

Abstract
Progression through mitosis is controlled by protein degradation that is mediated by the anaphase-promoting complex/cyclosome (APC/C) and its associated specificity factors. In budding yeast, APC/CCdc20 promotes the degradation of the Pds1p anaphase inhibitor at the metaphase-to-anaphase transition, whereas APC/CCdh1 promotes the degradation of the mitotic cyclins at the exit from mitosis. Here we show that Pds1p has a novel activity as an inhibitor of mitotic cyclin destruction, apparently by preventing the activation of APC/CCdh1. This activity of Pds1p is independent of its activity as an anaphase inhibitor. We propose that the dual role of Pds1p as an inhibitor of anaphase and of cyclin degradation allows the cell to couple the exit from mitosis to the prior completion of anaphase. Finally, these observations provide a novel regulatory paradigm in which the sequential degradation of two substrates is determined by the substrates themselves, such that an early substrate inhibits the degradation of a later one.
Keywords: Budding yeast, metaphase-to-anaphase transition, mitosis, cyclin degradation
Successful progression through a cell’s life cycle requires that the various cell cycle steps occur in a precise order. Often, this order is not determined by an obligate substrate/product mechanism that governs processes such as virus assembly or metabolic pathways, but rather by complex regulatory mechanisms. For example, under certain artificial conditions, cells may exit mitosis without previously undergoing anaphase, suggesting that these two key events are not linked by a product/substrate relationship. However, because under normal conditions cells invariably initiate anaphase before exiting mitosis, there must be a regulatory mechanism that determines the order of these events.
The metaphase-to-anaphase transition and the exit from mitosis are regulated by protein degradation. In both cases, protein degradation occurs via the ubiquitin pathway and requires the participation of a specific ubiquitin ligase, known as the anaphase-promoting complex or the cyclosome (APC/C) (King et al. 1995; Sudakin et al. 1995). The APC/C is a conserved multisubunit complex (Yu et al. 1998; Zachariae et al. 1998b). All APC/C substrates known to date contain a nine amino acid motif called the destruction box (Glotzer et al. 1991). The destruction box is essential for the APC/C-mediated ubiquitination, and its removal results in a protein that is resistant to APC/C-dependent degradation.
The activity of the APC/C at the metaphase-to-anaphase transition is required for the degradation of an anaphase inhibitor (Cohen-Fix and Koshland 1997b). The best-characterized anaphase inhibitors are the Pds1 protein (Pds1p) of budding yeast (Cohen-Fix et al. 1996; Yamamoto et al. 1996a,b) and the Cut2 protein (Cut2p) of fission yeast (Funabiki et al. 1996b). Recently, vertebrate anaphase inhibitors have been identified (M. Kirschner, pers. comm.). Stable forms of the anaphase inhibitors, generated by mutations in their destruction boxes, cause a metaphase arrest, indicating that their degradation is essential for normal progression through mitosis. The molecular function of these proteins is unknown, but it is speculated that they are inhibitors of one or more activators of the metaphase-to-anaphase transition. Pds1p and Cut2p have been shown to bind the Esp1 and Cut1 proteins, respectively, and Esp1p has been shown to activate sister chromatid separation (Funabiki et al. 1996a; Ciosk et al. 1998; Kumada et al. 1998). The Esp1 and Cut1 proteins share homology to each other and to several vertebrate gene products (McGrew et al. 1992). Although the different anaphase inhibitors do not have extensive similarity at the primary sequence level, they do share unusual charged amino acid distribution, indicating they may be related but rapidly divergent. Taken together, these families of anaphase inhibitors and activators most likely reflect a highly conserved mechanism of regulating the metaphase to anaphase transition.
At the exit of mitosis, the APC/C mediates the degradation of the several proteins, including the mitotic cyclins (Peters 1998). The latter contributes to the inactivation of the cyclin-dependent kinase, Cdk1, which, in turn, triggers the exit from mitosis. In budding yeast, Cdk1 inactivation can also be achieved by the presence of the Cdk1 inhibitor Sic1p (Schwob et al. 1994). Sic1p is normally active only late in mitosis, but ectopic expression of Sic1p at metaphase results in premature exit of mitosis, without a proper anaphase (Amon 1997). This result demonstrates the importance for proper timing of Cdk1 inactivation, and suggests that cyclin degradation at metaphase would also lead to Cdk1 inactivation and premature exit from mitosis. Thus, there must be a mechanism that restricts cyclin degradation to late mitosis.
Given that cyclin degradation must not take place until cells are ready to exit mitosis, the APC/C that promotes the degradation of the anaphase inhibitor at the metaphase-to-anaphase transition cannot simultaneously also promote cyclin destruction. In budding yeast, at least two different forms of APC/C exist that can be distinguished by their association with different auxiliary factors, the Cdc20 protein (Cdc20p) that promotes Pds1p ubiquitination (Visintin et al. 1997), and the Cdh1/Hct1 protein (Cdh1p) that promotes cyclin ubiquitination (Schwab et al. 1997). Cdh1p associates with the APC/C later in the cell cycle than Cdc20p (Fang et al. 1998; Zachariae et al. 1998a), and this association requires that Cdh1p will be dephosphorylated in a Cdc14p-dependent process (Visintin et al. 1998; Zachariae et al. 1998a; Jaspersen et al. 1999). What controls the timing of Cdh1p dephosphorylation is unknown. Nonetheless, progression through mitosis appears to be regulated by two different APC/C forms, one that is responsible for Pds1p degradation at the metaphase-to-anaphase transition, and the other that is responsible for the degradation of the mitotic cyclins and other APC/C substrates at the exit of mitosis.
Because the proper order of Pds1p and mitotic cyclin degradation is essential for the successful progression through mitosis, there is likely to be a regulatory mechanism that controls the activation of the APC/CCdh1 form. This mechanism may include proteins such as the Cdc5 polo-like kinase, the Cdc15 kinase, and the Cdc14 phosphatase, all of which are essential for the Clb2p degradation at the exit of mitosis, and act as a trigger for the initiation of cyclin destruction (Visintin et al. 1998, 1999; Morgan 1999; Shou et al. 1999). Here we show that Pds1p is not only an anaphase inhibitor, but also an inhibitor of this trigger, thereby coupling the exit from mitosis to the execution of anaphase. Our observations further imply that the temporal degradation of APC/C substrates may be determined by the substrates themselves, such that an early substrate (i.e. Pds1p) inhibits the degradation of a later substrate (e.g, the mitotic cyclins).
Results
Pds1p inhibits Clb2p degradation
An analysis of the timing of degradation of Pds1p and Clb2p (the major budding yeast mitotic cyclin) revealed that Pds1p degradation precedes that of Clb2p degradation by ∼20 min (Fig. 1 cf. A and C; Michaelis et al. 1997). This is consistent with the requirement for Pds1p degradation at the metaphase-to-anaphase transition and for Clb2p degradation at the exit of mitosis, and it correlates with the duration of anaphase, which lasts ∼20 min (Palmer et al. 1989; Straight et al. 1997). This difference in timing of degradation could have been due to a difference in the ubiquitination or degradation rates of the two proteins. If Pds1p and Clb2p degradation were independent events that differed only in the rate of ubiquitination, one would expect that inhibiting Pds1p degradation by mutating Pds1p itself should not affect the rate of Clb2p degradation. However, we have observed previously that in the presence of nondegradable Pds1p, cells neither initiated anaphase, nor exited mitosis (Cohen-Fix et al. 1996). As the exit from mitosis is controlled by Clb2p degradation, this observation raised the possibility that Pds1p is inhibiting Clb2p degradation, thus providing a mechanism for the temporal order of degradation of the two proteins.
Figure 1.
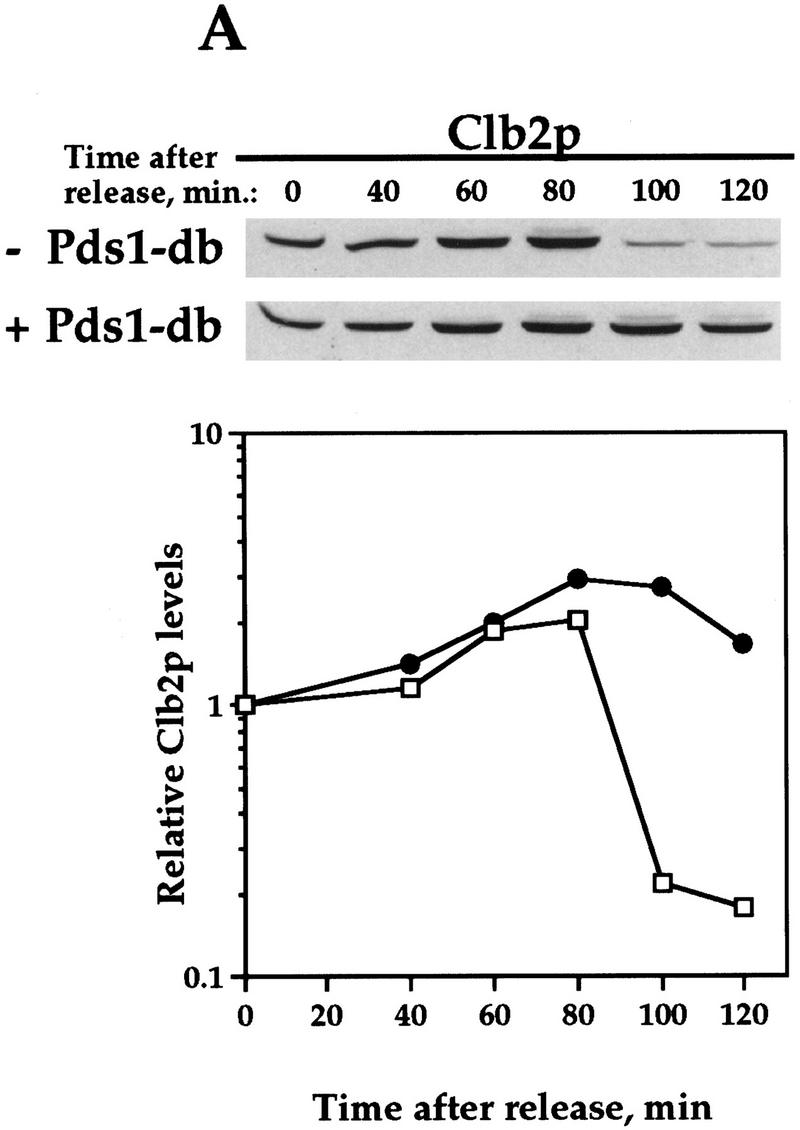
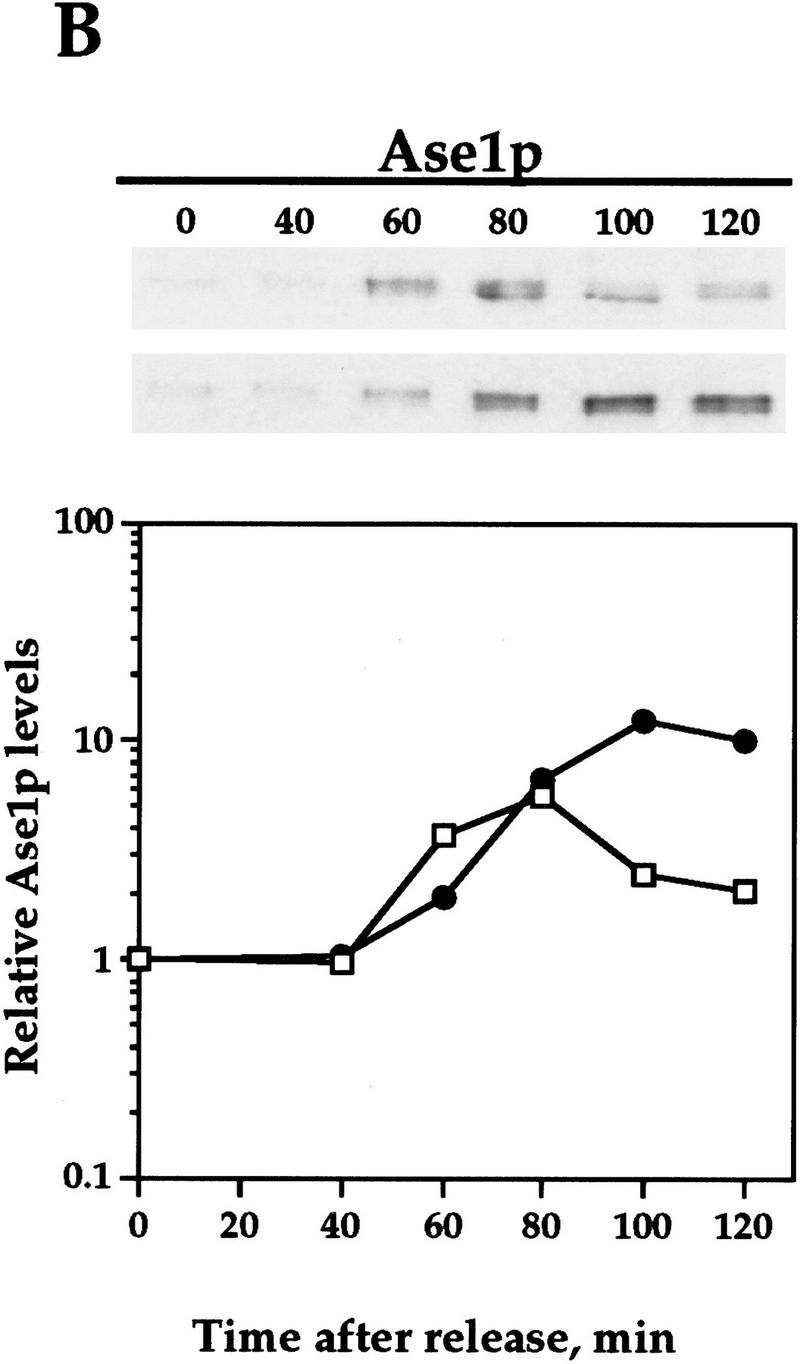
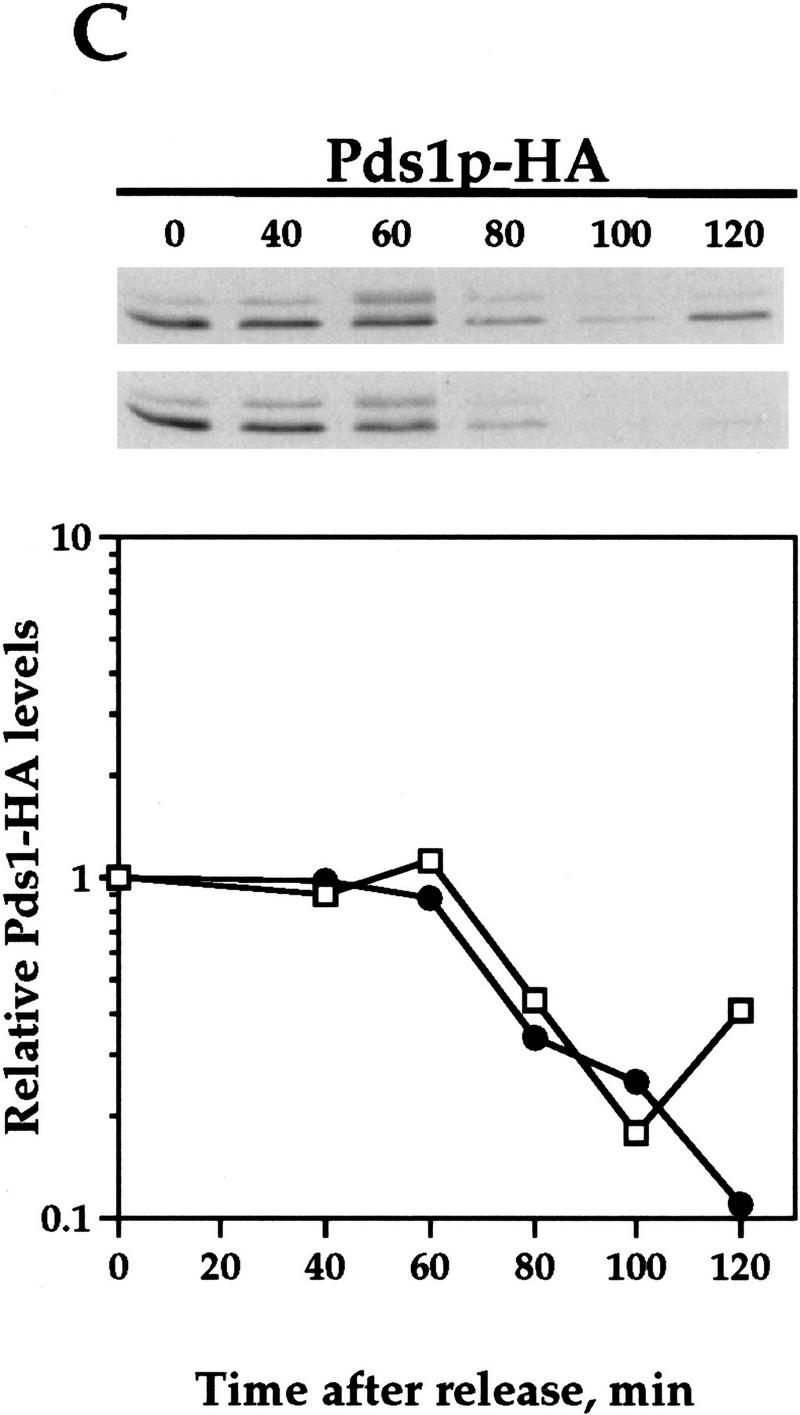
Pds1-db inhibits the degradation of the APC/CCdh1 substrates Clb2p and Ase1p. Wild-type cells (OCF1565) and cells containing GAL1–pds1-db (OCF1566) were grown at 30°C in YPR and arrested in S phase with HU (0.2 m final concentration). Following the arrest, the expression of GAL1–pds1-db was induced for 30 min by the addition of galactose (2% final concentration). The HU and galactose were then washed away and both cultures were released into YPD. Identical results were obtained when the cultures were released into YPRG (data not shown). Samples were taken at the indicated time points and processed for Western blot analysis as described in Materials and Methods. Time 0 indicates the time at which the cells were released from the S-phase arrest. The ability of Pds1p-db to inhibit Clb2p degradation was observed in several yeast backgrounds, including W303 (shown here) and A364a (see below). Western blot analyses were performed with antibodies against Clb2p (A), Ase1p (B), or HA (to detect Pds1–HA, C). The relative protein levels were determined by densitometry scanning (see Materials and Methods). (−Pds1-db, □) (+Pds1-db, ●) In the control cells (−Pds1-db), the levels of the HA-tagged wild-type Pds1p increased after 120 min following the release because these cells have exited mitosis and entered a new cell cycle (data not shown). This increase is not observed in cells expressing the Pds1-db mutant because they remain in metaphase, where the wild-type Pds1p is continuously degraded.
To test whether Clb2p degradation is regulated by Pds1p, we examined the levels of Clb2p in synchronized cells containing a stable form of Pds1p (henceforth Pds1-db) that is resistant to APC/C-mediated degradation because of a mutation in its destruction box (Cohen-Fix et al. 1996). For this experiment, cells were staged in S phase with hydroxyurea (HU), and released from the arrest after a 30-min induction of Pds1-db expression from a galactose inducible promoter. In the control cells that did not express Pds1-db, Clb2p levels began to drop between 80 and 100 min following the release (Fig. 1A). In contrast, in cells that contained Pds1-db, the degradation of Clb2p was inhibited (Fig. 1A). This activity of Pds1p could be extended to other APC/CCdh1p substrates, as Pds1p-db also inhibited the degradation of Ase1p (Fig. 1B), a microtubule-associated protein that was shown previously to be degraded at the exit of mitosis in an APC/CCdh1p-dependent manner (Juang et al. 1997). This inhibitory effect by Pds1-db was specific for APC/CCdh1p substrates, because under the same conditions, Pds1-db had no effect on the degradation rate of wild-type, HA-tagged Pds1p (Fig. 1C; Cohen-Fix et al. 1996). It has been shown previously that in vivo an abundant destruction box-containing peptide can cause a mitotic arrest, presumably by competing for the degradation of APC/C substrates (Yamano et al. 1998). However, because Pds1-db does not contain a functional destruction box, and because it did not inhibit wild-type Pds1p degradation, this simple type of competitive inhibition can be ruled out. Moreover, the ability of Pds1p-db to inhibit cyclin degradation is cell cycle dependent (see below), further excluding the possibility of simple competitive inhibition. Thus, Pds1p appears to be a specific inhibitor of APC/CCdh1p-substrate degradation. We have consistently observed that Pds1p can inhibit cyclin degradation for at least 3 hr (see below), after which time the levels of Clb2p begin to drop, and this is accompanied by the appearance of a-nucleated cells (data not shown). At this point we do not know whether this is due to an active bypass of or adaptation to the presence of Pds1p, or whether this is the terminal phenotype of dying cells.
The inhibition of Clb2p degradation by Pds1p is independent of the mitotic checkpoint
One of the conditions under which cyclin degradation is inhibited is when the mitotic/spindle assembly checkpoint is activated (Amon 1999). Under these conditions, cell cycle progression is halted at metaphase, and in budding yeast both Pds1p and Clb2p remain stable. At least one component of the spindle assembly checkpoint, Mad2p, has been shown to directly inhibit cyclin ubiquitination (Li et al. 1997; Fang et al. 1998). Therefore, it was formally possible that the inhibitory activity of Pds1p toward cyclin degradation is mediated indirectly by causing a cellular defect that activated the checkpoint. To test this possibility, we examined whether Pds1p can inhibit Clb2p degradation in cells that are defective in activating the spindle assembly checkpoint as a result of the absence of Mad2p. As seen in Figure 2, Pds1-db could still inhibit Clb2p degradation (A,B) and cell cycle progression (C) in the absence of Mad2p, indicating that the inhibitory effect of Pds1p is independent of the spindle assembly checkpoint. This conclusion is further supported by the observation that Pds1-db does not inhibit the degradation of wild-type Pds1p (Cohen-Fix et al. 1996); had Pds1-db induced a checkpoint response, one would expect that the inhibition of APC/C activity would block wild-type Pds1p degradation as well.
Figure 2.



Pds1p’s ability to inhibit Clb2p degradation is independent of the spindle damage checkpoint protein Mad2p. Wild-type, GAL1–pds1-db, mad2-1, and mad2-1 GAL1–pds1-db cells (strains VG906-1A, VG1369, VG1346-10D, and OCF2101, respectively), were grown in YPR and arrested in HU as described in the legend to Fig. 1. The expression of GAL1–pds1-db was induced for 30 min, after which the cells were released from the HU arrest into fresh YPRG medium containing the α mating factor, to inhibit cell cycle progression in the following G1. Samples were taken at the indicated time points and processed for Western blot analysis (A,B) or cell morphology (C), as described in Materials and Methods. (B) The relative Clb2p levels as detected by Western blot analysis (A) in the different strains, as indicated. (C) The distribution of cell types in the mad2-1 and mad2-1 GAL1–pds1-db cultures as scored by DAPI staining. The percentage of large budded, single nucleated cells (squares) or anaphase cells (circles), in the absence (open symbols) or presence (solid symbols) of Pds1-db, are shown.
Clb2p does not inhibit Pds1p degradation
The regulatory mechanism that is emerging from the results presented above is that the sequential order of degradation of APC/C substrates may be determined by the substrates themselves, such that an early substrate, that is, Pds1p, inhibits the degradation of a later substrate, for example, Clb2p. One predication from such a mechanism is that a later substrate would not inhibit the degradation of an earlier one. It has been shown previously that the presence of nondegradable Clb2p (Clb2-db) results in a late anaphase arrest (Surana et al. 1993), but it was not known whether under these conditions Pds1p was still being degraded at the metaphase-to-anaphase transition. To test whether Pds1p’s degradation is affected by Clb2-db, cells containing the clb2-db gene under a galactose-inducible promoter were arrested in S phase as described above, and then released, with and without inducing the expression of Clb2-db (Fig. 3). As expected, the presence of Clb2-db in the induced culture caused a cell cycle arrest in late mitosis (data not shown). However, Clb2-db had no effect on the rate of Pds1p degradation (Fig. 3A,B) or on the timing of the metaphase-to-anaphase transition (Fig. 3C). Therefore, it is not that any stable APC/C target can inhibit the degradation of another APC/C substrate, and this further supports the specificity of Pds1p’s ability to inhibit Clb2p degradation in mitosis.
Figure 3.



Clb2p does not inhibit Pds1p degradation in mitosis. A strain carrying PDS1–HA and a single copy of GAL1–clb2-dbΔ (OCF1557) was grown at 30°C in YPR and arrested in S phase with HU, as described. The culture was then split, and galactose was added to one of the new cultures to induce the expression of Clb2-db. An hour later, the HU was washed out. The uninduced culture was released into YPD, whereas the induced culture was released into YPRG. Samples for protein analysis and cell morphology were taken at the indicated time points, and processed as described in Materials and Methods. (A,B) Western blot analysis of Pds1–HA (left) and Clb2p (right), and the calculated relative Pds1p–HA levels (B) in the presence and absence of Clb2-db. Time 0 indicates the time point at which the cells were released from the S-phase arrest. The galactose induction started 60 min prior to the release (indicated as −60 min.). This strain also carries a deletion of the endogenous CLB2 gene, and as a result no Clb2p can be seen in the noninduced culture. The increase in the Pds1p–HA levels 120 min after the release (A) is due to the presence of cells that have entered a new cell cycle, whereas in the induced culture, cells remain blocked at late anaphase (data not shown). (C) Cell type distribution in the presence or absence of Clb2-db, as scored by DAPI staining. The percentage of large budded, single nucleated cells (squares) or anaphase cells (circles), in the absence (open symbols) or presence (solid symbols) of Pds1-db are shown.
Pds1p does not inhibit Clb2p degradation in G1
The most direct way by which Pds1p could inhibit Clb2p degradation is by inhibiting APC/C’s activity. If that were the case, one would expect that whenever Pds1p is present, Clb2p would be stabilized. In G1 phase, Clb2p is highly unstable and does not accumulate even when expressed from a strong inducible promoter (Amon et al. 1994). This is not the case in cells defective for APC/C function, in which the expression of Clb2p from an inducible promoter in G1-arrested cells leads to a dramatic increase in the protein level (Amon et al. 1994; see below). If Pds1p was a direct inhibitor of the APC/C form that is responsible for Clb2p degradation, then the presence of Pds1p in G1 should mimic APC/C inactivation.
To test this possibility, we compared the accumulation of Clb2p expressed from a galactose inducible promoter in the presence or absence of Pds1-db. As a control, we examined Clb2p’s accumulation in cells arrested in mitosis (in which Clb2p should be stable regardless of Pds1p’s presence), and in G1-arrested cells that were defective for APC/C function because of a conditional mutation in the CDC16 gene. All strains were arrested in G1 or mitosis, and the expression of Clb2p and Pds1-db was induced for 1 hr by the addition of galactose (Fig. 4). This induction was sufficient to allow for Clb2p accumulation in mitotic cells (Fig. 4A), and in G1-arrested cells that were defective for APC/C function (Fig. 4A, the cdc16-123 strain). In contrast, Clb2p did not accumulate in G1-arrested wild-type cells or in G1-arrested cells expressing Pds1-db (Fig. 4A,B). Longer times of induced expression in G1 also did not lead to preferential Clb2p accumulation in the strain expressing Pds1-db (data not shown). Thus, the ability of Pds1p to inhibit Clb2p degradation is restricted to mitosis, and it suggests that Pds1p regulates the activation of APC/C, rather than APC/C itself. According to this model, by the time cells reach G1, the cyclin-specific APC/C form has already been activated, and Pds1p can no longer inhibit Clb2p degradation.
Figure 4.


Pds1p does not inhibit cyclin degradation in G1. The strains used were as follows: wild-type (OCF1517.0, WT1), GAL1–pds1-db (OCF1517.2), wild-type (K1534, WT2), and cdc16-123 (K4438). All strains carried a plasmid encoding for GAL–CLB2–HA (pWS944 in WT1 and pds1-db; pOC16 in WT2 and cdc16-123). All strains were grown at 23°C in nonselective medium (YPR), and arrested in G1 with α factor (10−6 m final concentration), or in mitosis with nocodazole (15 μg/ml final concentration). When all cultures were arrested, they were shifted to 37°C, to inactivate the cdc16-123 gene product. Thirty minutes later, while maintaining the temperature at 37°C, dextrose (D) or galactose (G) were added (2% final concentration). The samples shown here were taken 1 hr later and processed for Western blot analysis as described in Materials and Methods. (A) The levels of Clb2p–HA with and without the galactose induction, in mitosis (top) and in G1 (bottom), detected by using anti-HA antibodies. (B) The levels of β-tubulin in each of the samples from the G1 arrested cultures, to show equal protein loading in all lanes. (C) Pds1-db in G1 and mitosis, in the presence of dextrose (D) or galactose (G), demonstrating that the lack of Clb2p stabilization in G1 in the GAL–pds1-db strain is not due to the lack of Pds1p-db induction.
Pds1p inhibits Cdh1p dephosphorylation
The activation of APC/C toward cyclin ubiquitination involves the association of APC/C with Cdh1p (Schwab et al. 1997; Visintin et al. 1997). Zachariae et al. (1998a) have recently demonstrated that this association occurs during mitosis, and that Cdh1p must be dephosphorylated for this association to take place. Because our results indicate that Pds1p inhibits cyclin degradation by inhibiting APC/C activation prior to G1, it could be doing so by preventing Cdh1p dephosphorylation in mitosis. To test this possibility, we examined the phosphorylation state of an HA-tagged Cdh1p following the release from an S-phase arrest in the presence or absence of Pds1-db. In S phase, Cdh1p was phosphorylated (Fig. 5, time 0; Zachariae et al. 1998a). Following the release from the S-phase arrest in the absence of Pds1-db, Cdh1p became dephosphorylated, and Clb2p was degraded (Fig. 5, left). In contrast, when cells were released from the S-phase arrest in the presence of Pds1-db, Cdh1p did not undergo dephosphorylation and consequently Clb2p was not degraded (Fig. 5, right). Thus, Pds1p appears to inhibit Clb2p degradation by inhibiting Cdh1p dephosphorylation, thus preventing the formation of the Cdh1p-associated form of APC/C that is responsible for cyclin degradation.
Figure 5.

Pds1-db inhibits the dephosphorylation of Cdh1p. Wild-type (OCF 2108) and GAL–pds1-db (OCF 2109) cells, both carrying an integrated copy of CDH1–HA, were grown at 30°C in YPR and arrested in S phase with HU, as described above. When cells were arrested, galactose was added to both cultures to induce the expression of Pds1-db in the OCF2109 strain. Thirty minutes later, the HU was washed away and the cells were released into fresh YPRG medium containing the α-mating pheromone (10−6 m final concentration), to capture cells in the following G1 phase. Samples were taken at the indicated time points and processed for Western blot analysis as described in Materials and Methods. Cdh1p–HA was detected with anti-HA antibodies, and Clb2p was detected with anti-Clb2p antibodies. The phosphorylation state of Cdh1p can be determined by the reduced migration of the phosphorylated Cdh1p forms (p) on SDS–polyacrylamide gels (Zachariae et al. 1998a).
Inhibition of cyclin degradation by Pds1p is independent of Pds1p’s inhibitory activity on anaphase initiation
One could imagine that Clb2p degradation is a direct consequence of anaphase initiation, and therefore Pds1p could prevent Clb2p degradation indirectly by inhibiting the metaphase-to-anaphase transition. Anaphase initiation requires the activity of Esp1p, the only known target of Pds1p (Ciosk et al. 1998). Therefore, if Pds1p’s ability to inhibit cyclin degradation stemmed from Pds1p’s inhibitory action on Esp1p, it would be expected that inactivating Esp1p directly should also inhibit cyclin degradation. esp1-1 mutant cells are defective in anaphase initiation in that sister chromatid separation and spindle elongation fail to take place, but these cells still exit mitosis, exhibiting a cut phenotype (McGrew et al. 1992). As the esp1-1 cells exit the aberrant mitosis, the H1 kinase activity of Cdk1 drops (Surana et al. 1993), consistent with cyclin degradation at that stage. However, Cdk1 H1 kinase levels can decrease because of the presence of the Sic1p inhibitor, without a reduction in the amount of the Cdk1-associated Clb2p (Schwab et al. 1997). To test directly whether cyclin degradation is dependent on the prior execution of anaphase, we examined the levels of Clb2p in esp1-1 mutant cells undergoing mitosis under nonpermissive conditions. To insure that the esp1-1 gene product was inactivated, the staging and temperature shift were done in G1 rather than S phase. esp1-1 mutant cells reached mitosis about 20 min later than wild-type cells, as indicated by flow cytometry analysis (data not shown) and the delay in the appearance of Clb2p (Fig. 6A). Still, Clb2p was degraded in the esp1-1 cells as they exited mitosis, albeit with a significant delay, indicating that inhibiting anaphase initiation is not sufficient to inhibit cyclin degradation. Hence, for Pds1p to inhibit cyclin degradation it must do something in addition to its known function as an anaphase inhibitor. Whether Pds1p inhibits Clb2p degradation via a protein other than Esp1p is currently unknown, but the finding that esp1-1 mutant cells delay in degrading Clb2p suggests that Esp1p may be required for efficient Clb2p degradation (see Discussion).
Figure 6.




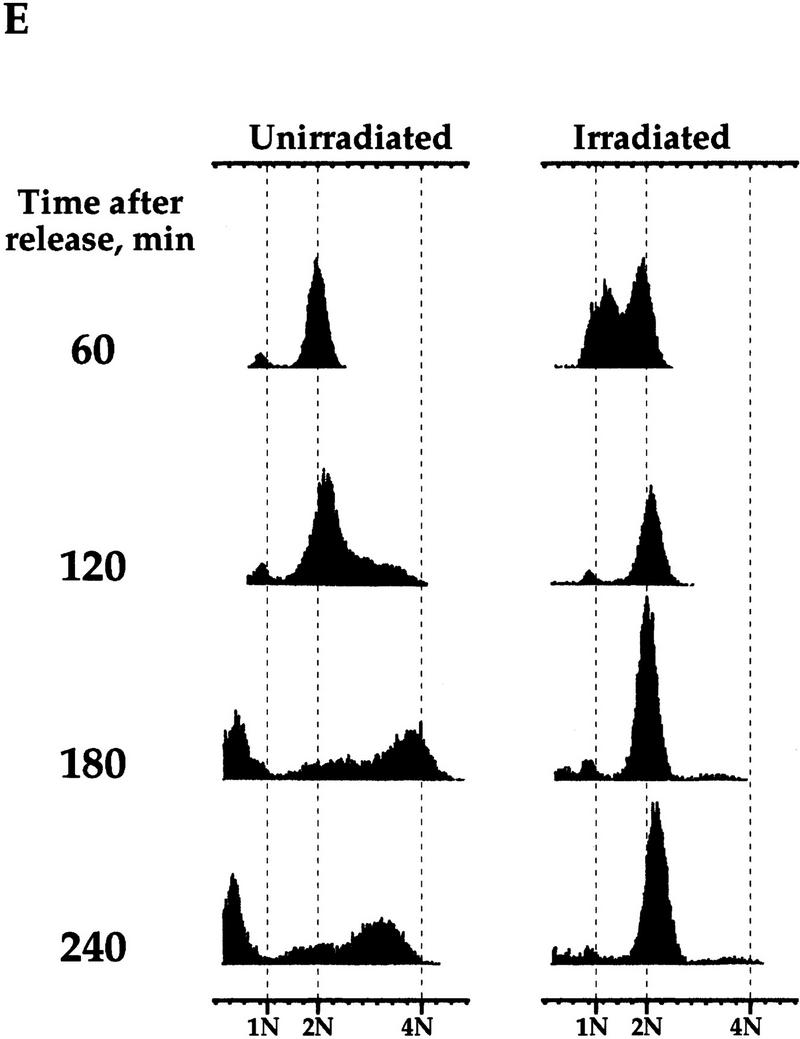
The regulation of Clb2p degradation by Pds1p is independent of sister chromatid cohesion. (A) Wild-type (K1534) and esp1-1 mutant cells (OCF2122-2A) were grown at 23°C and arrested in G1 with α factor. When the arrest was complete, the cultures were shifted to 37°C for 30 min to inactivate the esp1-1 gene product, and then released from the arrest at the nonpermissive temperature. Samples were taken at the indicated time points and analyzed for the presence of Clb2p by Western blot analysis with anti-Clb2p antibodies. (B,C,D) esp1-1 (OCF1561-6C) and esp1-1 GAL–pds1-db (OCF2111-3A) cells were grown at 23°C in YPR and arrested in G1 with α factor, as described above. When all cultures were arrested, galactose was added to induce the expression of Pds1-db, and 30 min later the cultures were shifted to 37°C. After the temperature shift, the α factor was washed out, and the cells were released into fresh YPRG that was prewarmed to 37°C. Samples were collected at the indicated time points and processed for Western blot analysis (B,C) and cell morphology (D) as described in Materials and Methods. (□) −Pds1-db; (●) +Pds1-db in C and D. (B) The levels of Clb2p in the wild-type and Pds1-db strains. (C) The relative Clb2p levels as calculated from the Western blot shown in B. (D) Postmitotic cells were defined as any cell type other than shmooed cells and large budded cells with a single nucleus. At the last time point of the control cells (−Pds1-db), this category included unbudded, single nucleated cells (9%), anucleated unbudded cells (8%), cells with two buds and a single nucleus, which presumably represent cells that rebudded without undergoing cytokinesis or nuclear division (10%), and large budded cells with two grossly unequal DNA masses (3%). (E) esp1-1 cells (2788) were grown in YPD and arrested in G1 with α factor as described above. The culture was concentrated, and either left untreated or exposed to γ irradiation [4 krads, (Cohen-Fix and Koshland 1997a)]. Both cultures were incubated at 37°C for 30 min prior to being released from the G1 arrest. Cells were then maintained at 37°C for the remainder of the experiment. Samples were collected at the indicated time point and processed for DNA content by flow cytometry. In the unirradiated culture, the appearance of DNA peaks >2N and <1N (180 min after release) was accompanied by the appearance of cells containing little or no nuclear DNA (data not shown), typical of cells exiting mitosis without undergoing anaphase (McGraw et al. 1992).
If the ability of Pds1p to inhibit cyclin degradation is independent of its function as an anaphase inhibitor, one would expect that Pds1p could still inhibit cyclin degradation in the esp1-1 mutant. This was examined in two ways; one was by expressing Pds1p-db in the esp1-1 mutant under nonpermissive conditions and examining its ability to inhibit cyclin degradation. As can be seen in Figure 6, Pds1p-db did inhibit cyclin degradation (B,C) and exit from mitosis (D) in the esp1-1 mutant. The other way we examined the ability of Pds1p to inhibit cyclin degradation in the esp1-1 mutant was by testing whether esp1-1 mutant cells grown at the nonpermissive temperature can be arrested in mitosis in the presence of DNA damage. On induction of DNA damage, the DNA damage checkpoint pathway causes cells to arrest in mitosis. In this arrest, Pds1p is essential for preventing the metaphase-to-anaphase transition as well as the exit of mitosis and the degradation of the mitotic cyclins (Yamamoto et al. 1996b; Tinker-Kulberg and Morgan 1999). Hence, the induction of DNA damage is analogous to the presence of Pds1p-db. Irradiation of the esp1-1 mutant led to a mitotic arrest, whereas nonirradiated esp1-1 mutant cells exited mitosis, displaying a DNA content profile that is typical of the cut phenotype (Fig. 6E). Therefore, Pds1p’s ability to inhibit cyclin degradation, and hence block the exit from mitosis, is independent of its function as an anaphase inhibitor.
Discussion
Two transitions in mitosis are regulated by APC/C-mediated degradation, the metaphase-to-anaphase transition, when Pds1p is degraded, and the exit from mitosis, when the mitotic cyclins, Ase1p, and other proteins are degraded. The precocious activation of mitotic cyclin degradation can lead to the premature exit from mitosis, but under normal conditions this is likely to be prevented by regulatory mechanisms that couple the exit of mitosis to the completion of earlier processes. Here we show that Pds1p, which was formerly known to inhibit anaphase initiation, is also involved in inhibiting the degradation of the mitotic cyclins Clb2p and Ase1p. The ability of Pds1p to inhibit Clb2p degradation is restricted to mitosis, is independent of the mitotic/spindle damage checkpoint pathway, and does not occur in the G1 phase of the cell cycle. Similar observations were also made by Tinker-Kulberg and Morgan (1999). In addition, we found that Pds1p inhibited Cdh1p dephosphorylation, a prerequisite for Cdh1p’s association with APC/C and consequently cyclin ubiquitination. Taken together, our results suggest that Pds1p is an inhibitor of mitotic cyclin degradation, and it does so by specifically inhibiting the activation of the APC/C form that is required for the exit of mitosis, namely APC/CCdh1p.
Until now, the only known molecular function for Pds1p in mitosis was to inhibit the metaphase-to-anaphase transition by associating with Esp1p, an activator of anaphase initiation (Cohen-Fix et al. 1996; Yamamoto et al. 1996a; Ciosk et al. 1998). The ability of Pds1p to inhibit Cdh1p dephosphorylation and cyclin destruction, functions that are required at the end of mitosis, raises two questions: Is this activity independent of Pds1p’s function as an anaphase inhibitor, and, if this is a novel activity, is it mediated via a protein or proteins other than Esp1p? One could have easily imagined that Pds1p has only one primary function as an anaphase inhibitor, and that cyclin destruction is a consequence of anaphase initiation. However, it has been shown previously that despite their inability to initiate proper anaphase, esp1-1 mutant cells can exit mitosis (McGrew et al. 1992), and we demonstrated here that this exit from mitosis is accompanied by cyclin degradation. Therefore, blocking anaphase initiation is not sufficient to inhibit cyclin degradation, and hence the ability of Pds1p to inhibit cyclin degradation must represent a novel function. Consistent with this interpretation, in the esp1-1 mutant cells, Pds1p-db could still inhibit cyclin degradation, and the presence of DNA damage, which induces a Pds1p-dependent mitotic arrest, blocked the exit from mitosis. On the basis of these observations, we suggest that Pds1p not only regulates anaphase initiation, but is also independently an inhibitor of cyclin degradation.
The issue of whether Pds1p acts via a protein or proteins other than Esp1p is more complicated. Esp1p has not been shown previously to be involved in regulating cyclin degradation, and the fact that the mitotic cyclins are degraded in the esp1-1 mutant suggests that Pds1p may inhibit cyclin degradation through a protein other than Esp1p. However, we cannot rule out the possibility that at the nonpermissive temperature, the esp1-1 gene product is defective in promoting sister chromatid separation, but has partially retained a putative activity involved in regulating cyclin degradation. In fact, Tinker-Kulberg and Morgan (1999) have proposed that Esp1 is an activator of Clb2p degradation, because overexpression of Esp1p results in precocious cyclin degradation, in a Pds1p-independent manner. In addition, esp1-1 mutant cells exhibit a delay in initiating Clb2 degradation (this report; Tinker-Kulberg and Morgan 1999). This delay is not due a general retardation of cell cycle progression (this report), nor is it likely to be due to the activation of the mitotic checkpoint, as esp1-1 mutant cells degrade Pds1p with the same kinetics as wild-type cells (Ciosk et al. 1998). We have also noted that when the esp1-1 gene product was inactivated after S phase, there was a significant delay in the timing of cyclin degradation, without an apparent defect in chromosome segregation (O. Cohen-Fix, unpubl.). This observation is consistent with the interpretation that Esp1p has two functions, one in promoting sister chromatid separation and the other in inducing Clb2 degradation. If Esp1p is a positive regulator of cyclin degradation, then both Pds1p and Esp1p are involved in this novel regulatory pathway.
Our results define a new regulatory pathway for controlling the timing of Cdk1 inactivation (Fig. 7). In this pathway, Pds1p acts upstream of a group of proteins that function as a trigger in the inactivation of Cdk1 at the end of mitosis (Visintin et al. 1998,1999; for review, see Morgan 1999; Shou et al. 1999). This trigger may include the Cdc14, Cdc15, and Cdc5 proteins, and perhaps also Esp1p. Pds1p could associate with and inhibit one or more of the components of the trigger, thereby preventing it from firing. A previously known regulatory mechanism for Cdk1 inactivation involves the Cdk1 inhibitor Sic1 (Schwob et al. 1994). It is important to note that these two regulatory pathways differ in two important ways. Whereas Sic1p is regulated by this trigger (Visintin et al. 1998), our results suggest that Pds1p is an inhibitor of the trigger. In addition, Sic1p’s role is limited to the exit of mitosis, whereas the Pds1p pathway couples the completion of an early event (anaphase) to the execution of a later one (exit from mitosis).
Figure 7.

Pds1p is an inhibitor of a molecular trigger that regulates the timing of mitotic cyclin degradation. In addition to its function as an anaphase inhibitor, Pds1p also affects the timing of cyclin degradation, possibly by inhibiting the activity of one or more proteins that act as a trigger in the activators of cyclin ubiquitination (see text for details).
Although our model provides the molecular basis for the temporal order of events in mitosis, it does not explain the observed lag between the initiation of Pds1p degradation and that of Clb2p degradation. This lag period coincides with the length of anaphase, and it allows cells to complete anaphase before exiting mitosis. The cause of this lag is unknown, but it is possible that cyclin degradation is inhibited by a subset of the Pds1p molecules that are not degraded at the metaphase-to-anaphase transition. According to this model, the Pds1p responsible for inhibiting sister chromatid separation is degraded at the time of anaphase initiation. A separate pool that regulates the exit from mitosis remains resistant to degradation, possibly due to its association with components of the trigger, thereby reducing its affinity to APC/C. This Pds1p would eventually get degraded, thereby allowing for anaphase to be completed before cyclin degradation induces the exit of mitosis. Consistent with this possibility, we do observe very low levels of Pds1p in cells that are arrested late in mitosis by the inactivation of the Cdc15 protein (Cohen-Fix et al. 1996; data not shown).
Another source for the lag between the time of Pds1p degradation and the initiation of cyclin degradation is that cyclin degradation itself may have a built-in time-delay mechanism. As mentioned earlier, Cdh1p association with APC/C depends on Cdh1p’s dephosphorylation. It has been shown that Cdk1 itself is responsible for the phosphorylation of Cdh1p (Zachariae et al. 1998a; Jaspersen et al. 1999). Therefore, there is a balance between the phosphorylation and dephosphorylation rates of Cdh1p, and this balance is shifted toward the phosphorylated state as long as the trigger is inhibited by the presence of Pds1p. Once Pds1p is gone, the trigger is fired, and the dephosphorylation reaction of Cdh1p is activated. This, in turn, initiates a positive feedback loop for cyclin degradation; as more dephosphorylated Cdh1p associates with APC/C, the rate of cyclin degradation increases, resulting in overall lower Cdk1 activity, which would further shift Cdh1p toward the dephosphorylated state. This positive feedback loop can allow cyclin degradation to build up gradually once Pds1p is degraded, thereby providing the time period needed for cells to progress though anaphase.
There are likely to be multiple, overlapping control mechanisms that regulate progression through mitosis, and the built-in timing mechanism for cyclin degradation proposed here may be sufficient to insure the proper timing of exit from mitosis, even when Pds1p is absent. However, the absence of Pds1p does cause a marked reduction in cell viability, and this is greatly exacerbated under growth conditions that are less than optimal (e.g., intracellular damage), when progression through the cell cycle must be further delayed or even stopped (Yamamoto et al. 1996a). Hence, the regulatory role played by Pds1p in coupling the metaphase-to-anaphase transition to the exit of mitosis is crucial for enabling cells to adapt to changing environmental conditions.
Materials and methods
Strains and plasmids
The strains used were as follows: Strains OCF1517.0, OCF1517.2, K1534, and K4438 were described previously (Cohen-Fix et al. 1996); OCF1565, as OCF1517.0 but also PDS1–HA::URA3; OCF1566, as 1517.2 but also PDS1–HA::URA3; OCF2108, as K1534 but also CDH1–HA::TRP1; OCF2109, as OCF1517.2, but also CDH1–HA::TRP1; VG906-1A, MATa bar1 trp1 leu2; VG1369, as VG906-1A but also LEU2::GAL1–pds1–mdb; VG1346-10D, MATa bar1 mad2-1 leu2 trp1; OCF2101, as VG1346-10D but also LEU2::GAL1–pds1–mdb; OCF1557, MATa ade2 leu2 his3 clb2::LEU2 TRP1:: GAL–clb2–db PDS1–HA::URA3; OCF1561-6C, MATa lys2 leu2 ura3 esp1-1; OCF2111-3A, MATa esp1-1 LEU2::GAL1–pds1–mdb; K2788 MATa esp1-1 ura3 leu2 lys2; OCF2122-2A, MATa bar1 esp1-1 leu2. The plasmids used were as follows: pWS944, a YCplac22-based vector carrying GAL10–CLB2–HA (a generous gift from Bruce Futcher, Cold Spring Harbor Laboratory); pOC16 is the same as pWS944, but in a Ycplac111 backbone.
Cell cycle arrest and release
YPD, YPR, and YPRG medium contained 1% yeast extract, 2% bactopeptone, and 2% glucose (YPD), raffinose (YPR), or raffinose and galatose (YPRG). Cell cycle arrests were performed with hydroxyurea (HU) (Sigma, 0.2 m final concentration) or α-factor (Sigma, 5 μm final concentration). Release from the cell cycle arrest was done by washing cultures in fresh medium, at the appropriate temperature. In the case of release from the α-factor-induced arrest, the medium also contained Pronase (Sigma, 0.1 mg/ml final concentration).
Western blot analysis
Western blot analysis was carried out as described (Cohen-Fix et al. 1996). The primary antibodies used were 12CA5 against the HA epitope (BAbCO, Richmond, CA); rabbit anti-Clb2p (a generous gift from David Morgan, UCSF); rabbit anti-Ase1p (a generous gift from David Pellman, Dana-Farber Cancer Institute, Boston, MA); rabbit anti-β tubulin, raised against amino acids 438–457 of yeast β-tubulin; and rabbit anti-Pds1p. Quantification of band intensity was by an Agfa Arcus 2 scanner, and NIH Image version 1.61. To account for possible differences in sample loading and increased protein amounts as cells progressed through the cell cycle, relative protein levels were normalized to a protein band that was constant throughout the cell cycle. Values are presented as a fraction of the protein amount at the first time point shown, and were graphed on a log scale to reflect the magnitude in protein level changes, that were up to 100-fold.
Other methods
Flow cytometry analysis was done as described (Yamamoto et al. 1996a). γ irradiation was done as described (Cohen-Fix and Koshland 1997a).
Acknowledgments
We thank Vincent Guacci, Amgelica Amon, Kim Nasmyth, Bruce Futcher, and Wolfgang Seufert for strains and plasmids, David Morgan for anti-Clb2p antibodies, and David Pellman for anti-Ase1p antibodies. We also thank David Morgan and Rachel Tinker-Kulberg for communicating results prior to publication, and Chris Hardy, Michael Lichten, Alex Strunnikov, April Robins, and members of the Cohen-Fix and Koshland laboratories for stimulating discussions and comments on the manuscript. This work was funded in part by a National Institutes of Health (NIH) grant (GM18382-02) to O.C.F. D.K. is supported by an NIH grant (GM1718), and is an investigator with the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL ornacf@helix.nih.gov; FAX (301) 402-0053.
References
- Amon A. Regulation of B-type cyclin proteolysis by Cdc28-associated kinases in budding yeast. EMBO J. 1997;16:2693–2702. doi: 10.1093/emboj/16.10.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— The spindle checkpoint. Curr Opin Genet Dev. 1999;9:69–75. doi: 10.1016/s0959-437x(99)80010-0. [DOI] [PubMed] [Google Scholar]
- Amon A, Irniger S, Nasmyth K. Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell. 1994;77:1037–1050. doi: 10.1016/0092-8674(94)90443-x. [DOI] [PubMed] [Google Scholar]
- Ciosk R, Zachariae W, Michaelis C, Shevchenko A, Mann M, Nasmyth K. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell. 1998;93:1067–1076. doi: 10.1016/s0092-8674(00)81211-8. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Koshland D. The anaphase inhibitor of Saccharomyces cerevisiae Pds1p is a target of the DNA damage checkpoint pathway. Proc Natl Acad Sci. 1997a;94:14361–14366. doi: 10.1073/pnas.94.26.14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— The metaphase-to-anaphase transition: Avoiding a mid-life crisis. Curr Opin Cell Biol. 1997b;9:800–806. doi: 10.1016/s0955-0674(97)80080-4. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Peters JM, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes & Dev. 1996;10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes & Dev. 1998;12:1871–1883. doi: 10.1101/gad.12.12.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funabiki H, Kumada K, Yanagida M. Fission yeast Cut1 and Cut2 are essential for sister chromatid separation, concentrate along the metaphase spindle and form large complexes. EMBO J. 1996a;15:6617–6628. [PMC free article] [PubMed] [Google Scholar]
- Funabiki H, Yamano H, Kumada K, Nagao K, Hunt T, Yanagida M. Cut2 proteolysis required for sister-chromatid seperation in fission yeast. Nature. 1996b;381:438–441. doi: 10.1038/381438a0. [DOI] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Morgan DO. Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol. 1999;9:227–236. doi: 10.1016/s0960-9822(99)80111-0. [DOI] [PubMed] [Google Scholar]
- Juang YL, Huang J, Peters JM, McLaughlin ME, Tai CY, Pellman D. APC-mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science. 1997;275:1311–1314. doi: 10.1126/science.275.5304.1311. [DOI] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. Cell 81: 279–288. 1995. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. [DOI] [PubMed] [Google Scholar]
- Kumada K, Nakamura T, Nagao K, Funabiki H, Nakagawa T, Yanagida M. Cut1 is loaded onto the spindle by binding to Cut2 and promotes anaphase spindle movement upon Cut2 proteolysis. Curr Biol. 1998;8:633–641. doi: 10.1016/s0960-9822(98)70250-7. [DOI] [PubMed] [Google Scholar]
- Li Y, Gorbea C, Mahaffey D, Rechsteiner M, Benezra R. MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. Proc Natl Acad Sci. 1997;94:12431–12436. doi: 10.1073/pnas.94.23.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrew JT, Goetsch L, Byers B, Baum P. Requirement for ESP1 in the nuclear division of Saccharomyces cerevisiae. Mol Biol Cell. 1992;3:1443–1454. doi: 10.1091/mbc.3.12.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35–45. doi: 10.1016/s0092-8674(01)80007-6. [DOI] [PubMed] [Google Scholar]
- Morgan DO. Regulation of APC and the exit from mitosis. Nat Cell Biol. 1999;1:E47–E53. doi: 10.1038/10039. [DOI] [PubMed] [Google Scholar]
- Palmer RE, Koval M, Koshland D. The dynamics of chromosome movement in the budding yeast Saccharomyces cerevisiae. J Cell Biol. 1989;109:3355–3366. doi: 10.1083/jcb.109.6.3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM. SCF and APC: The yin and yang of cell cycle regulated proteolysis. Curr Opin Cell Biol. 1998;10:759–768. doi: 10.1016/s0955-0674(98)80119-1. [DOI] [PubMed] [Google Scholar]
- Schwab M, Lutum AS, Seufert W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 1997;90:683–693. doi: 10.1016/s0092-8674(00)80529-2. [DOI] [PubMed] [Google Scholar]
- Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Shou W, Seol JH, Shevchenko A, Baskerville C, Moazed D, Chen ZW, Jang J, Charbonneau H, Deshaies RJ. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell. 1999;97:233–244. doi: 10.1016/s0092-8674(00)80733-3. [DOI] [PubMed] [Google Scholar]
- Straight AF, Marshall WF, Sedat JW, Murray AW. Mitosis in living budding yeast: Anaphase A but no metaphase plate. Science. 1997;277:574–578. doi: 10.1126/science.277.5325.574. [DOI] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–197. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surana U, Amon A, Dowzer C, McGrew J, Byers B, Nasmyth K. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO J. 1993;12:1969–1978. doi: 10.1002/j.1460-2075.1993.tb05846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinker-Kulberg, R.L. and D.O. Morgan. 1999. Pds1 and Esp1 control both anaphase and mitotic exit in normal cells and following DNA damage. Genes & Dev. (This issue). [DOI] [PMC free article] [PubMed]
- Visintin R, Prinz S, Amon A. CDC20 and CDH1: A family of substrate-specific activators of APC- dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk- dependent phosphorylation. Mol Cell. 1998;2:709–718. doi: 10.1016/s1097-2765(00)80286-5. [DOI] [PubMed] [Google Scholar]
- Visintin R, Hwang ES, Amon A. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus . Nature. 1999;398:818–823. doi: 10.1038/19775. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Guacci V, Koshland D. Pds1p is required for faithful execution of anaphase in the yeast, Saccharomyces cerevisiae. J Cell Biol. 1996a;133:85–97. doi: 10.1083/jcb.133.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s) J Cell Biol. 1996b;133:99–110. doi: 10.1083/jcb.133.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano H, Tsurumi C, Gannon J, Hunt T. The role of the destruction box and its neighbouring lysine residues in cyclin B for anaphase ubiquitin-dependent proteolysis in fission yeast: Defining the D-box receptor. EMBO J. 1998;17:5670–5678. doi: 10.1093/emboj/17.19.5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Peters JM, King RW, Page AM, Hieter P, Kirschner MW. Identification of a cullin homology region in a subunit of the anaphase- promoting complex. Science. 1998;279:1219–1222. doi: 10.1126/science.279.5354.1219. [DOI] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 1998a;282:1721–1724. doi: 10.1126/science.282.5394.1721. [DOI] [PubMed] [Google Scholar]
- Zachariae W, Shevchenko A, Andrews PD, Ciosk R, Galova M, Stark MJ, Mann M, Nasmyth K. Mass spectrometric analysis of the anaphase-promoting complex from yeast: Identification of a subunit related to cullins. Science. 1998b;279:1216–1219. doi: 10.1126/science.279.5354.1216. [DOI] [PubMed] [Google Scholar]