravenous injections. The horizontal bar represents the time of intracerebroventricular administration. *p < 0.05 versus baseline (one-way ANOVA for repeated measures and Dunnett's test).
Blockade of nicotine-induced excitation of dopamine neurons in vitro by noncannabinoid fatty acid ethanolamides
We next asked whether modulation of nicotine effects by PPAR-α could be studied in brain slices containing the mesencephalon by using whole-cell patch-clamp recordings. The effect of nicotine was studied on posterior VTA DA neurons. Figure 4A (top) shows a typical action potential of a representative DA neuron, when recorded in the current-clamp mode, with its typical low threshold, broad action potential, and prominent afterhyperpolarization. The second derivative of this action potential originates the waveform (Fig. 4A, bottom) that has been used for cell identification of DA neurons in many in vivo extracellular studies (Grace and Bunney, 1983, 1984; Ungless et al., 2004), and qualitatively corresponds to the typical action potential recorded in vivo and shown in Figure 1A. DA neurons recorded under current-clamp mode displayed an average frequency of 1.8 ± 0.1 Hz (n = 102) and fired spontaneously in a clock-like, single-spike mode. Consistent with the literature (Pidoplichko et al., 1997), DA neurons responded to bath-applied nicotine (1 μm, 2 min) with a transient excitation of discharge rate (∼40%) (Fig. 4B–D). This excitation peaked (137 ± 12.8% of baseline, n = 6), and was statistically significant (F(6,41) = 8.03, p < 0.0001, one-way ANOVA), during the first minute of application. Under voltage-clamp mode (Vholding= −70 mV), nicotine caused a transient inward current of 40.3 ± 5.6 pA (n = 6) (Fig. 4F), due to rapid activation and desensitization of nAChRs (Pidoplichko et al., 1997). We next examined the effects of the three different FAEs (i.e., mAEA, OEA and PEA) on nicotine induced excitation of VTA DA neurons. These drugs were applied for 5 min to assess whether they per se modulated spontaneous firing of DA neurons, and then coapplied with nicotine (1 μm). mAEA was tested at two different concentrations, 30 nm and 1 μm. The lower concentration of mAEA did not significantly affect spontaneous discharge rate of DA neurons or modulate nicotine-evoked excitation (the peak of nicotine with mAEA was 147.7 ± 13.7% of baseline firing rate, p = 0.58, t test, vs nicotine alone) (Fig. 4E,G). Since this concentration of mAEA might have been too low, we next tested a concentration of 1 μm, which itself significantly enhanced firing rate of DA neurons to 370.6 ± 110% of baseline level (F(5,125) = 21.74, n = 6, p = 0.0001, one-way ANOVA for repeated measures; data not shown). We asked whether mAEA-induced stimulation was due to activation of CB1 and/or TRPV1 receptors. The CB1 receptor antagonist AM281, at a concentration (500 nm) that fully reverses activation of CB1 receptors by maximal concentrations of agonists (Melis et al., 2004), had no effect on mAEA-induced stimulation of DA neuron firing rate (F(1,72) = 0.67, n = 5, p = 0.4365, two-way ANOVA; data not shown). However, this stimulation was completely blocked by the TRPV1 receptor antagonist capsazepine (10 μm, F(1,171) = 8.13, n = 5–6, p = 0.019, two-way ANOVA; data not shown). This observation is consistent with other studies showing that TRPV1 agonists stimulate DA neuron activity by enhancing glutamatergic synaptic transmission onto DA neurons (Marinelli et al., 2005). To isolate the agonistic activity of mAEA at CB1 receptors, or possibly at PPAR-α, and avoid TRPV1-induced excitation of DA cells that could mask or confound the effects of the subsequent application of nicotine, we applied nicotine in the presence of capsazepine. Under these circumstances, excitatory effects of nicotine were unmodified compared with controls (151.4 ± 13.5% of baseline, t = 0.77, n = 6, p = 0.4563, t test) (Fig. 4G), suggesting that CB1 receptor stimulation did not affect nicotine-induced excitation of DA neuron firing and that mAEA did not activate PPAR-α (see below).
Figure 4.

Activation of dopamine neurons by nicotine is prevented by OEA and PEA in vitro. A, Typical action potential waveform of a dopamine neuron recorded under current-clamp mode (top) and its second derivative (bottom). B, Representative traces of a dopamine neuron spontaneous activity during baseline level (top), nicotine application (nic, 1 μm for 2 min; middle), and wash out (bottom). C, Rate histogram depicting an example of the effect of nicotine on dopamine neuron firing rate. The horizontal bar represents the time of nicotine application. D, Time course of the excitatory effect of nicotine on dopamine neuron discharge rate (30 s bins). E, OEA (3 μm) and PEA (10 μm), but not mAEA (30 nm), blocked nicotine-induced activation of dopamine neurons. The dashed and the solid bars represent the times of fatty acid ethanolamide (OEA, PEA, or mAEA) or nicotine application, respectively. F, The bar graph shows that OEA (3 μm and PEA 10 μm) also blocked nicotine-induced inward currents (Iholding) when dopamine neurons were recorded under the voltage-clamp mode (Vhold= −70 mV). The inset shows that nicotine (black line) caused a 47 pA inward current under voltage-clamp mode, which was completely abolished in the presence of OEA (3 μm; OEA's effect on Iholding is superimposed in light gray for comparison). The horizontal bar represents the time of nicotine application. G, Bar graph summarizing the actions of mAEA (30 nm and 1 μm), OEA (3 μm), and PEA (10 μm) on nicotine-induced enhancement of dopamine neuron discharge rate (average of the first minute of nicotine perfusion). mAEA at 1 μm was perfused in the presence of capsazepine (CPZ, 10 μm) to prevent the vanilloid-induced excitation of dopamine neurons. H, Dose-effect relationship of OEA's blockade of nicotine-induced excitation of dopamine neurons. Numbers above bars indicate the n values for each group of experiments. Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01.
Next, we tested two different noncannabinoid FAEs, OEA and PEA. Interestingly, OEA (0.3, 1 and 3 μm) dose-dependently prevented nicotine-induced excitation (97.44 ± 5% and 92.01 ± 7% of baseline at 1 and 3 μm, respectively; 1 μm: n = 5, t = 2.65, p = 0.01; 3 μm: n = 7, t = 3.22, p = 0.04) (Fig. 4H), without affecting DA neuron spontaneous activity during preapplication (Fig. 4E). The effects of OEA were mimicked by PEA (10 μm, 101.9 ± 3% of baseline, n = 6, t = 2.66, p = 0.01) (Fig. 4E,G). Consistently, under voltage-clamp mode (Vholding= −70 mV), the nicotine-induced inward current was completely abolished when nicotine was perfused in the presence of OEA (3 μm, 5 min preapplication: −0.3 ± 3.4 pA, n = 6, t = 7.13, p = 0.0004, paired t test) (Fig. 4F), or PEA (10 μm, 5 min preapplication: −0.6 ± 7.5 pA, N = 5, t = 4.442, p = 0.001, paired t test) (Fig. 4F). During preapplication, OEA and PEA did not induce inward or outward currents onto DA neurons (data not shown).
Based on these results, we expected that the PPAR-α antagonist MK886 would block the actions of OEA and PEA on nicotine-induced excitation. As predicted, when OEA or PEA were coapplied with MK886 (300 nm), nicotine's effects on firing rate of DA neurons were restored (155.8 ± 16.6% and 163.8 ± 11.3% of baseline in the presence of OEA and PEA, respectively; OEA+MK886 vs OEA alone: F(2,323) = 7.59, n = 8, p = 0.004, two-way ANOVA; PEA+MK886 versus PEA alone: F(1,228) = 4.84, n = 8, p = 0.04, two-way ANOVA) (Fig. 5A,B,C). MK886 when perfused either alone or in combination with OEA/PEA did not alter spontaneous firing rate of VTA DA neurons (Fig. 5C). However, MK886 significantly enhanced nicotine-induced activation of DA neurons (207 ± 27% of baseline, MK886+nicotine vs nicotine: t = 2.167, n = 7, p < 0.05, t test) (Fig. 5B).
Figure 5.
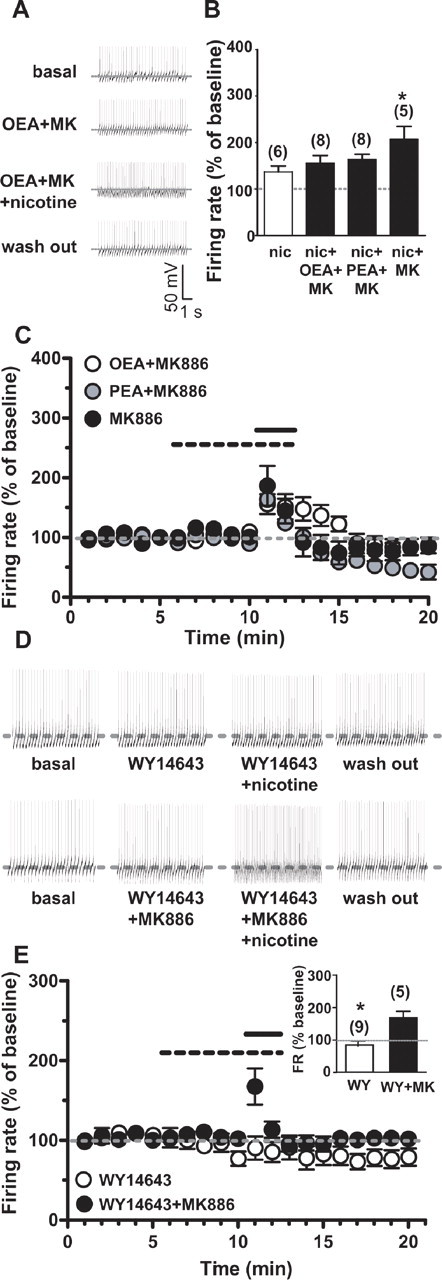
OEA and PEA block nicotine activation of dopamine neurons through a PPAR-α-mediated mechanism. A, Representative traces of the spontaneous activity of a dopamine neuron during baseline (top), OEA (3 μm) plus the PPAR-α antagonist MK886 (0.3 μm) preapplication (5 min, second panel), subsequent nicotine application (1 μm, 2 min, third panel), and wash out (bottom). B, Bar graph illustrating the effect of MK886 on nicotine-induced activation of VTA DA neurons and on OEA- and PEA-mediated inhibition of nicotine excitation (average of the first minute of nicotine perfusion). Note that activation of dopamine neurons by nicotine was fully restored when either OEA or PEA were coapplied with MK886. Notably, MK886 itself significantly potentiated nicotine-induced excitation. C, Time course of the effect of MK886 (0.3 μm), alone or in combination with either OEA or PEA, on nicotine induced excitation. The dashed bar represents the time of fatty acid ethanolamide (OEA, PEA) plus MK886 or MK886 alone application. The solid bar represents the time of nicotine application. D, Representative traces of dopamine neuron firing rate showing that the PPAR-α agonist WY14643 (300 nm) mimicked the actions of OEA and PEA by preventing nicotine-induced excitation (top), which was then restored by the coapplication of MK886 (bottom). E, Time course of the effect of nicotine on dopamine neuron firing rate in the presence of WY14643 (open symbols) or WY14643 plus MK886 (closed symbols). The dashed and the solid bars represent the times of PPAR-α agonist/antagonist or nicotine application, respectively. In the inset, the bar graph summarizes the effects of WY14643 (WY) on nicotine-induced excitation of dopamine neuron firing rate (FR) with or without MK886. Numbers above bars indicate the n values for each group of experiments. Data are expressed as mean ± SEM. *p < 0.05.
Next, we determined whether the synthetic PPAR-α agonist WY14643 would alter the effects of nicotine on DA cells. WY14643 (300 nm) was per se ineffective on DA neuronal firing rate, but fully prevented nicotine-induced excitation (83.7 ± 14.7% of baseline, n = 9, t = 2.54, p = 0.02) (Fig. 5D,E). The effect of WY14643 was also reversed by MK886 (167.7 ± 22.7% of baseline; WY14643+MK886 vs WY14643 alone: F(1,228) = 5.30, n = 5, p < 0.05, two-way ANOVA) (Fig. 5D,E), confirming the role of PPAR-α in the modulation of DA neuron responses to nicotine.
Mechanisms downstream of PPAR-α activation in the modulation of nicotine effects: involvement of tyrosine kinases
Although it is well established that PPAR-α regulates gene expression (Berger and Moller, 2002), the effects of OEA, PEA and WY14643 observed in the present study were fairly rapid in onset, thus ruling out gene induction as a possible mechanism, and suggesting a more likely nongenomic (Gardner et al., 2005) mechanism occurring in such a short time scale. Among many diverse pathways, we chose to investigate the regulation of tyrosine kinases, because PPAR-α agonists have been shown to activate several tyrosine kinases, such as the Src family kinase (SFK) (Gardner et al., 2005), which phosphorylates and negatively regulates α7 nAChRs (Charpantier et al., 2005).
We hypothesized that phosphorylation of nAChRs could account for PPAR-α mediated inhibition of nicotine effects. To explore this possibility, we incubated slices with the general tyrosine kinase inhibitor genistein (10 μm), which has indirect effects on nAChRs arising from the inhibition of intracellular phosphorylation pathways. Experiments were conducted under voltage-clamp mode on nicotine-induced inward currents, since genistein had aspecific channel blocker properties which led to a complete blockade of action potential generation (data not shown). Genistein was able to prevent OEA blockade of nicotine effects and restored nicotine-evoked inward currents (37.8 ± 4.4 pA, n = 6, t = 6.79, p < 0.0001) (Fig. 6A,B), demonstrating that inhibition of tyrosine kinases reverses the effect of PPAR-α activation.
Figure 6.
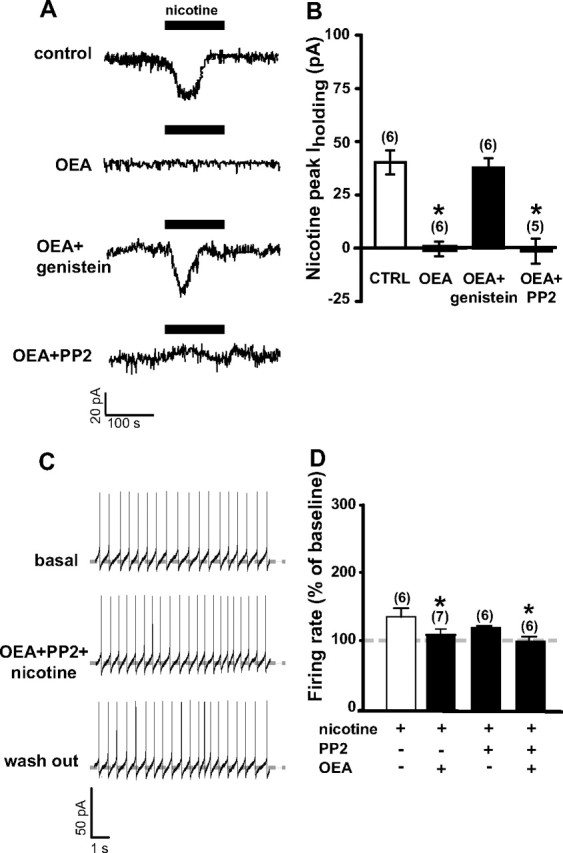
Tyrosine kinases, but not Src kinase, inhibition prevent OEA-induced blockade of nicotine effects. A, In voltage-clamp mode, nicotine (1 μm, 2 min) caused a 43 pA inward current (top) that is blunted by OEA preapplication (3 μm, 5 min; second panel). Pretreatment with the general tyrosine kinase inhibitor genistein (10 μm, 5 min) fully blocked OEA actions by restoring nicotine-induced inward current (third panel). However, pretreatment with the selective Src kinase inhibitor PP2 (10 μm, bottom) failed to prevent OEA actions on nicotine-induced change in holding current (Iholding). B, Bar graph summarizing the effects of OEA per se and in the presence of either kinase inhibitor on nicotine-induced inward current. C, Representative traces of dopamine neuron firing rate showing that PP2 failed to prevent OEA's action on nicotine-induced excitation (middle). D, Bar graph summarizing the effect of OEA on nicotine-induced enhancement of dopamine neuron discharge rate (average of the first minute of nicotine perfusion) alone or in the presence of PP2. Numbers above bars indicate the n values for each group of experiments. Data are expressed as mean ± SEM. *p < 0.05.
To investigate which tyrosine kinase phosphorylates and negatively modulates nAChRs, we focused on SFKs, on the basis of previous reports highlighting the role of SFK in the regulation of α7 nAChRs (Charpantier et al., 2005). We predicted that inhibition of SFK would reverse the effects of OEA. To test this hypothesis, slices were incubated (1 h) and continuously perfused with the SFK inhibitor PP2 (10 μm). This treatment did not change electrophysiological features of recorded DA neurons (data not shown). However, PP2 failed to reverse OEA blockade of nicotine's effects on DA cells under both voltage- and current-clamp modes. Indeed, in the presence of PP2, OEA abolished nicotine-induced inward currents (−1.4 ± 5.7, n = 5, t = 0.16, p > 0.5) (Fig. 6A,B) as well as the nicotine-induced enhancement of firing rate (100.9 ± 7.1% of baseline, F(1,108) = 0.06, n = 6, p = 0.8, two-way ANOVA) (Fig. 6C,D), suggesting that SFK is not involved in the negative modulation of nAChRs by PPAR-α agonists.
Discussion
The present study revealed that naturally occurring noncannabinoid FAEs can modulate the responses of VTA DA neurons to nicotine via PPAR-α, possibly by phosphorylation of nAChRs. This is the first evidence of an important functional role of this family of nuclear receptor transcription factors in the brain. It also highlights the role of FAEs, devoid of cannabinoid actions, in the regulation of neuronal functions.
Centrally mediated effects of the noncannabinoid FAEs have been poorly characterized, although OEA and PEA might constitute an independent endocannabinoid-like system. This view is supported by the findings that their synthesis and inactivation occurs independently of “classic” endocannabinoids, although in a similar on demand manner (Hansen et al., 1995; Stella and Piomelli, 2001; Mackie and Stella, 2006). The molecular targets underlying their pharmacological effects have remained elusive until the discovery of their agonistic actions at PPAR-α (Fu et al., 2003). These nuclear receptors are ubiquitously distributed in the CNS (Moreno et al., 2004), but their roles in neuronal physiology, or in pathophysiological mechanisms of brain disorders, are largely unknown.
Indirect enhancement of brain FAEs levels obtained by blockade of their major hydrolyzing enzyme, FAAH, by URB597 (Kathuria et al., 2003) has been reported to produce antidepressant, anxiolytic and analgesic effects in rodents (Kathuria et al., 2003; Gobbi et al., 2005; Piomelli et al., 2006; Russo et al., 2007). All of these effects are prevented by treatment with CB1 receptor antagonists, and have been ascribed to increased AEA levels, thus suggesting that augmented levels of OEA and PEA do not contribute significantly. However, a PPAR-α antagonist was recently reported to block the peripheral analgesic effects of URB597, suggesting that analgesia may be mediated by FAEs binding at PPAR-α (Jhaveri et al., 2008). In the present experiments, we discovered that inhibition of FAAH, rather than blockade of CB1 receptors, suppresses nicotine-induced activation of DA neurons. The lack of effect by rimonabant was unexpected in light of recent reports that CB1 antagonists decrease DA release evoked by nicotine in the nucleus accumbens (Cohen et al., 2002; Cheer et al., 2007). One can argue, however, that the suppression by rimonabant of evoked DA release may be independent from the inhibition of firing activity of DA neurons in the VTA, and may be an effect primarily involving their terminal regions or local circuits within the nucleus accumbens. Hence, presynaptically located nAChRs potently regulate DA release in the striatum, including the nucleus accumbens (Zhou et al., 2001). The effect of URB597 was even more surprising, since it contradicts the notion that the endocannabinoid system exerts a facilitatory effect on nicotine reward and addiction. Consistent with the present results, recent findings indicate that URB597 prevents the development of nicotine-induced CPP, acquisition of nicotine self-administration and nicotine-induced reinstatement in both CPP and self-administration models of relapse (Scherma et al., 2008).
Here, we found that the effects of URB597 were not entirely dependent on CB1 receptor stimulation, since nicotine-induced increases in DA neuron bursting were not reversed by rimonabant or AM251, whereas increases in firing rate were. The PPAR-α antagonist MK886 reversed URB597's blockade of nicotine-induced bursting in DA neurons, suggesting that FAEs, other than AEA, play a role in antagonizing the effects of nicotine. More importantly, OEA, but not mAEA, blocked the effects of nicotine on DA neurons in vivo. These results were substantiated by the findings that both OEA and PEA, but not mAEA, completely prevented nicotine-induced excitation of DA neurons in vitro. Additionally, OEA and PEA actions via PPAR-α were confirmed by the antagonism exerted by MK886, and by the observation that the PPAR-α agonist WY14643 mimicked the actions of noncannabinoid FAEs. Although AEA has been reported to display binding affinity for PPAR-α (Sun et al., 2006, 2007), our results are not consistent with those findings, since mAEA had no effects on nicotine-induced excitation of DA neurons, contrary to OEA and PEA. However, the studies of Sun et al. (2006, 2007) were performed in HeLa cells transiently transfected with PPAR-α, thus other investigations are necessary to confirm the binding properties of AEA at PPAR-α under more physiological conditions and, more importantly, in neurons. Remarkably, the analgesic properties of mAEA are fully preserved in PPAR-α knock-out mice, suggesting a PPAR-α-independent mechanism of action, whereas those of OEA and PEA are abolished (LoVerme et al., 2006).
Studies on recombinant or native nAChRs expressed in Xenopus oocytes or in mouse thalamic synaptosomes, respectively, have demonstrated that AEA (Oz et al., 2003; Spivak et al., 2007; Butt et al., 2008) or fatty acids (Butt et al., 2002; Barrantes, 2004) can modulate nAChR function as noncompetitive antagonists. Our results tend to exclude this possibility. In fact, they strongly support the notion that OEA and PEA effects are specifically mediated by PPAR-α, since they are blocked by the selective synthetic antagonist and mimicked by the agonist. Additionally, we found that mAEA did not alter nicotine-induced DA neuron excitation, making its action as nAChR antagonist unlikely.
As mentioned above, rimonabant revealed a significant component mediated by CB1 receptors in the effects of URB597. This piece of evidence is apparently difficult to reconcile with the results obtained with mAEA. Hence, mAEA does not significantly modulate nicotine's effects either in vivo or in vitro, arguing against an involvement of CB1 receptors in the modulation of the excitatory actions of nicotine on VTA DA neurons. There are possible explanations for this discrepancy: first, the effects of URB597 are due to the combination of CB1- (by AEA) and PPAR-α- (by OEA and PEA) mediated effects. CB1 receptors and PPAR-α may share opposing or reinforcing intracellular pathways (including modulation of protein kinases) (for review, see Alexander and Kendall, 2007). Second, URB597 enhances brain levels of endogenously released FAEs in a discrete and region-specific manner, and may influence the release of other endocannabinoids as well, including 2-arachidonoylglycerol (Di Marzo and Maccarrone, 2008; Maccarrone et al., 2008). Conversely, exogenously applied mAEA induces the activation of CB1 receptors throughout the brain. The third possible explanation is a differential involvement of TRPV1 channels, which may be activated by AEA (following URB597 administration) or by mAEA itself. However, our results in vitro tend to exclude the possibility that TRPV1 receptors play a significant role in the modulation of nicotine effects, since their activation by mAEA, or blockade by the selective antagonist capsazepine did not change nicotine-induced excitation of DA neurons.
We investigated also the mechanism by which PPAR-α may modulate the effects of nicotine. Due to the rapid onset of agonist actions, we hypothesized that this could be a nongenomic effect. Hence, PPARs exert pleiotropic effects on many different intracellular pathways, including protein kinases (Gardner et al., 2005). It was recently shown that the functional properties of α7 nAChRs depend on the tyrosine phosphorylation status of the receptor, being the result of a balance between SFKs and tyrosine phosphatases (Charpantier et al., 2005), which negatively or positively modulate nAChR-mediated currents, respectively. Additionally, phosphorylation/dephosphorylation of tyrosine residues in nAChRs controls the number of functional surface receptors (Cho et al., 2005).
Interestingly, we found that the effects of the tyrosine kinase inhibitor genistein were consistent with the idea that PPAR-α-mediated nAChR phosphorylation could account for the blockade of neuronal responses to nicotine, although at this stage we cannot identify the specific tyrosine kinase involved. A proposed mechanism is displayed in Figure 7. A constitutive interaction between PPAR-α and tyrosine kinases is also possible, and may tonically control the ratio of phosphorylated/dephosphorylated nAChRs, as indicated by the enhanced effects of nicotine in the presence of the PPAR-α antagonist MK886. It cannot be excluded that OEA and PEA may be endogenous modulators of acetylcholine transmission, since stimulation of muscarinic receptors was shown to stimulate the biosynthesis of OEA and PEA (Stella and Piomelli, 2001), suggesting the intriguing possibility of a reciprocal control between acetylcholine and FAEs.
Figure 7.

Schematic diagram illustrating the proposed mechanism of PPAR-α activation, and modulation of responses of DA neurons to nicotine, by the noncannabinoid fatty acid ethanolamides OEA and PEA. Their action is mimicked by the synthetic PPAR-α agonist WY14643 and blocked by the PPAR-α antagonist MK886. URB597 enhances brain levels of OEA and PEA in vivo by inhibiting their major catabolizing enzyme, FAAH. It is proposed that activated PPAR-α stimulate the activity of tyrosine kinases (Tyr Kin) through a nongenomic mechanism. Tyrosine kinases, in turn, induce the phosphorylation (P) of nAChR, which reduces their responses to the agonists, or promotes rapid internalization. Genistein, a general tyrosine kinase inhibitor, blocks the effects of PPAR-α activation. ↓, activation; ⊥, inhibition.
This novel mechanism of regulation of nAChRs by PPAR-α may represent a new therapeutic avenue for the discovery of medications to support patients during nicotine abstinence. Our data demonstrate that the actions of OEA are not restricted to the periphery and suggest that modulation of neuronal responses to nicotine by OEA may represent an interesting extension of its peripheral anorexic properties. PPAR-α agonists, such as fibrates, are well established medications clinically used in the treatment of lipid metabolism disorders. Their central effects are considered negligible due to their poor ability to cross the blood brain barrier. However, changes in brain lipid metabolism and/or composition, or modifications of the levels of endogenous lipid signaling molecules may exert unsuspected actions on neurotransmitter functions, which might be exploited therapeutically.
Footnotes
This work was supported by a grant from the Italian Ministry of University (PRIN 2005), Philip Morris USA and Philip Morris International, and by the Intramural Research Program of National Institute on Drug Abuse, National Institutes of Health, and Department of Health and Human Services.
References
- Alexander SP, Kendall DA. The complications of promiscuity: endocannabinoid action and metabolism. Br J Pharmacol. 2007;152:602–623. doi: 10.1038/sj.bjp.0707456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrantes FJ. Structural basis for lipid modulation of nicotinic acetylcholine receptor function. Brain Res Brain Res Rev. 2004;47:71–95. doi: 10.1016/j.brainresrev.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- Butt C, Alptekin A, Shippenberg T, Oz M. Endogenous cannabinoid anandamide inhibits nicotinic acetylcholine receptor function in mouse thalamic synaptosomes. J Neurochem. 2008;105:1235–1243. doi: 10.1111/j.1471-4159.2008.05225.x. [DOI] [PubMed] [Google Scholar]
- Butt CM, Hutton SR, Marks MJ, Collins AC. Bovine serum albumin enhances nicotinic acetylcholine receptor function in mouse thalamic synaptosomes. J Neurochem. 2002;83:48–56. doi: 10.1046/j.1471-4159.2002.01135.x. [DOI] [PubMed] [Google Scholar]
- Cahill K, Ussher M. Cannabinoid type 1 receptor antagonists (rimonabant) for smoking cessation. 2007 Cochrane Database Syst Rev:CD005353. [Google Scholar]
- Castañé A, Berrendero F, Maldonado R. The role of the cannabinoid system in nicotine addiction. Pharmacol Biochem Behav. 2005;81:381–386. doi: 10.1016/j.pbb.2005.01.025. [DOI] [PubMed] [Google Scholar]
- Charpantier E, Wiesner A, Huh KH, Ogier R, Hoda JC, Allaman G, Raggenbass M, Feuerbach D, Bertrand D, Fuhrer C. Alpha7 neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and Src-family kinases. J Neurosci. 2005;25:9836–9849. doi: 10.1523/JNEUROSCI.3497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheer JF, Wassum KM, Sombers LA, Heien ML, Ariansen JL, Aragona BJ, Phillips PE, Wightman RM. Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J Neurosci. 2007;27:791–795. doi: 10.1523/JNEUROSCI.4152-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CH, Song W, Leitzell K, Teo E, Meleth AD, Quick MW, Lester RA. Rapid upregulation of alpha7 nicotinic acetylcholine receptors by tyrosine dephosphorylation. J Neurosci. 2005;25:3712–3723. doi: 10.1523/JNEUROSCI.5389-03.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen C, Perrault G, Voltz C, Steinberg R, Soubrié P. SR141716, a central cannabinoid (CB(1)) receptor antagonist, blocks the motivational and dopamine-releasing effects of nicotine in rats. Behav Pharmacol. 2002;13:451–463. doi: 10.1097/00008877-200209000-00018. [DOI] [PubMed] [Google Scholar]
- D'Agostino G, La Rana G, Russo R, Sasso O, Iacono A, Esposito E, Raso GM, Cuzzocrea S, Lo Verme J, Piomelli D, Meli R, Calignano A. Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator-activated receptor-alpha agonist, modulates carrageenan-induced paw edema in mice. J Pharmacol Exp Ther. 2007;322:1137–1143. doi: 10.1124/jpet.107.123265. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Maccarrone M. FAAH and anandamide: is 2-AG really the odd one out? Trends Pharmacol Sci. 2008;29:229–233. doi: 10.1016/j.tips.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Schwieler L, Engberg G. Excitatory and inhibitory responses of dopamine neurons in the ventral tegmental area to nicotine. Synapse. 2002;43:227–237. doi: 10.1002/syn.10044. [DOI] [PubMed] [Google Scholar]
- Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther. 2005;313:352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Forget B, Hamon M, Thiebot MH. Cannabinoid CB1 receptors are involved in motivational effects of nicotine in rats. Psychopharmacology (Berl) 2005;181:722–734. doi: 10.1007/s00213-005-0015-6. [DOI] [PubMed] [Google Scholar]
- Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez De Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- Gardner OS, Dewar BJ, Graves LM. Activation of mitogen-activated protein kinases by peroxisome proliferator-activated receptor ligands: an example of nongenomic signaling. Mol Pharmacol. 2005;68:933–941. doi: 10.1124/mol.105.012260. [DOI] [PubMed] [Google Scholar]
- Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci U S A. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons–1. Identification and characterization. Neuroscience. 1983;10:301–315. doi: 10.1016/0306-4522(83)90135-5. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci. 1984;4:2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen HS, Lauritzen L, Strand AM, Moesgaard B, Frandsen A. Glutamate stimulates the formation of N-acylphosphatidylethanolamine and N-acylethanolamine in cortical neurons in culture. Biochim Biophys Acta. 1995;1258:303–308. doi: 10.1016/0005-2760(95)00134-x. [DOI] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Robinson I, Garle MJ, Patel A, Sun Y, Sagar DR, Bennett AJ, Alexander SP, Kendall DA, Barrett DA, Chapman V. Inhibition of fatty acid amide hydrolase and cyclooxygenase-2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator-activated receptor-alpha in a model of inflammatory pain. Neuropharmacology. 2008;55:85–93. doi: 10.1016/j.neuropharm.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Goldberg SR. Rimonabant, a CB1 antagonist, blocks nicotine-conditioned place preferences. Neuroreport. 2004;15:2139–2143. doi: 10.1097/00001756-200409150-00028. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Goldberg SR. Cannabinoid CB1 receptor antagonists as promising new medications for drug dependence. J Pharmacol Exp Ther. 2005;312:875–883. doi: 10.1124/jpet.104.077974. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Forget B, Aubin HJ, Goldberg SR. Blocking cannabinoid CB1 receptors for the treatment of nicotine dependence: insights from pre-clinical and clinical studies. Addict Biol. 2008;13:239–252. doi: 10.1111/j.1369-1600.2008.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- LoVerme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther. 2006;319:1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Rossi S, Bari M, De Chiara V, Fezza F, Musella A, Gasperi V, Prosperetti C, Bernardi G, Finazzi-Agrò A, Cravatt BF, Centonze D. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat Neurosci. 2008;11:152–159. doi: 10.1038/nn2042. [DOI] [PubMed] [Google Scholar]
- Mackie K, Stella N. Cannabinoid receptors and endocannabinoids: evidence for new players. Aaps J. 2006;8:E298–306. doi: 10.1007/BF02854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Pascucci T, Bernardi G, Puglisi-Allegra S, Mercuri NB. Activation of TRPV1 in the VTA excites dopaminergic neurons and increases chemical- and noxious-induced dopamine release in the nucleus accumbens. Neuropsychopharmacology. 2005;30:864–870. doi: 10.1038/sj.npp.1300615. [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereu G, Yoon KW, Boi V, Gessa GL, Naes L, Westfall TC. Preferential stimulation of ventral tegmental area dopaminergic neurons by nicotine. Eur J Pharmacol. 1987;141:395–399. doi: 10.1016/0014-2999(87)90556-5. [DOI] [PubMed] [Google Scholar]
- Moreno S, Farioli-Vecchioli S, Cerù MP. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience. 2004;123:131–145. doi: 10.1016/j.neuroscience.2003.08.064. [DOI] [PubMed] [Google Scholar]
- Oz M, Ravindran A, Diaz-Ruiz O, Zhang L, Morales M. The endogenous cannabinoid anandamide inhibits alpha7 nicotinic acetylcholine receptor-mediated responses in Xenopus oocytes. J Pharmacol Exp Ther. 2003;306:1003–1010. doi: 10.1124/jpet.103.049981. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 3. San Diego: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, Dasse O, Monaghan EP, Parrott JA, Putman D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597) CNS Drug Rev. 2006;12:21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez de Fonseca F, Navarro M, Gómez R, Escuredo L, Nava F, Fu J, Murillo-Rodríguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. An anorexic lipid mediator regulated by feeding. Nature. 2001;414:209–212. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- Russo R, Loverme J, La Rana G, Compton TR, Parrott J, Duranti A, Tontini A, Mor M, Tarzia G, Calignano A, Piomelli D. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther. 2007;322:236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- Scherma M, Panlilio LV, Fadda P, Fattore L, Gamaleddin I, Le Foll B, Justinová Z, Mikics E, Haller J, Medalie J, Stroik J, Barnes C, Yasar S, Tanda G, Piomelli D, Fratta W, Goldberg SR. Inhibition of anandamide hydrolysis by URB597 reverses abuse-related behavioral and neurochemical effects of nicotine in rats. J Pharmacol Exp Ther. 2008;327:482–490. doi: 10.1124/jpet.108.142224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoaib M. The cannabinoid antagonist AM251 attenuates nicotine self-administration and nicotine-seeking behaviour in rats. Neuropharmacology. 2008;54:438–444. doi: 10.1016/j.neuropharm.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Solinas M, Justinova Z, Goldberg SR, Tanda G. Anandamide administration alone and after inhibition of fatty acid amide hydrolase (FAAH) increases dopamine levels in the nucleus accumbens shell in rats. J Neurochem. 2006;98:408–419. doi: 10.1111/j.1471-4159.2006.03880.x. [DOI] [PubMed] [Google Scholar]
- Solinas M, Tanda G, Justinova Z, Wertheim CE, Yasar S, Piomelli D, Vadivel SK, Makriyannis A, Goldberg SR. The endogenous cannabinoid anandamide produces delta-9-tetrahydrocannabinol-like discriminative and neurochemical effects that are enhanced by inhibition of fatty acid amide hydrolase but not by inhibition of anandamide transport. J Pharmacol Exp Ther. 2007;321:370–380. doi: 10.1124/jpet.106.114124. [DOI] [PubMed] [Google Scholar]
- Solinas M, Goldberg SR, Piomelli D. The endocannabinoid system in brain reward processes. Br J Pharmacol. 2008;154:369–383. doi: 10.1038/bjp.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak CE, Lupica CR, Oz M. The Endocannabinoid Anandamide Inhibits the Function of {alpha}4beta2 Nicotinic Acetylcholine Receptors. Mol Pharmacol. 2007;72:1024–1032. doi: 10.1124/mol.107.036939. [DOI] [PubMed] [Google Scholar]
- Stella N, Piomelli D. Receptor-dependent formation of endogenous cannabinoids in cortical neurons. Eur J Pharmacol. 2001;425:189–196. doi: 10.1016/s0014-2999(01)01182-7. [DOI] [PubMed] [Google Scholar]
- Stolerman IP, Shoaib M. The neurobiology of tobacco addiction. Trends Pharmacol Sci. 1991;12:467–473. doi: 10.1016/0165-6147(91)90638-9. [DOI] [PubMed] [Google Scholar]
- Suardíaz M, Estivill-Torrús G, Goicoechea C, Bilbao A, Rodríguez de Fonseca F. Analgesic properties of oleoylethanolamide (OEA) in visceral and inflammatory pain. Pain. 2007;133:99–110. doi: 10.1016/j.pain.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Sun Y, Alexander SP, Kendall DA, Bennett AJ. Cannabinoids and PPARalpha signalling. Biochem Soc Trans. 2006;34:1095–1097. doi: 10.1042/BST0341095. [DOI] [PubMed] [Google Scholar]
- Sun Y, Alexander SP, Garle MJ, Gibson CL, Hewitt K, Murphy SP, Kendall DA, Bennett AJ. Cannabinoid activation of PPAR alpha; a novel neuroprotective mechanism. Br J Pharmacol. 2007;152:734–743. doi: 10.1038/sj.bjp.0707478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Magill PJ, Bolam JP. Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science. 2004;303:2040–2042. doi: 10.1126/science.1093360. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]