

The crystal structure of the N-terminal fragment of the short nonmuscle α-tropomyosin has been determined at a resolution of 0.98 Å.
Keywords: coiled coils, actin-binding proteins, tropomodulin-binding proteins, cytoskeletal proteins
Abstract
Tropomyosin (TM) is an elongated two-chain protein that binds along actin filaments. Important binding sites are localized in the N-terminus of tropomyosin. The structure of the N-terminus of the long muscle α-TM has been solved by both NMR and X-ray crystallography. Only the NMR structure of the N-terminus of the short nonmuscle α-TM is available. Here, the crystal structure of the N-terminus of the short nonmuscle α-TM (αTm1bZip) at a resolution of 0.98 Å is reported, which was solved from crystals belonging to space group P31 with unit-cell parameters a = b = 33.00, c = 52.03 Å, α = β = 90, γ = 120°. The first five N-terminal residues are flexible and residues 6–35 form an α-helical coiled coil. The overall fold and the secondary structure of the crystal structure of αTM1bZip are highly similar to the NMR structure and the atomic coordinates of the corresponding Cα atoms between the two structures superimpose with a root-mean-square deviation of 0.60 Å. The crystal structure validates the NMR structure, with the positions of the side chains being determined precisely in our structure.
1. Introduction
Tropomyosin (TM) is an elongated two-chain α-helical coiled coil (for a review, see Gunning et al., 2005 ▶). More than 40 long and short TM isoforms are encoded by four genes utilizing alternative splicing of nine exons. The distribution of TM isoforms varies in different tissues and changes during development. TM molecules bind along the actin filament. TM stabilizes actin filaments by preventing their severing and depolymerization; it regulates actin dynamics and the interactions of actin with other actin-binding proteins.
Important binding sites are localized in the N-terminus of TM. It forms a complex with the C-terminus of TM that allows TMs to form a long rod along the actin filament. It also contains the tropomodulin (Tmod) binding site (for a review, see Kostyukova, 2008 ▶). When Tmod binds to the TM at the pointed end of the actin filament it forms a tight cap that prevents polymerization/depolymerization of the filament and stabilizes its length. There are four Tmod isoforms (Tmod1–4); Tmod1 can be found both in muscle and nonmuscle cells. Its interaction with nonmuscle TMs, namely short nonmuscle α-TM, is important for cytoskeleton formation in cells, e.g. during neuron development (Gunning et al., 2005 ▶) or the assembly of the membrane skeleton in erythrocytes (Sung et al., 2000 ▶).
To study the structure and function of the N-terminus of short nonmuscle α-TM, a chimeric peptide was designed (Greenfield et al., 2001 ▶). It contained 19 N-terminal residues of the short α-TM encoded by exon 1b. To stabilize the coiled-coil structure, the peptide contained an additional 18 C-terminal residues of the GCN4 leucine-zipper domain. Structures of this peptide alone and in a complex with the C-terminal TM fragment have been solved by NMR (Greenfield et al., 2001 ▶, 2009 ▶).
Attempts to solve the structure of complexed TM and Tmod peptides using NMR were not successful. We attempted to crystallize the complex of these peptides. We used Tmod1 and short nonmuscle α-TM peptides in crystallization trials. However, we found that although this complex was sufficiently tight, with a K d of 0.09 ± 0.02 µM (Kostyukova et al., 2007 ▶), the crystals that we obtained contained only the TM fragment. Here, we report the crystal structure of the short nonmuscle α-TM peptide, αTm1bZip, at a resolution of 0.98 Å, which is the highest resolution obtained for a structure of a tropomyosin fragment.
2. Materials and methods
2.1. Sample preparation
A Tmod1 fragment (residues 1–38) corresponding to the first TM-binding site (Tmod1s1) and an N-acetylated chimeric TM peptide (αTm1bZip) were synthesized by the Tufts University Core Facility (Boston, Massachusetts, USA). The quality of the synthetic peptides was confirmed using mass spectroscopy; the molecular weights of the peptides were the same as those predicted. The peptide concentrations were determined using a BCA protein-assay kit (Thermo Fisher Science, Rockford, Illinois, USA) or by measuring their difference spectra in 6 M guanidine–HCl between pH 12.5 and 7.0 (Edelhoch, 1967 ▶) using extinction coefficients of 2357 per tyrosine and 830 per tryptophan (Fasman, 1989 ▶). Circular-dichroism measurements were made using an Aviv model 400 spectropolarimeter (Lakewood, New Jersey, USA) as described previously (Kostyukova et al., 2007 ▶).
2.2. Crystallization
The complex between the Tmod1 fragment (residues 1–38) and the αTM1bZip peptide was obtained by mixing equimolar amounts of the two components in 10 mM Tris–HCl pH 7.0. The final concentration of the complex was 10 mg ml−1. Screening of crystallization conditions was carried out by the sitting-drop vapour-diffusion method (150 nl protein solution mixed with 150 nl reservoir solution and equilibrated against 120 µl reservoir solution) at 293 K in 96-well plates using a Mosquito crystal nanolitre liquid handler (TTP LabTech) and the following screening kits: Crystal Screen and Crystal Screen 2 from Hampton Research, and Wizard I, II and III and Cryo I and II from Emerald BioSystems. Crystals were obtained using condition No. 30 of Wizard III (0.17 M ammonium sulfate, 25.5% PEG 4000, 15% glycerol; Fig. 1 ▶ a). Further optimization was performed using the microseeding technique. A crystal seed stock was produced from the original crystal using a Seed Bead kit (Hampton Research). The stock was diluted 1:300 with 0.17 M ammonium sulfate, 10% PEG 4000. Improved single crystals were obtained using the hanging-drop vapour-diffusion method in drops consisting of 2 µl complex solution and 2 µl diluted crystal seed solution equilibrated against 1 ml 0.17 M ammonium sulfate, 10% PEG 4000 (Fig. 1 ▶ b). To check the crystal content, several crystals were washed extensively in reservoir solution, dissolved in gel-loading buffer and loaded onto SDS–PAGE, which showed that the crystals contained the αTM1bZip fragment only.
Figure 1.

Crystals of αTM1bZip (a) obtained in 0.17 M ammonium sulfate, 25.5% PEG 4000, 15% glycerol and (b) obtained after microseeding in 0.17 M ammonium sulfate, 10% PEG 4000. Scale bar, 0.1 mm.
2.3. X-ray data collection, analysis and structure solution
Before data collection, crystals were soaked for several minutes in cryoprotectant solution consisting of the respective reservoir solution supplemented with 20% glycerol. Crystals were mounted in a nylon loop and flash-cooled in liquid nitrogen. A complete native X-ray diffraction data set was collected on beamline BL41XU at SPring-8 using a MAR225HE detector. The data were integrated and scaled with MOSFLM (Leslie, 2006 ▶) and SCALA (Evans, 2006 ▶) from the CCP4 package (Winn et al., 2011 ▶). Detection and analysis of crystal twinning was performed in phenix.xtriage from the PHENIX software package (Adams et al., 2002 ▶). Twinning correction was performed with the CCP4 program DETWIN using the twinning operator −h, −k, l. The crystals belonged to space group P31, with unit-cell parameters a = b = 33.00, c = 52.03 Å, α = β = 90, γ = 120° and one protein molecule (one dimer) in the asymmetric unit. The structure was solved by molecular replacement with Phaser (McCoy et al., 2007 ▶) using model 1 (residues 7–36) of the NMR structure of αTM1bZip as a search model (PDB entry 1ihq; Greenfield et al., 2001 ▶). The model was further refined in REFMAC5 (Murshudov et al., 2011 ▶). The final model consisted of 60 amino-acid residues and 91 water molecules; the crystallographic R factor was 0.167 and R free was 0.171 (Table 1 ▶). The structure was validated in PROCHECK (Laskowski et al., 1993 ▶) and deposited in the PDB under code 3azd.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data statistics | |
| Space group | P31 |
| Unit-cell parameters (Å, °) | a = b = 33.00, c = 52.03, α = β = 90.0, γ = 120.0 |
| Wavelength (Å) | 0.8 |
| No. of images | 280 |
| Resolution (Å) | 15.73–0.98 (1.03–0.98) |
| Completeness (%) | 98.6 (100.0) |
| Total reflections | 253608 (35877) |
| Unique reflections | 36372 (5431) |
| Multiplicity | 7.0 (6.6) |
| Rmerge† (%) | 10.7 (52.4) |
| Mean I/σ(I) | 11.3 (3.1) |
| Refinement statistics | |
| Resolution range (Å) | 14.29–0.98 |
| R factor | 0.167 |
| Rfree | 0.171 |
| No. of atoms (protein/water) | 504/91 |
| Mean B factor (Å2) | 13.56 |
| R.m.s. deviations from ideal | |
| Bond lengths (Å) | 0.019 |
| Bond angles (°) | 2.415 |
| Ramachandran plot (%) | |
| Most favoured | 94.6 |
| Additionally allowed | 5.4 |
| Disallowed | 0.0 |
R
merge = 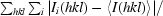
 , where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of Ii(hkl) for all i measurements.
, where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of Ii(hkl) for all i measurements.
3. Results and discussion
When the first crystals were obtained by screening, we analyzed complex formation under the same solution conditions using circular dichroism. The peptides (10 µM) were able to form a complex in the crystallization conditions. Of the two peptides, the TM peptide is well folded and the Tmod fragment is disordered. The Tmod fragment acquires structure upon complex formation; however, it is not clear whether part of it remains unstructured. SDS–PAGE of the crystals revealed that they contained TM peptide only. We think that the TM peptide started crystallizing and this changed the equilibrium between the complex and the individual components by removing one of the peptides from the reaction mixture. This caused further dissociation of the complex, resulting in the gradual growth of crystals composed of TM peptide only.
The structure of the chimeric peptide that contains the N-terminus of the long muscle α-TM has been solved by both NMR and X-ray crystallography at 2 Å resolution (Greenfield et al., 1998 ▶; Brown et al., 2001 ▶), but no crystal structure of the N-terminus of the short nonmuscle α-TM is available. The peptide we used for crystallization, αTM1bZip, was acetylated at the N-terminus as in the native protein, while the peptide used for NMR studies, GlyTM1bZip, was not acetylated but had an additional N-terminal Gly.
The Ramachandran plot generated with the program PROCHECK revealed no residues within disallowed regions (Table 1 ▶). There are 53 nonglycine residues (94.6%) in the most favoured regions and three residues in additionally allowed regions (5.4%). There is clear electron density for residues 6–35, which form an α-helical coiled coil (Fig. 2 ▶). No electron density was seen for the first five residues or residues 36–37. Therefore, we concluded that these residues are flexible in αTM1bZip. In the TM part of the chimeric peptide residues Leu6, Val9, Ile13 and Leu16 in the a and d positions of the heptad repeat have packing typical of a coiled coil. The N-terminal residues are also flexible in the NMR structure (Greenfield et al., 2001 ▶) and this is significantly different from the long TM. In the long TM the N-terminus is shorter by five residues and the coiled-coil domain starts from the first N-terminal residue (Greenfield et al., 1998 ▶).
Figure 2.

(a) Structure of αTM1bZip, with the chains shown in different colours. (b) Unbiased simulated-annealed F o − F c OMIT map at a contour level of 2σ calculated after deleting residues 19–22 in chain B. (c) Ribbon model of the chimeric TM peptides in the crystal (blue) and NMR model 1 (red) structures. The images were prepared with PyMOL (DeLano, 2002 ▶)
The overall fold and secondary structure of the crystal structure of αTM1bZip are highly similar to the NMR structure and the atomic coordinates of the corresponding Cα atoms between the two structures superimpose with a root-mean-square deviation of 0.60 Å (for model 1 of PDB entry 1ihq; Fig. 2 ▶ c). The high resolution and low B factors for individual atoms (the highest B factor is 17.55 Å2) allow definition of the exact positions of all side chains. Cα–Cα′ distances between the residues forming the coiled coil vary from 5.89 to 6.42 Å. The longest distance is between the leucines at position 6. From the NMR structure it was concluded that the packing of the side chains of Leu6 and Val9 is looser than that of Ile13 and Leu16 (Greenfield et al., 2001 ▶). The Cβ–Cβ′ distances (calculated as an average for ten NMR structures) are 6.01 (35) Å for Leu6, 5.19 (34) Å for Val9, 4.02 (6) Å for Ile13 and 3.76 (26) Å for Leu16. However, the corresponding distances in our structure are 5.82 Å for Leu6, 4.51 Å for Val9, 4.9 Å for Ile13 and 3.96 Å for Leu16. Therefore, in our structure it is only the packing of the side chain of Leu6 that is considerably less tight; it is therefore more exposed. The tighter packing of our structure may be a consequence of the N-terminal acetylation.
In general, the crystal structure validated the NMR structure of the N-terminus of short nonmuscle α-TM, with the positions of the side chains being determined precisely in our structure.
Supplementary Material
PDB reference: tropomyosin N-terminal fragment, 3azd
Acknowledgments
This work was supported by direct funding provided by OIST and in part by National Institutes of Health grant GM081688 (to ASK). The authors are grateful to Dr S.-Y. Park for his help in obtaining beam time and Dr N. Greenfield for discussion.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Brown, J. H., Kim, K.-H., Jun, G., Greenfield, N. J., Dominguez, R., Volkmann, N., Hitchcock-DeGregori, S. E. & Cohen, C. (2001). Proc. Natl Acad. Sci. USA, 98, 8496–8501. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Edelhoch, H. (1967). Biochemistry, 6, 1948–1954. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Fasman, G. D. (1989). Practical Handbook of Biochemistry and Molecular Biology, p. 84. Boca Raton: CRC Press.
- Greenfield, N. J., Huang, Y. J., Palm, T., Swapna, G. V., Monleon, D., Montelione, G. T. & Hitchcock-DeGregori, S. E. (2001). J. Mol. Biol. 312, 833–847. [DOI] [PubMed]
- Greenfield, N. J., Kotlyanskaya, L. & Hitchcock-DeGregori, S. E. (2009). Biochemistry, 48, 1272–1283. [DOI] [PMC free article] [PubMed]
- Greenfield, N. J., Montelione, G. T., Farid, R. S. & Hitchcock-DeGregori, S. E. (1998). Biochemistry, 37, 7834–7843. [DOI] [PubMed]
- Gunning, P. W., Schevzov, G., Kee, A. J. & Hardeman, E. C. (2005). Trends Cell Biol. 15, 333–341. [DOI] [PubMed]
- Kostyukova, A. S. (2008). Cell. Mol. Life Sci. 65, 563–569. [DOI] [PMC free article] [PubMed]
- Kostyukova, A. S., Hitchcock-Degregori, S. E. & Greenfield, N. J. (2007). J. Mol. Biol. 372, 608–618. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Sung, L. A., Gao, K.-M., Yee, L. J., Temm-Grove, C. J., Helfman, D. M., Lin, J. J. & Mehrpouryan, M. (2000). Blood, 95, 1473–1480. [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: tropomyosin N-terminal fragment, 3azd