

Abstract
A crucial virulence factor for intracellular Mycobacterium tuberculosis survival is Protein kinase G (PknG), a eukaryotic-like serinethreonine protein kinase expressed by pathogenic mycobacteria that blocks the intracellular degradation of mycobacteria in lysosomes. Inhibition of PknG results in mycobacterial transfer to lysosomes. Withania somnifera, a reputed herb in ayurvedic medicine, comprises a large number of steroidal lactones known as withanolides which show various pharmacological activities. We describe the docking of 26 withanferin and 14 withanolides from Withania somnifera into the three dimensional structure of PknG of M. tuberculosis using GLIDE. The inhibitor binding positions and affinity were evaluated using scoring functions- Glidescore. The withanolide E, F and D and Withaferin - diacetate 2 phenoxy ethyl carbonate were identified as potential inhibitors of PknG. The available drug molecules and the ligand AX20017 showed hydrogen bond interaction with the aminoacid residues Glu233 and Val235. In addition to Val235 the other amino acids, Gly237, Gln238 and Ser239 are important for withanolide inhibitor recognition via hydrogen bonding mechanisms.
Background
Mycobacterium tuberculosis continues to kill millions of people yearly worldwide and tuberculosis continues to be a major health problem. Despite the existence of effective chemotherapies, no new drugs have come to the market in >40 years. In addition the rise in drug resistance among M. tuberculosis strains is becoming a severe threat to public health, illustrated by the recent emergence of extensively drug-resistant tuberculosis (XDR-TB) that has caused a mortality of >98% [1]. Virulence of pathogenic mycobacteria is related to their capacity to survive for prolonged times within macrophage phagosomes [2, 3], whereas, normally internalized and phagocytosed bacteria are rapidly degraded within phagolysosomes, and pathogenic mycobacteria have evolved to block lysosomal delivery [3]. One strategy by which pathogenic mycobacteria prevent lysosomal maturation is through the activity of mycobacterial protein kinase G (PknG), a eukaryotic-like serine/threonine protein kinase that is not required for mycobacterial growth per se but is essential for survival within host macrophages [4, 5]. This PknG blocks the intracellular degradation of mycobacteria in lysosomes [6]. It is involved in host cell signal transduction capable of modulating host macrophage trafficking pathways [7]. Importantly, mycobacteria overexpressing a kinase-dead mutant of PknG are rapidly transferred to lysosomes and killed, demonstrating that PknG kinase activity is crucial for mycobacterial survival [8]. The fact that PknG is translocated into the host cytosol suggests that compounds aimed at blocking PknG activity do not require transport across the only limited permeable mycobacterial cell wall [4]. Together, these findings make PknG an attractive and promising drug target. Indeed, blocking PknG kinase activity by a specific inhibitor results in rapid transfer of mycobactera to lysosomes and killing of the intracellularly residing bacilli. PknG is a multidomain protein (Figure 1) consisting of a rubredoxin, kinase and tetratricopeptide repeat domain and show that the rubredoxin domain is essential for the activity of PknG. The structure reveals that the inhibitor-binding pocket is shaped by a unique set of amino acid residues that, in combination, is not found in any human kinase [8, 9]. This structural information provides the basis for the identification of novel compounds aimed at blocking the proliferation of M tuberculosis from W. somnifera.
Figure 1.
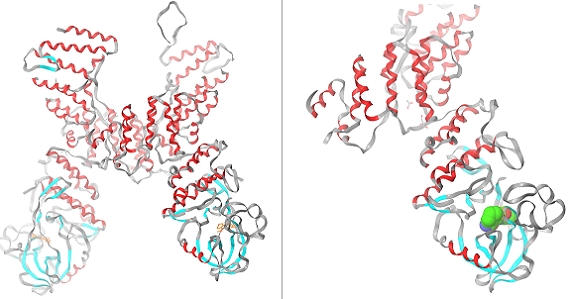
Structure of the PknG-AX20017 complex (PDB ID 2PZI).
Plants are rich in a wide variety of secondary metabolites such as tannins, terpenoids, alkaloids, flavonoids etc. which have been found to have antimicrobial properties. Withania somnifera (L) Dunal (Aswaghanda or Indian “Ginseng”) is widely used in ayurvedic medicines and is consumed as a dietary supplement around the world [10]. The major bioactive compounds of Ashwagandha are steroidal lactones compounds known as withanolides. They also contain several alkaloids, withanosides, reducing sugars, few flavonoids, tannins etc. [10]. These compounds have shown to possess a remarkable range of therapeutic properties. Other investigations indicated that leaf extracts of W. somnifera also has antibacterial properties [11]. The withanolides are C28-steroidal lactones built on an intact or rearranged ergostane framework, in which C-22 and C-26 are approximately oxidized to form a six-membered lactone ring. The basic structure (Figure 2) is designated as the withanolide skeleton. There are many novel structural variants of withanolides with modifications either of the carbocyclic skeleton or the side chain and these have often been described as modified withanolides or ergostan-type steroids related to withanolides. Hence, the present study was aimed to identify the potential PknG inhibitors from W. somnifera by molecular docking studies using Glide (Schrödinger).
Figure 2.
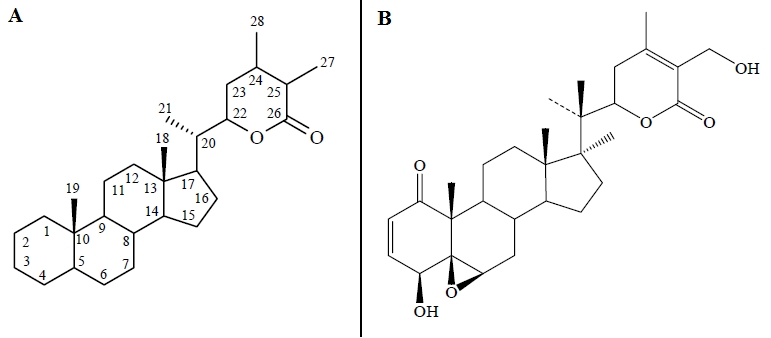
(A) Basic structure of Withanolides, (B) Basic structure of Withaferin A
Methodology
Selection of docking molecules
A set of 26 withaferin and 14 withanolide derivatives were downloaded from pubchem database [ http://pubchem.ncbi.nlm.nih.gov/] for the docking studies. Similarly the commercially available drugs namely Ciproflaxin, Kanamycin, Isoniazid, Ethionamide, Prothionamide, Pyrazinamide, Amino salicylic acid, Cycloserine, Streptomycin and Ethambutol were taken from the literature. All the ligands were built using Maestro build panel. The collected ligands were prepared by using LigPrep [Schrodinger] which uses MMFF 94s force field and gave the corresponding low energy 3D conformers of the ligands. These ligands were neutralized and checked for their ADME properties using QikProp (Schrodinger). QikProp helps in analyzing the pharmacokinetics and pharmacodynamics of the ligand by assessing the drug like properties.
Target propein preparation
The three dimensional crystal structure of PknG from M. tuberculosis in Complex with Tetrahydrobenzothiophene AX20017 (PDB Id: 2PZI) was downloaded from the Protein Data Bank (PDB) ( http://www.rcsb.org) (Figure 1). Before docking the ligands into the protein active site, the protein was prepared using protein preparation wizard of Schrodinger's molecular docking software. In this protein preparation all water molecules and hetero atoms were removed. Hydrogen atoms were added to the protein, including the necessary to define the correct ionization and tautomeric states of amino acid residues such as Asp, Ser, Glu, Arg and His. The active site of the protein was defined for generating the grid. The screened ligands were then docked into the prepared grid, for which [standard precision mode] was selected. No constraints were defined.
Docking
Docking was carried out using GLIDE (Grid-Based Ligand Docking with Energies) software. Glide searches for favourable interaction between one or more ligand molecules and a receptor molecule. The combination of position and orientation of a ligand relative to receptor, along with its conformation in flexible docking, is referred to as a ligand pose. The ligand poses that Glide generates, pass through a series of hierarchical filters that evaluate the ligands interaction with the receptor. The initial filters test the spatial fit of the ligand to the defined active site, and examine the complementarity of ligand-receptor interactions using a grid-based method patterned after the empirical ChemScore function. Poses that pass these initial screens enter the final stage of the algorithm, which involves evaluation and minimization of a grid approximation to the OPLS-AA nonbonded ligand-receptor interaction energy. Final scoring is then carried out on the energy-minimized poses. GlideScore is based on the ChemScore, but includes a stericclash term and adds buried polar atoms devised by Schrodinger to penalize electrostatic mismatches: Gscore=0.065*vdW + 0.130*Coul + Lipo + Hbond + BuryP + RotB + Site.
Results and Discussion
Out of 40 withanolide derivative compounds, only 10 compounds showed better interaction with the target protein. The docking confirmation with best GlideScore had been analyzed.
Binding mode of available antituberculosis drug molecules
The GlideScore values of available drug molecules range between -4.40 and -6.91 (Table 1, see Table 1). The drug Kanamycin and Isoniazid showed the better interaction than the other drug molecules and the GlideScore values are -6.91 and -6.63. The main chain amino acids Glu233 and Val235 form hydrogen bonds, with the drug molecules.
Binding mode of compound AX20017
The compound AX20017 inhibitor is bound deep within a narrow pocket formed by the inter lobe cleft of the kinase domain as reported in the x-ray crystallographic structure [9] and the G-Score value is -5.6 (Table 2, see Table 2). The main chain Glu233:O and Val235:NH of PknG form hydrogen bonds with inhibitor (Figure 3).
Figure 3.
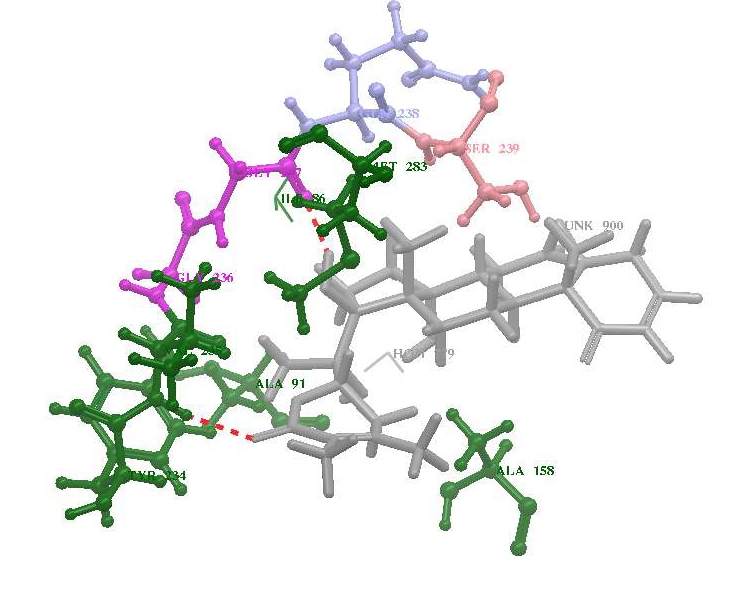
Potential docking pose for the ligands (A) withanolide D, (B) withanolide E and (C) withanolide F within the active site of PknG
Binding mode of bioactive compounds from W. somnifera
The binding efficiency of the bioactive compounds from W. somnifera is shown in Table 2 (see Table 2) and the G-score value is ranging between 4.58 to 7.86. Out of 22 compounds tested, Withanolide E, F and D showed the best G-Score value of 7.86, 7.69, and 7.63 respectively than the withaferin and currently existing drug molecules. More negative GlideScore value indicates the better interaction of the inhibitor with the target protein. A comparison of the different docking poses of compounds suggests that the compounds namely withanolide E, F and D showed fairly potent activity against PknG with the more negative G-Score value than the available drug molecules The bioactive compounds was positioned in a similar orientation like Ax20017 and forming hydrogen bonding with the residues Glu233, Val235, Gly237, Gln238, Ser239, Lys241, Ile292 and Ser293 of the kinase domain as shown in Table 2 (see Table 2). The calculated distances between these residues and the ligand molecule ranged from 2.671 to 3.429 Å. The presence of strong hydrogen bond interactions between the oxygen atom of the ligands and Val235 was observed. This hydrogen-bonding network places the inhibitor such that van der Waals contacts are made with the side chains of Ala158, Ile165, Val179, Lys181, Met232, Ile292 and Asp293 of PknG. These results indicate that the predicted nonbonding interactions and hydrophobic chains to accommodate the ligands are similar to the experimental data obtained from crystal structure as reported in the earlier studies [8]. The ADME properties indicated that withanolides obey the Lipinski's rule of five (Table 3, see Table 3) the basic rules to be followed by a candidate drug molecule.
Conclusion
Withanolide D, E and F show better with PknG based on GlideScore and interaction with the active site residues in comparison with the existing drug molecules. This data provides molecular insights to the consideration of Withanolides as potential candidates against the PknG target in M. tuberculosis.
Supplementary material
Acknowledgments
We thank Tamil Nadu State Council for Science and Technology, for research funding.
Footnotes
Citation:Aishwarya & Santhi, Bioinformation 7(1): 1 - 4 (2011)
References
- 1.MR Masjedi, et al. Clin Infect Dis. 2006;43:841. [Google Scholar]
- 2.L Nguyen, J Pieters. Trends Cell Biol. 2005;15:269. doi: 10.1016/j.tcb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 3.DG Russell. Nat Rev Mol Cell Biol. 2001;2:569. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 4.A Walburger, et al. Science. 2004;304:1800. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- 5.L Nguyen, et al. J Bacteriol. 2005;187:5852. doi: 10.1128/JB.187.16.5852-5856.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.ST Cole, et al. Nature. 2001;409:1007. [Google Scholar]
- 7.J Gatfield, J Pieters. Adv Immunol. 2003;81:45. doi: 10.1016/s0065-2776(03)81002-7. [DOI] [PubMed] [Google Scholar]
- 8.N Scherr, et al. Proc Natl Acad Sci U S A. 2007;104:12151. [Google Scholar]
- 9.D Tiwari, et al. J Biol Chem. 2009;284:27467. doi: 10.1074/jbc.M109.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.B Mahesh, S Satish. World J Agric Sci. 2008;4:839. [Google Scholar]
- 11.L Davis, G Kuttan. J Ethnopharmacol. 2000;71:193. doi: 10.1016/s0378-8741(99)00206-8. [DOI] [PubMed] [Google Scholar]
- 12.M Owais, et al. Phytomedicine. 2005;12:229. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.