

Abstract
Astrocytes are the most abundant cells in the brain, playing vital roles in neuronal survival, growth, and function. Understanding the mechanism(s) regulating astrocyte proliferation will have important implications in brain development, response to injury, and tumorigenesis. Cyclin B1 is well known to be a critical regulator of mitotic entry via its interaction with cyclin-dependent kinase 1. In rat astrocytes, we now show that the mRNA binding protein cytoplasmic polyadenylation element binding protein 1 (CPEB1) is associated with cyclin B1 mRNA and that this interaction is enriched at the centrosome. In addition, if growth-arrested astrocytes are stimulated to divide, CPEB1 is phosphorylated and cyclin B1 mRNA is polyadenylated, both hallmarks of CPEB1 activation, resulting in an increase in cyclin B1 protein. CPEB1 binding to mRNA initially inhibits translation; therefore, removing CPEB1 from mRNA should result in an increase in translation due to derepression. Indeed, when we either knocked down CPEB1 protein with siRNA or sequestered it from endogenous mRNA by expressing RNA containing multiple CPEB1 binding sites, cyclin B1 protein was increased and cell proliferation was stimulated. Our data suggest a mechanism wherein CPEB1 is bound and represses cyclin B1 mRNA translation until a signal to proliferate phosphorylates CPEB1, resulting in an increase in cyclin B1 protein and progression into mitosis. Our results demonstrate for the first time a role for CPEB1 in regulating cell proliferation in the brain.
Introduction
Astrocytes are the most abundant cells in the CNS and play a critical role in neuronal survival, growth, and functional integrity. Astrocytes provide nutritional support and protection for neurons and regulate metabolic homeostasis in brain. Astrocytes also play an important role in the cellular response to brain injury, proliferating to contain the damage site in a process known as reactive gliosis (Sofroniew and Vinters, 2010). In addition, the most common type of primary brain tumor is the astrocyte-derived glioblastoma, of which the most aggressive form (glioblastoma multiforma) has an expected survival time of ∼1 year after diagnosis (Kanu et al., 2009). Clearly, understanding the mechanism(s) regulating the proliferation of astrocytes will be important in controlling the outcome of traumatic brain injury and in designing therapeutics to combat glioblastoma.
New protein synthesis is an essential process in the regulation of numerous physiological processes, including cell proliferation, differentiation, migration, and cellular adaptation to extracellular environment. Historically, transcription factors were considered the primary regulators of gene expression. However, this notion has been challenged in the past decade with the identification of mRNA-specific binding proteins and micro-RNA species that target specific mRNA(s). Translational regulation of protein expression has several advantages over transcriptional regulation, such as rapidity of response and the ability to spatially localize the response by targeting mRNA to cellular subdomains (Kiebler and Bassell, 2006).
Cytoplasmic polyadenylation element binding protein 1 (CPEB1) is one such mRNA-specific binding protein that has important roles in cell cycle regulation in germ cells as well as in experience-induced synaptic changes in neurons (Tay and Richter, 2001; McEvoy et al., 2007). CPEB1 binds to cytoplasmic polyadenylation elements (CPEs) in 3′-untranslated region (3′-UTR) of target mRNA. When bound, CPEB1 forms a multiprotein complex that represses the translation; however, when CPEB1 is phosphorylated on threonine 171 (T171), the bound mRNA is polyadenylated and translation is initiated (Mendez et al., 2000a).
The molecular mechanism of CPEB1 regulation in cell cycle regulation has been best studied in Xenopus oocytes (Stebbins-Boaz et al., 1996). Here, CPEB1 phosphorylation initiates translation of c-mos then cyclin B1 (Mendez et al., 2002), and cyclin B1 synthesis is required for progression into mitosis (Minshull et al., 1989; Murray and Kirschner, 1989). There is convincing evidence that CPEB1 plays a critical role in oocyte cell cycle regulation; however, there is only one report of CPEB mediating this process in somatic (HeLa) cells (Novoa et al., 2010).
CPEB1 is also present in astrocytes where it regulates the synthesis of β-catenin protein at the leading edge of migrating astrocytes (Jones et al., 2008), perhaps paralleling the function of CPEB1 in neuronal growth cones where it also regulates the synthesis of β-catenin (Kundel et al., 2009). In that earlier study, we noticed what looked like an inhibition of cell division when we blocked the activation of CPEB-mediated translation (Jones et al., 2008). In the present study we examine the possibility that CPEB1 may have a role in cell cycle regulation in astrocytes and through the regulation of cyclin B1 mRNA.
Materials and Methods
Materials.
DMEM/F12, fetal bovine serum (FBS), penicillin/streptomycin, and 0.25% trypsin-EDTA were purchased from Invitrogen. Agarose, cytochalasin B, EDTA, IGEPAL CA-630, lithium chloride, nocodazole, PIPES, potassium hydroxide, sucrose, trichloroacetic acid, Tween 20, 2-mercaptoethanol, and trypsin were purchased from Sigma. CPEB1 siRNA and PMSF were from Santa Cruz Biotechnology. ECL Western blotting detection reagents were obtained from GE Healthcare. SuperScript II reverse transcriptase, TRIzol reagent, and TRIzol LS reagent were from Invitrogen. DNase Ι was purchased from Roche. T4 RNA ligase was from Promega. Protein G-agarose was obtained from Millipore Corporation. Taq polymerase and deoxyribonucleotide triphosphate (dNTP) were from Takara. Actinomycin D, cycloheximide, and protease inhibitor cocktail were from Calbiochem. Cordycepin was purchased from Tocris Bioscience.
The following antibodies were purchased from the companies listed: anti-β-actin, anti-GFAP, anti-α-tubulin were from Sigma; phospho-histone H3 antibody was from Millipore, cyclin B1 antibody, Aurora A kinase antibody, and γ-tubulin antibody were from Abcam; CPEB1 antibody and pCPEB antibody were from Santa Cruz Biotechnology or were kindly donated by Dr. Thomas Soderling (Vollum Institute, Portland, OR); and FMRP antibody from Millipore.
Culture of rat primary astrocytes.
Rat primary astrocyte cell culture was prepared as described previously (Shin et al., 2001). Briefly, cortices were prepared from newborn (2∼3 day old) Sprague Dawley rats of both sexes. The cortices were dissociated by incubating in 0.1% trypsin-containing DMEM/F12 and then passed through 135 μm nylon mesh. Cells were plated onto poly-l-lysine (10 μg/ml)-coated 75 cm2 culture flask and maintained for 7 d in DMEM/F12 supplemented with 10% FBS. Cells were trypsinized (0.25% trypsin and 0.02% EDTA) and replated onto poly-d-lysine (10 μg/ml)-coated 100 mm plates. Five to seven days later, cells were used for experiments. The cultures contained mainly astrocytes (>95%) and microglia (<5%), as determined by immunocytochemistry using cell type-specific antibodies, i.e., anti-GFAP for astrocytes and anti-isolectin B4 or OX-42 for microglia (data not shown).
Western blot analysis.
Cells were washed twice with PBS and lysed with 2× SDS-PAGE sample buffer. An aliquot containing 50 μg of total protein was separated by 10% SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked with 1 μg/ml polyvinyl alcohol in PBS including 0.2% Tween 20 for 1 h. The membranes were incubated with first antibody overnight at 4°C and then with peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) for 2 h at room temperature. Specific bands were detected using the ECL system (GE Healthcare) and exposed to Bio-Rad electrophoresis image analyzer.
RT-PCR.
Total RNA was isolated from astrocytes using TRIzol reagent (Invitrogen), and 1 μg of total RNA was converted to cDNA using a SuperScript II reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Specific DNA bands were amplified by PCR. The amplified DNA products were resolved by 1.0% agarose gel electrophoresis and visualized by staining with ethidium bromide and exposed to a Bio-Rad electrophoresis image analyzer. All primers were purchased from Invitrogen. The primers used in this analysis are as follows:
CPEB1, 5′-CAGGAACTTCTGTGTGGCCATCTT-3′ (forward) and 5′- TTCACTTAGGCCATCTGGGCTCAG-3′ (reverse);
Cyclin B1, 5′-AAAGGCGTAACTCGAATGGA-3′ (forward) and 5′-CCGACCTTTTATTGAAGAGCA-3′ (reverse);
β-actin, 5′-TGTCACCAACTGGGACGATA-3′ (forward) and 5′-TCTCAGCTGTGGTGGTGAAG-3′ (reverse);
FMRP, 5′-TTGGTACCTTGCACACATCA-3′ (forward) and 5′-AAGTTAGCGCCTTGCTGAAT-3′ (reverse);
GFAP, 5′-AAGAACTGGATCTCCTCCTC-3′ (forward) and 5′-TAGCTACATCGAGAAGGTCC-3′ (reverse); and
GAPDH, 5′-TCCCTCAAGATTGTCAGCAA-3′ (forward) and 5′-AGATCCACAACGGATACATT-3′ (reverse).
Transient transfection.
Transient transfection in astrocytes was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions with minor modifications. Briefly, construct (CPEB-GFP, 3-CPE, CPEB1 siRNA) and Lipofectamine were mixed in serum- and antibiotics-free media. The DNA (RNA)-Lipofectamine complex was added to the 60–70% confluent cells. After 3 h of incubation, the medium was changed back to growth medium-containing serum and allowed to recover for 24 h at 37°C. Sequences for the 3CPE experiments were as follows:
3CPE control RNA (SCPE):
ACGUUAUUAUUGAAUUGUUAUUAUUGAAUUGUUAUUAUUGAAUU;
3CPE construct (3CPE):
ACGUUUUUAAUGAAUUGUUUUUAAUGAAUUGUUUUUAAUGAAUU.
Cell proliferation (MTT) assay.
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay is based on the ability of a mitochondrial dehydrogenase enzyme from viable cells to cleave the tetrazolium rings of the pale yellow MTT and form a dark blue formazan crystals which is largely impermeable to cell membranes, thus resulting in its accumulation within healthy cells. Solubilization of the cells by the addition of a detergent results in the dissolution of the crystals. The number of surviving cells is directly proportional to the level of the formazan product created. The color can then be quantified using a simple colorimetric assay. The results were read on a multiwell scanning spectrophotometer (ELISA reader, TECAN) at 590 nm. Used on sister cultures plated at the same density, this is routinely used for a measure of cell survival/proliferation (van de Loosdrecht et al., 1994).
Sucrose gradient centrifugation.
Sucrose gradient centrifugation was performed according to the reported method (Mitchison and Kirschner, 1986) with minor modification. Rat primary astrocytes (80% confluent) were treated with nocodazole (10 μg/ml) and cytochalasin B (5 μg/ml) for 90 min. Cells were washed with PBS, 0.1× PBS plus 8% sucrose and then 8% sucrose. Cells were lysed with LB buffer (1 mm Tris-HCl, 0.1% 2-ME, 0.5% IGEPAL CA-630, 1 mm PMSF, 1 mm protease inhibitor cocktail) for 15 min and sedimented at 1500 × g for 5 min. The supernatants were filtered through a 37 μm mesh. The cell lysates were added to 2 ml of 60%, 2 ml of 50%, 2 ml of 40%, and 2 ml of 30% sucrose gradient and were centrifuged with SW 41 Ti swinging bucket rotor using a Beckman Optima XL-100K preparative ultracentrifuge at 26,000 rpm for 60 min (Beckman Instruments). Sucrose solutions were made in PE buffer (10 mm PIPES, 1 mm EDTA, 8 mm 2-ME, pH to 7.2 with KOH). After centrifugation, samples were fractionated by dropwise collection of each 500 μl aliquot. From top lysate fraction to 60% sucrose fraction, we used sequential numbering from 1 to 18. Each fraction was diluted 1:5 with PE buffer and then precipitated using trichloroacetic acid. Samples were incubated at −20°C for 2 h. After supernatants were removed, samples were washed by acetone and 2× SDS-PAGE sample buffer was used for Western blot analysis. The centrosomal fraction was found in the middle of a 60% fraction. For RNA isolation, each fraction was diluted 1:5 with DEPC-treated water and then precipitated by lithium chloride. Samples were incubated at −20°C for 24 h. After supernatants were removed, samples were prepared with TRizol LS reagent for RNA isolation.
Poly(A) tail assay.
To test the poly(A) tail (PAT) length of cyclin B1 mRNA, the PAT assay was performed according to the reported method (Rassa et al., 2000) with minor modification. Briefly, total RNA was isolated from cultured astrocytes, and 1 μg of total RNA was used for the PAT assay. RNA was ligated at 37°C for 30 min with 50 pmol of primer GB-135 (5′-P-GGTCACCTTGATCTGAAGC-NH2-3′) in a 10 μl volume using T4 RNA ligase (Promega). GB-135 contained a 3′ amino modification to block ligation at this end. The reaction was boiled for 5 min and cooled to room temperature. Reverse transcription was performed at 45°C for 45 min in a 50 μl reaction using 50 pmol of primer GB-136 (5′-GCTTCAGATCAAGGTGACCTTTTT-3′), 5 μl of dNTP (Takara), 1 μl of SuperScript II reverse transcriptase (Invitrogen), 5 μl of 0.1 m DTT, and 10 μl of 5× RT buffer. The reaction was boiled for 5 min, cooled to room temperature, and used in PCR. Resulting PCR products were analyzed on 2.0% agarose gel, visualized by staining with ethidium bromide, and exposed to a Bio-Rad electrophoresis image analyzer. Maximum poly(A) tail length was determined relative to DNA standards by subtracting the size of each primer and the distance to the site of polyadenylation from the maximum size of the PCR product. All primers were purchased from Invitrogen.
Immunoprecipitation.
Rat primary astrocytes (80% confluent) were lysed with LB buffer (1 mm Tris-HCl, 0.1% 2-ME, 0.5% IGEPAL CA-630, 1 mm PMSF, 1 mm protease inhibitor cocktail) for 15 min and sedimented at 1500 × g for 5 min. The supernatants were filtered through 37 μm mesh and incubated with 50 μl of CPEB1 antibody (Santa Cruz Biotechnology) for 24 h at 4°C under agitation. After incubation, 50 μl of protein G-agarose (Millipore) was added and incubated for 24 h at 4°C under agitation. Samples were then centrifuged and the supernatant was removed. The pelleted beads were washed in LB buffer three times before dissociation from the beads by 2× SDS-PAGE sample buffer for Western blot analysis. For RT-PCR, 1 ml of TRIzol LS reagent (Invitrogen) was added for RNA isolation. As a negative control, all procedures were performed exactly the same without CPEB1 antibody.
Immunocytochemistry.
Cultured rat primary astrocytes on cover glass (Fisher Scientific) were washed and fixed with 4% paraformaldehyde at 4°C for 2 h. For detection of centrosomal cyclin B1, cells were fixed with 70% cold ethanol in PBS at 4°C for 2 h. The cells were treated with 0.3% Triton X-100 for 15 min at room temperature and blocked for 30 min with blocking buffer (1% BSA, 5% FBS in PBS) at room temperature. The cells were incubated overnight at 4°C with primary antibodies against cyclin B1 (mouse, 1:1000) or γ-tubulin (rabbit, 1:500) and washed in buffer (0.1% BSA, 0.5% FBS in PBS). Secondary antibodies conjugated with tetramethylrhodamine ethyl ester (anti-mouse) or FITC (anti-rabbit) were diluted in blocking buffer and incubated for 2 h at room temperature. After three washes, the cover glass was mounted in Vectashield (Vector Laboratories) and viewed with a confocal microscope (TCS-SP, Leica).
FACS analysis.
Rat primary astrocytes were transfected with CPEB1 siRNA (catalog no. sc-37756, Santa Cruz Biotechnology) and allowed to recover for 24 h. Cells were harvested with trypsin and centrifuged for 3 min. Then, cells were fixed in 70% ethanol for at least 30 min. Cells were pelleted and resuspended in PBS with 100 μl of RNase A (100 μg/ml) and incubated for 20 min at 37°C. Then cells were stained with 400 μl of propidium iodide (50 μg/ml) for 30 min and the cell cycle was measured by FACS analyzer (FACSCalibur, Becton Dickinson).
Statistical analysis.
Data were expressed as the mean ± SEM and analyzed for statistical significance using one-way ANOVA followed by the Newman–Keuls test as a post hoc test; a value of p < 0.05 was considered significant.
Results
CPEB1 expression in rat primary astrocytes
CPEB1 expression in rat primary astrocytes was reported recently (Jones et al., 2008), and we confirm these results (Fig. 1A,B). Furthermore, we show that CPEB1 is expressed throughout the astrocyte but enriched in the cytosol surrounding the nucleus (Fig. 1C). However, we noticed that in the small population of astrocytes caught in the act of cell division at the time of fixation, CPEB1 was strikingly localized to the centrosome (Fig. 1D). In addition, the kinase that phosphorylates CPEB1 in astrocytes, Aurora A kinase (Jones et al., 2008), is also localized to the centrosome (Fig. 1D). To confirm this observation, we used a sucrose density gradient to biochemically isolated the centrosome fraction derived from pure astrocytes cultures (Mitchison and Kirschner, 1986). We used the enrichment of γ-tubulin to identify the centrosomal fractions that were isolated around the middle of the 60% sucrose layer in fractions 16 and 17 (Fig. 2). Consistent with the immunolocalization (Fig. 1D), CPEB1, cyclin B1, and Aurora A were all enriched in the centrosome fractions (Fig. 2). In contrast, the majority of β-actin, used as a negative control, was restricted to the upper region of the gradient. In addition, two other RNA-binding proteins, FMRP and Pumilio, were not present in the centrosome fraction (Fig. 2A).
Figure 1.
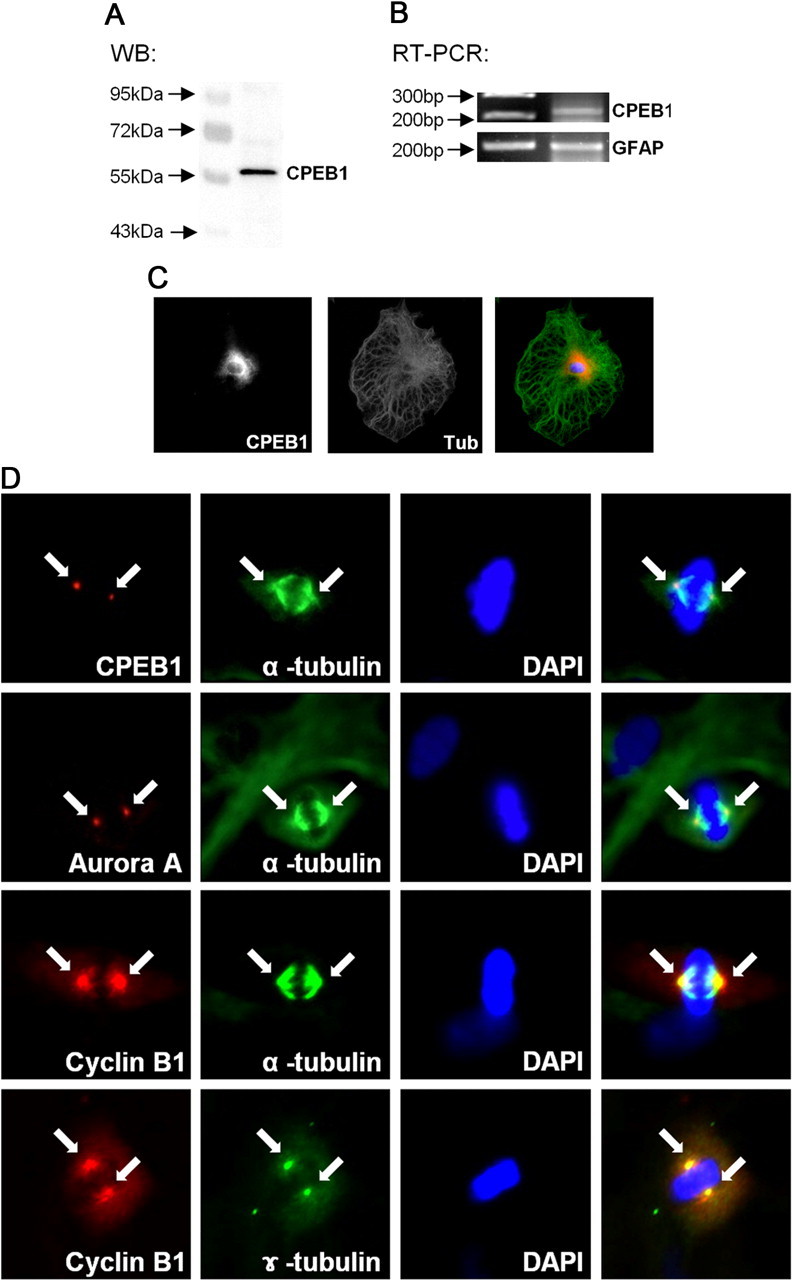
Localization of CPEB1 to the centrosome during mitosis A, Western blot analysis of rat primary astrocytes. Total cell lysates of rat primary astrocytes were subjected to Western blot analysis using an antibody against CPEB1. B, Expression of CPEB1 mRNA was confirmed by RT-PCR. C, Rat primary astrocytes were cultured on glass coverslips and fixed with 4% paraformaldehyde. Analysis of CPEB1 immunoreactivity (CPEB) revealed cytosolic expression with perinuclear enrichment. For comparison, astrocytes were also stained with α-tubulin (Tub), which was distributed throughout the cell. An overlay with tubulin in green and CPEB1 in red is shown on the right. D, Immunocytochemistry was performed to investigate centrosomal localization of CPEB1, Aurora A kinase, and cyclin B1 protein. Actively dividing rat primary astrocytes were fixed and stained for CPEB1 immunoreactivity (CPEB, red) as described above. Condensed DNA was stained with DAPI (blue), and the mitotic spindle was visualized with α-tubulin (green). Note CPEB1 immunoreactivity on centrosome. Aurora A kinase (red) immunoreactivity was also enriched in centrosome during mitosis. This localization mimics that for cyclin B1 expression in dividing astrocytes and directly overlays with γ-tubulin (green) staining, confirming that it is at the centrosome.
Figure 2.
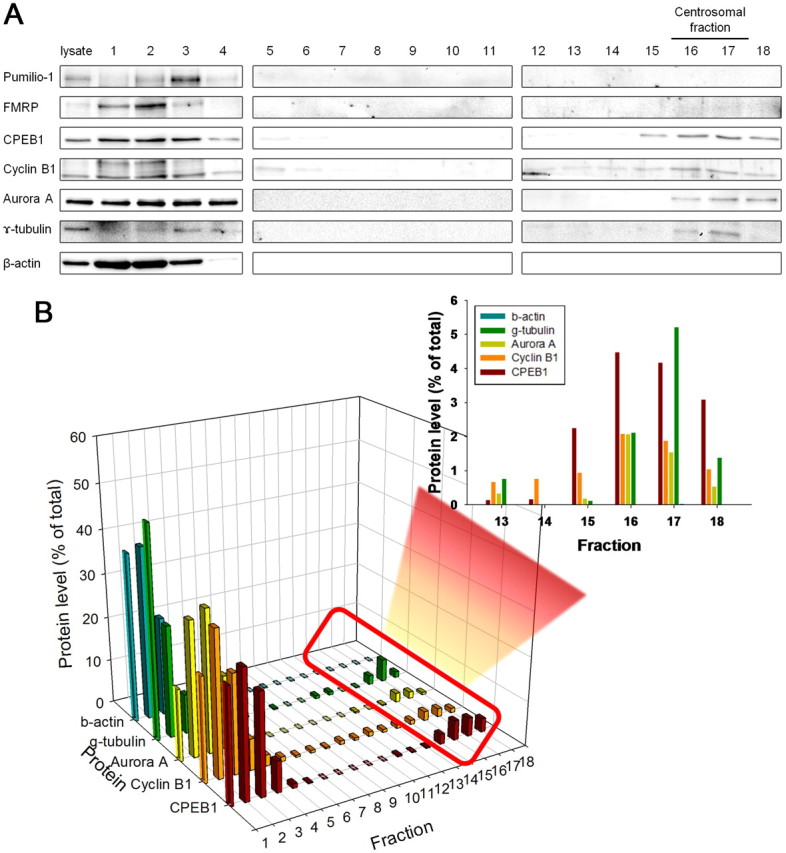
CPEB1, cyclin B1, and Aurora A comigrate with the centrosomal fraction In a biochemical fractionation of a pure astrocytes cell culture, CPEB enriches in the centrosomal fraction, but another mRNA-binding protein FMRP does not. Eighteen fractions were isolated from a sucrose gradient (see Materials and Methods) and were analyzed for Western blots using antibodies shown on the left. From top lysate fraction to 60% sucrose fraction, we used sequential numbering from 1 to 18, each lane loaded with equal volume. A, The fractions containing the centrosome (16, 17) are indicated by an enrichment in γ-tubulin. Cyclin B1, CPEB1, and Aurora A kinase were present in the centrosomal fractions. This was in contrast to β-actin and FMRP. B, Quantification of the data shown in A. Data were averaged from three experiments.
Cyclin B1 mRNA was localized to centrosome and binds CPEB1
Next, we examined whether cyclin B1 mRNA was enriched in centrosomal fraction of rat primary astrocytes. RNA isolation from each fraction followed by RT-PCR using primers specific for cyclin B1, FMRP, and GAPDH showed differential enrichment throughout the gradient (Fig. 3A). In one of the centrosomal fractions, cyclin B1 mRNA was present in relatively high abundance. However, the mRNAs encoding GAPDH and FMRP were found in fractions 3–5 and were not present in the centrosome fractions (Fig. 3A), indicating a specific localization of cyclin B1 mRNA to the centrosome. Previous work in oocytes has implicated CPEB1 in the translational regulation of cyclin B1 mRNA via the direct binding of CPEB1 to the CPE domain in the 3′-UTR (de Moor and Richter, 1999; Groisman et al., 2000; Tay et al., 2000). We confirm that this relationship exists in astrocytes by immunoprecipitation (IP) using an antibody specific against CPEB1. Analysis of the CPEB1-IP reveals that our CPEB1 antibody efficiently immunoprecipitates CPEB1 protein from whole-cell lysates (Fig. 3B) or centrosome fraction (Fig. 3D). We next isolated RNA from the CPEB1-IPs and performed RT-PCR with primers specific for cyclin B1 to determine whether cyclin B1 mRNA is in the CPEB1 complex. The results shown in Figure 3, C and E, indicate that cyclin B1 mRNA specifically associates with CPEB1 protein in whole-cell lysates and centrosome fraction, while the non-CPE-containing mRNAs encoding GFAP, β-actin, and GAPDH do not.
Figure 3.

Cyclin B1 mRNA associated with CPEB1 and was enriched in the centrosomal fraction. A, Cellular fractionation as in Figure 3, although this time it was processed for mRNA retrieval and RT-PCR analysis. Cyclin B1 mRNA is enriched in fraction 17, the centrosome fraction. The mRNA encoding GAPDH and FMRP localize to a much higher fraction. Image represents one experiment but is typical of the results obtained in three experiments. B, CPEB1-IP followed by Western blot analysis demonstrates that there was no significant interaction of CPEB1 with β-actin and γ-tubulin. C, CPEB1-IP followed by mRNA extraction and RT-PCR analysis reveals a specific interaction between CPEB1 protein and cyclin B1 mRNA. D, Western blot (WB) of a CPEB1-IP from the centrosomal fractions (16 and 17) shows specific pull down of CPEB1 protein. E, RNA isolation from the CPEB1-IP in D followed by RT-PCR showing cyclin B1 mRNA association with CPEB1 in the centrosome fraction. Control lanes (IgG) in B–E contain nonspecific IgG antibodies for the IP that were processed identically (n = 4).
Cell proliferation induces a phosphorylation of CPEB1
The association of CPEB1 with cyclin B1 mRNA suggests that one possible role of CPEB1 in astrocytes is to regulate the expression of cyclin B1 during cell division. To begin to address this, we arrested cells in culture by removing serum from the medium. We then added 10% FBS back to the medium to stimulate cell division and examined the phosphorylation state of CPEB1 by Western blotting using a phospho-specific CPEB1 antibody (kindly provided by Dr. Thomas Soderling). The antibody specifically recognizes phosphorylation of CPEB1 at T171, which is thought to be critical to the activation of CPEB1 (Atkins et al., 2004). Stimulation of cell proliferation induced a rapid phosphorylation of CPEB1 (starting ∼2 min poststimulus) that peaked 15–30 min after stimulation (Fig. 4A). Total CPEB1 levels remained constant during the first hour after stimulation but declined after 12 h of serum reintroduction (Fig. 4B). The decrease in CPEB1 levels after stimulation could be the result of proteosome-mediated degradation that is similar to that detected during oocyte maturation (Mendez et al., 2002).
Figure 4.

Cell proliferation induces a phosphorylation of CPEB1 A, Rat primary astrocytes were grown to roughly 60% confluence and then arrested by depleting serum from the medium for 24 h. Proliferation was stimulated by the addition of 10% FBS back to the medium. At each time points, cells were harvested and the phospho-CPEB1 (pCREB1) level was detected by Western blot analysis. B, The total amount of CPEB1 in astrocytes grown in culture and then arrested in serum-free medium (SF) for 9 h was compared to that of cells incubated in SF medium for 9 h followed by 12 h in 10% FBS-containing medium. Each lane was loaded with equal protein; β-actin was used as a loading control. (*p ≤ 0.05, n = 4).
Cyclin B1 mRNA is polyadenylated following FBS stimulation
As indicated above, CPEB1 phosphorylation on T171 can result in the cytoplasmic polyadenylation of bound mRNAs. To determine whether cyclin B1 mRNA is regulated in this manner, we examined the size of the cyclin B1 poly(A)-tail following readmission of serum into the medium using a poly(A) tail assay (PAT assay; see Materials and Methods). Rat primary astrocytes were growth arrested by serum depletion for 24 h and induced to proliferate by the addition of FBS into the medium. Cyclin B1 mRNA poly(A) tail length grew within 10 min of the introduction of FBS to the culture medium and remained elongated through 30 min before returning to baseline by 60 min (Fig. 5A). Moreover, the increase of cyclin B1 mRNA polyadenylation was associated with an increase in cyclin B1 protein expression, which also increased dramatically at around 10 min following the addition of FBS to the medium (Fig. 5B). These observations are consistent with a mechanism in which FBS stimulates CPEB1 phosphorylation, resulting in polyadenylation and translation of cyclin B1 mRNA. Therefore, we probed this mechanism further by using pharmacological inhibitors to transcription (actinomycin D), translation (cycloheximide), or polyadenylation (cordycepin).
Figure 5.
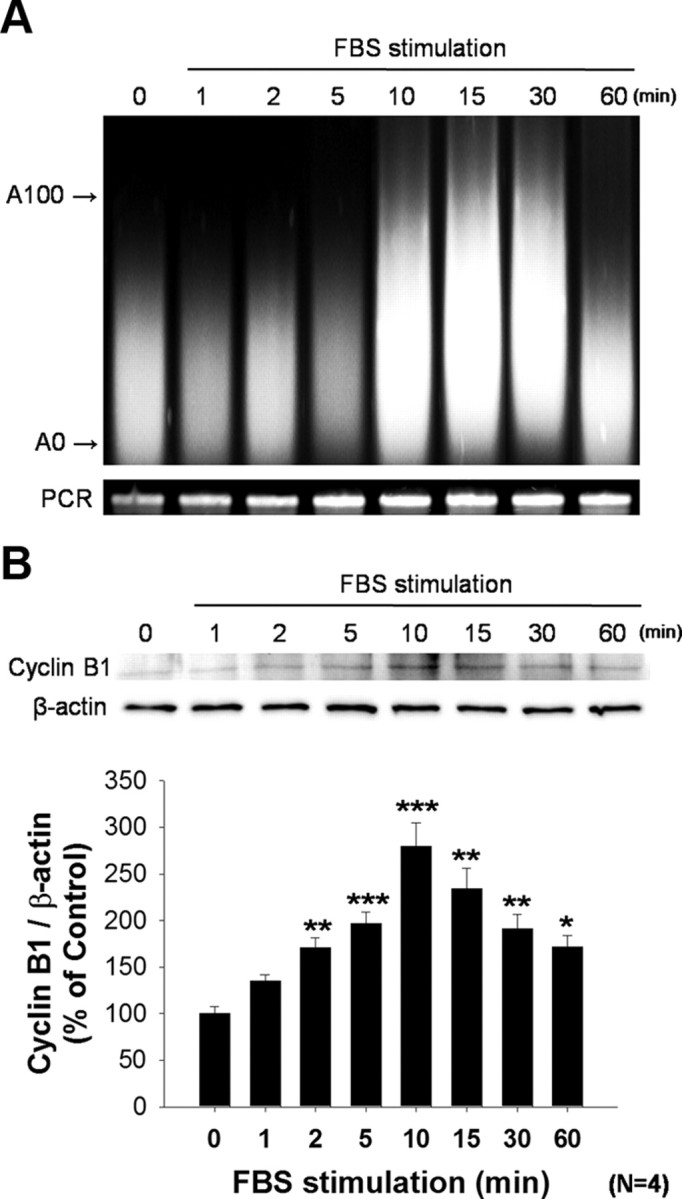
Cyclin B1 mRNA polyadenylation and translation following FBS stimulation Astrocytes were growth arrested by the removal of serum from the medium for 24 h and proliferation was induced by the addition of 10% FBS to the medium. A, Poly(A) tail assay to determine the length of cyclin B1 mRNA. By 10 min after FBS addition, the poly(A) tail length of cyclin B1 mRNA was increased dramatically. By 60 min the poly(A) tail size had returned to baseline. B, Following a similar time course, cyclin B1 protein levels were also increased following FBS stimulation. Quantification of these data indicates that an increase in protein levels can be detected as early as 2 min after FBS stimulation (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001, n = 4).
In cells that had been growth arrested and then stimulated with FBS in the presence of actinomycin D, we still detect both an increase in cyclin B1 mRNA polyadenylation and protein expression (Fig. 6A,B). This suggests that the increase in both poly(A) tail length and translation is occurring to mRNA already present at the time of stimulation. One would then predict that in the presence of cycloheximide, FBS would still be capable of inducing cyclin B1 polyadenylation, but that would not result in an increase in protein, as translation is inhibited. That is indeed what we see (Fig. 6A,B). To determine whether polyadenylation itself is responsible for the increase in cyclin B1 protein expression, we stimulated with FBS in the presence of cordycepin, an adenosine analog that once added to the poly(A) tail prevents further polyadenylation. Cordycepin blocks both FBS-induced polyadenylation of cyclin B1 mRNA (Fig. 6A) and cyclin B1 expression (Fig. 6B). This same experiment can be performed from RNA and protein extracted from the centrosomal fraction. Although the absolute levels of both RNA and protein are less than those detected in whole homogenates, the results are identical (Fig. 6C,D). In growth-arrested cells there is very little γ-tubulin or cyclin B1 in the centrosome fraction; however, both are recruited following FBS stimulation (Fig. 6D). The lack of γ-tubulin or cyclin B1 in the centrosomal fraction with either cycloheximide or cordycepin treatment may indicate a lack of recruitment of these components to the centrosome and thus a failure to progress into M phase.
Figure 6.

Increase in cyclin B1 protein is dependent upon polyadenylation of mRNA present at time of stimulation Pharmacological manipulation using either the transcription inhibitor [actinomycin D (Act); 50 μg/ml], polyadenylation inhibitor [cordycepin (Cor); 50 μm], or translation inhibitor [cycloheximide (Cyc); 50 μm] during FBS stimulation. A, PAT assay was performed on astrocytes either in serum-free condition or 30 min following FBS stimulation. The polyadenylation of cyclin B1 mRNA that increased with FBS stimulation was not blocked by either Act or Cyc but was inhibited by Cor. B, Expression of cyclin B1 protein in the same experiment examined by Western blot analysis. Quantification of the results shows that the increase in cyclin B1 protein with FBS stimulation is completely blocked by both Cor and Cyc. Before serum addition, no drug affected the levels of cyclin B1. (*p ≤ 0.05; **p ≤ 0.01, n = 4). C, Cells treated in a similar fashion and then fractioned to isolate the centrosomal fraction were analyzed by PAT assay. Polyadenylation of cyclin B1 mRNA in centrosomal fraction was also detected following FBS stimulation. Similar to whole-cell homogenates, Act and Cyc did not repress polyadenylation, while Cor completely repressed polyadenylation. D, The centrosomal fraction further analyzed by Western blot revealed an increase in cyclin B1 and γ-tubulin in the presence of Act that was similar to that of control (Con). However, Cor and Cyc completely blocked the increase in cyclin B1 synthesis seen with FBS stimulation in Con. A representative experiment was shown out of four similar experiments.
CPEB1 levels regulate cell cycle
CPEB1-bound mRNAs are thought to be in a translationally inhibited state until CPEB1 is phosphorylated (Wells, 2006). This suggests that one could activate translation of bound mRNA by simply removing CPEB1 from the mRNA and thus derepress translation. We employed two means by which to remove CPEB1 from its cognate mRNA. First, we depleted CPEB1 using siRNA; second, we transfected RNA into astrocytes that contain multiple copies of CPE sequences to sequester CPEB1 from its endogenous targets (Stebbins-Boaz et al., 1996; de Moor and Richter, 1999). We then determined the consequence of altering CPEB1 binding to endogenous mRNA on cell proliferation by using three methods: a biochemical marker for dividing cells (phospho-histone H3), an indicator of the total number of cells (MTT assay), and FACS analysis.
Using CPEB1 siRNA technology, we efficiently knocked down CPEB1 mRNA and protein levels in cultured astrocytes (Fig. 7A–C). Surprisingly, removing CPEB1 resulted in an increase in cell proliferation as measured by both an MTT assay (Fig. 7D) or by phospho-histone H3 levels (Fig. 7F). Consistent with this finding, when CPEB1 is knocked down by siRNA, cyclin B1 protein levels increase, suggesting that CPEB1 is repressing mRNA translation (Fig. 7G,H). FACS analysis of cells treated with siRNA for CPEB1 also reveal an increase in the number of cells in G2/M phase compared to control-treated cells (Fig. 7E). These data again suggest that the removal of CPEB1 from mRNA results in derepression of at least cyclin B1 mRNA.
Figure 7.
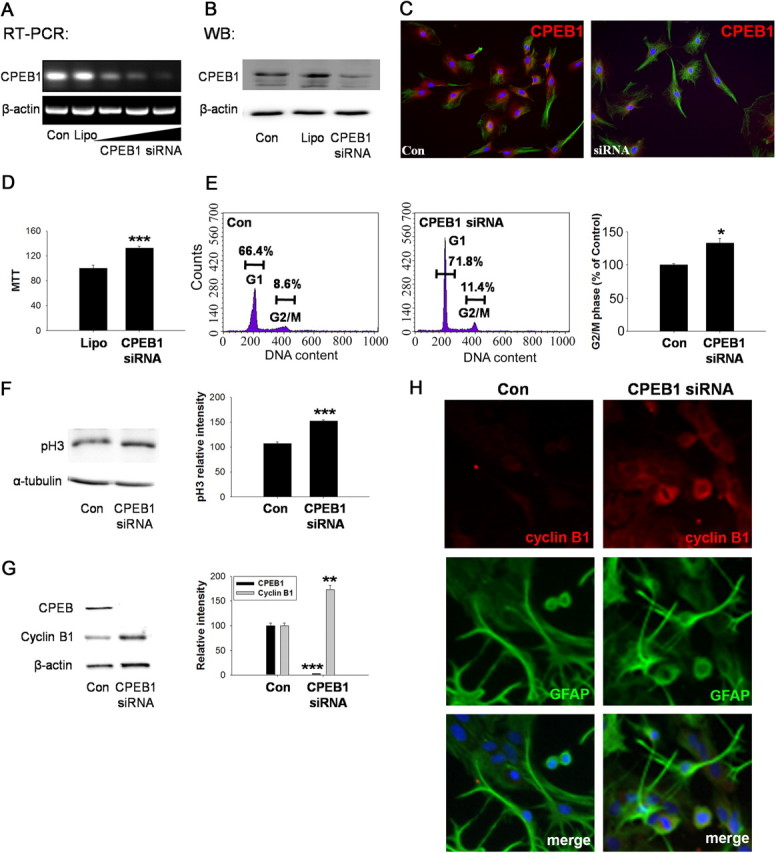
Knockdown of CPEB1 expression from astrocytes promotes cell cycle progression Astrocytes were transfected with CPEB1 siRNA for 3 h with Lipofectamine as described in Materials and Methods and allowed to recover for 24 h. A, The level of CPEB1 mRNA in rat primary astrocytes was determined by RT-PCR. Increasing the concentration of siRNA (indicated by sloped line under gel) decreased the level of CPEB1-encoding mRNA detected when compared to untransfected (Con) and Lipofectamine alone (Lipo) controls. B, The level of CPEB1 protein was similarly affected by siRNA expression. C, Immunocytochemical staining of CPEB1 reveals the loss of CPEB1 expression in siRNA-treated cells (right) compared to untransfected controls (left). D, Astrocyte number in culture was measured by MTT assay. siRNA transfection led to the increase in cell number compared to Lipo control (n = 4). E, Cell cycle analysis using FACS. Astrocytes treated with siRNA to CPEB1 as above were then processed for FACS analysis. Based upon propidium iodide staining, there is a significant increase in the proportion of cells in G2/M phase compared to controls (n = 4). F, Phospho-histone H3, a marker for dividing cells, was significantly increased after CPEB1 knockdown by siRNA (n = 4). G, Western blot analysis of CPEB1 knockdown demonstrates an increase in cyclin B1 protein levels compared to control (n = 4). H, Immunocytochemistry using an antibody cyclin B1 and GFAP in either untreated astrocytes (Con) or astrocytes treated with CPEB1 siRNA. The merged image shows cyclin B1, GFAP, and DAPI. *p ≤ 0.05**; p < 0.01; ***p ≤ 0.001.
To confirm this hypothesis, we overexpressed RNA encoding reiterative CPEB1 binding domains (3CPE) to sequester endogenous CPEB1 away from its cadre of target mRNAs. In primary astrocytes, expression of 3CPE resulted in a significant decrease in the cyclin B1 mRNA associated with CPEB1 (Fig. 8A), indicating that a dissociation with endogenous target mRNAs was indeed achieved. The dissociation of CPEB1 from cyclin B1 mRNA resulted in an increase in cyclin B1 expression and cell proliferation as measured by phospho-histone H3 levels (Fig. 8B,F). Expression of 3CPE also resulted in an increase in cyclin B1 associated with the centrosomal fractions (Fig. 8C) but did not change the level of CPEB1 or cyclin B1 mRNA associated with these fractions (Fig. 8C,D). Since some CPEB1 is still clearly associated with cyclin B1 mRNA (Fig. 8A), the simplest interpretation of these data is that the interaction of CPEB1 with cyclin B1 mRNA localized to the centrosome is resilient to manipulation by 3CPE, and the increase in cyclin B1 protein is recruited from the cytoplasm. However, there are other interpretations (see Discussion). FACS analysis of cells expressing 3CPE also indicates an enhanced cell proliferation (Fig. 8E).
Figure 8.

Expression of 3CPE increases cyclin B1 protein and induces proliferation. Astrocytes were either untreated (Con) or transfected with either 3CPE or a mutated (scrambled) version (SCPE) and assayed 24 h later. A, mRNA was detected by RT-PCR following IP with CPEB1 antibody. All data are expressed as mean ± SEM (n = 3). B, SCPE or 3CPE-expressing astrocytes were harvested and assayed for expression level of cyclin B1 and phospho-histone H3 in total cell lysates by Western blot. Quantification of Western blot data was normalized with α-tubulin (right). All data are expressed as mean ± SEM (n = 3). C, Centrosomal fractions (16 and 17; see Fig. 2) were isolated and the levels of CPEB1, cyclin B1, and γ-tubulin were determined by Western blot. Quantification of Western blot data was normalized with α-tubulin (right). All data are expressed as mean ± SEM (n = 3). D, The level of cyclin B1 mRNA in the centrosomal fraction was determined by RT-PCR. E, Astrocytes treated with 3CPE were processed for FACS analysis. Based upon propidium iodide staining, there is a significant increase in the proportion of cells in G2/M phase compared to controls (n = 4). F, Immunocytochemistry using an antibody cyclin B1 and GFAP in either untreated astrocytes (Con) or astrocytes treated with 3CPE. The merged image shows cyclin B1, GFAP, and DAPI. All data are expressed as mean ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001 (all compared to Con).
Since removal of CPEB1 resulted in cell proliferation, we next determined whether overexpression of CPEB1 would have the opposite response. Thus, astrocytes transfected with wild-type CPEB1-expressing constructs were examined using the same techniques described for siRNA analysis. Overexpression of CPEB1 in rat primary astrocytes (Fig. 9A,B) inhibited cell proliferation as measured by the MTT assay and the percentage of cells in G2/M phase (Fig. 9C,D), as well as phospho-H3 levels (Fig. 9E). Overexpression of CPEB1 also resulted in a decrease in cyclin B1 protein levels, again suggesting that unphosphorylated CPEB1 is acting as a translational repressor (Fig. 9F,G).
Figure 9.

Overexpression of CPEB1 inhibits cell cycle progression. Rat primary astrocytes were transfected with CPEB1-GFP for 3 h with Lipofectamine (Lipo) as described in Materials and Methods and allowed to recover for 24 h. A, The level of CPEB1 mRNA in rat primary astrocytes was determined by RT-PCR. B, The level of CPEB1 protein was detected by Western blot. C, The number of astrocytes in culture was measured by MTT assay and shown to significantly decrease when CPEB1 is overexpressed (n = 4). D, FACS analysis of astrocytes transfected with CPEB1-GFP show a decrease in the number of cells in G2/M phase when compared to untransfected controls (Con) (n = 4). E, Phospho-histone H3, a marker for dividing cells, was also decreased with CPEB1 overexpression (n = 4). F, Overexpression of CPEB1 also resulted in a decrease in the levels of cyclin B1 detected (n = 4). G, Immunocytochemistry using an antibody cyclin B1 and GFAP in either untreated astrocytes (Con) or astrocytes overexpressing CPEB1. The merged image shows cyclin B1, GFAP and DAPI. ***;p ≤ 0.001.
Discussion
Astrocyte proliferation is a hallmark of brain injury. Among other functions, this proliferation, or reactive gliosis, is thought to be responsible for an efficient inflammatory response (Bush et al., 1999) and a rapid restoration of the blood brain barrier (Faulkner et al., 2004). However, astrocyte proliferation will also repress functional recovery, as it prevents axonal regeneration (Menet et al., 2003). Therefore, elucidating the regulatory mechanisms controlling astrocytes proliferation will be vital in our understanding of the brain response to injury. In addition, since astrocytes are also the primary source of cancer in the brain, understanding the proliferative process might provide insight into the uncontrolled proliferation seen in tumorigenesis.
Several findings in the present study demonstrate that CPEB1-dependent mRNA translation can regulate cell cycle and proliferation of astrocytes: (1) CPEB1 colocalized to the centrosome with Aurora A kinase and cyclin B1 mRNA in astrocytes; (2) FBS-induced astrocyte proliferation induced a concomitant phosphorylation of CPEB1, polyadenylation of the CPEB1-bound mRNA encoding cyclin B1, and an increase in cyclin B1 protein; (3) removal of CPEB1 from endogenous mRNA (either by siRNA knockdown or expression of CPE-containing mRNA) induced an increase in cyclin B1 protein and cell proliferation; and (4) overexpression of unphosphorylated CPEB1 reduced cyclin B1 protein levels and suppressed cell division.
The regulation of mRNA translation is recognized as an important process for oocyte maturation and early embryogenesis (Bastock and St Johnston, 2008). One mechanism thought to be critical for oocyte progression into M phase is cytoplasmic polyadenylation and the subsequent activation of translation of cyclin B1 mRNA. This regulation of cyclin B1 mRNA is mediated by the binding of CPEB1 to CPEs within the 3′-UTR. CPEB1 is capable of repressing bound mRNA, keeping it translationally dormant until CPEB1 is phosphorylated, which frees the bound mRNA from repression and allows for efficient translation (de Moor and Richter, 1999). This mechanism of regulation is not unique to cyclin B1, although it is the best studied example of this mechanism in oocytes (Piqué et al., 2008). Indeed, CPEB-mediated regulation of the cell cycle was recently demonstrated in HeLa cells (Novoa et al., 2010). CPEB1 is also expressed in the brain, where it regulates mRNA translation in neurons following a wide range of neuronal stimuli including neurotrophins and glutamate (Wu et al., 1998; Shin et al., 2004; McEvoy et al., 2007; Kundel et al., 2009), suggesting it may play a role in several distinct signaling events.
CPEB1 was also recently identified in astrocytes, where it appears to play a role in astrocyte migration as it regulates the expression of β-catenin at the leading edge of migrating astrocytes (Jones et al., 2008). Considering the diverse function of CPEB1 in neurons and its ability to regulate the cell cycle in oocytes, we probed astrocytes to determine whether CPEB1-mediated mRNA translation could be playing a role in controlling cell cycle via its regulation of cyclin B1 mRNA. We show that CPEB1 protein and cyclin B1 mRNA are both enriched at the centrosome in astrocytes and that CPEB1 phosphorylation coincides with the polyadenylation and translation of cyclin B1 mRNA. Thus, it appears that CPEB1 localized to the leading edge of migrating astrocytes may play a role in their directed movement, while CPEB1 localized to the centrosome is regulating cell cycle proteins. How CPEB1 is localized to these distinct cellular domains is an interesting but as yet unexplored process.
The centrosome has several functions within the cell; among them are a microtubule organizing center and the recruitment of components critical for cell cycle progression during cell division (Hinchcliffe et al., 2001; Khodjakov and Rieder, 2001; Piel et al., 2001). Indeed, the centrosome is thought to act as a scaffold for cell cycle-regulating proteins and is important for several cell cycle transitions, including G1-S and G2-M (Doxsey et al., 2005). For example, Cdk1-cyclin B1 activation is the critical molecular event that triggers entry into mitosis (Nurse, 1990), and this activation is localized to the centrosome (Jackman et al., 2003). While the activation of Cdk1-cyclin B1 complex requires cdc25B phosphatase (Lindqvist et al., 2005), the recruitment of active Cdk1-cyclin B1 to the centrosome requires the activation of Aurora A kinase (Hirota et al., 2003). We show that CPEB1 is also recruited to the centrosome during mitosis along with the mRNA encoding cyclin B1. In addition, Aurora A kinase has been implicated in the phosphorylation of CPEB1 in astrocytes (Jones et al., 2008), oocytes (Mendez et al., 2000b) and neurons (McEvoy et al., 2007). This suggests a mechanism whereby the centrosome scaffolds CPEB1-bound cyclin B1 mRNA along with Aurora A kinase to enrich cyclin B1 levels and potentially spatially restrict Cdk1-cyclin B1 signaling to the centrosome.
We used serum deprivation to arrest cells in G0 or G1 phase before centrosome duplication in S phase (Hinchcliffe and Sluder, 2001; Cooper, 2003). Since γ-tubulin is recruited to the centrosome during prophase of mitosis (Khodjakov and Rieder, 1999), we used this as a marker for G2/M transition. Under serum deprivation conditions, γ-tubulin is not detected in the centrosome fraction; however, γ-tubulin is recruited to the centrosome following FBS addition to the culture, inducing cell cycle progression into M phase (Fig. 6D, compare lanes 1 and 5). Interestingly, cycloheximide suppressed the recruitment of γ-tubulin to the centrosome following FBS stimulation (Fig. 6D, lane 8), indicating that the G2/M transition, which is activated by Cdk1-cyclin B1 complex (Crasta et al., 2006), is not proceeding in the absence of new protein synthesis. Importantly, G2/M transition does occur in the presence of actinomycin D (Fig. 6D, lane 6), demonstrating that it is the activation of mRNA translation from existing mRNAs that mediates this transition. Furthermore, thee adenosine analog cordycepin, which blocks polyadenylation, also prevents G2/M transition, thus implicating CPEB1 in this process.
CPEB1 binding to mRNA has a dual function. First, it can repress mRNA translation through direct binding to the 3′-UTR and indirect binding to the 5′-cap via a complex of proteins, including eIF4E (Groppo and Richter, 2009). Second, CPEB1-bound mRNA can be translationally activated following the phosphorylation of CPEB1 at T171, and at least two kinases are capable of phosphorylating CPEB1: α-CaMKII and Aurora A (Mendez et al., 2000b; Atkins et al., 2004). Therefore, a knock-down of CPEB1 could result in a derepression of previously bound mRNA, which is consistent with our data showing an increase in cyclin B1 protein and stimulation of cell division. By the same logic, sequestration of CPEB1 away from its endogenous targets by expressing a noncoding, CPE-containing mRNA (3CPE) should have the same effect, and it did (Fig. 8). Conversely, overexpression of CPEB1 reduced cyclin B1 protein levels and suppressed proliferation, likely due to its ability to repress mRNA translation. Interestingly, 3CPE expression did not reduce the level of cyclin B1 mRNA at the centrosome. This was unexpected, as expression of a CPEB mutant protein lacking a microtubule binding domain did reduce cyclin B1 mRNA levels in the oocyte (Groisman et al., 2000), indicating that the mRNA localization was dependent upon CPEB. It is possible that in astrocytes cyclin B1 mRNA is anchored to the centrosome independently of CPEB1, but another plausible explanation is that the CPEB1-cyclin B1 mRNA at the centrosome is particularly stable and thus resistant to 3CPE disruption. Further analysis will be needed to distinguish these possibilities.
Of course global reduction or enhancement of CPEB1 levels will affect many mRNAs within the cell and could result in numerous phenotypes. For example, Burns and Richter (2008) describe a decrease in the translation efficiency of p53 mRNA upon CPEB1 knock down in fibroblasts, which could contribute to these cells bypassing senescence to resemble transformed cells. Together with our data, this would suggest that misregulation by CPEB1 could result in both increased cell proliferation and cells that do not senesce, a troubling combination.
In conclusion, the present findings suggest that cytoplasmic polyadenylation-dependent mRNA translation plays an important role in cell cycle regulation in astrocytes, mediated in part through the regulation of cyclin B1. These results provide additional insight into the regulation of astrocyte proliferation, and since Aurora A kinase is known to be upregulated in many forms of cancer (Saeki et al., 2009), they may provide insight into the uncontrolled proliferation seen in glioblastoma tumorigenesis.
Footnotes
The authors declare no conflict of interest or competing commercial interests.
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by Ministry of Education, Science and Technology Grant 331-2008-1-E00057 (C.Y.S.) and National Institutes of Health-National Institute of Neurological Disorders and Stroke Grant NS-064287-01A1 (D.G.W.).
References
- Atkins CM, Nozaki N, Shigeri Y, Soderling TR. Cytoplasmic polyadenylation element binding protein-dependent protein synthesis is regulated by calcium/calmodulin-dependent protein kinase II. J Neurosci. 2004;24:5193–5201. doi: 10.1523/JNEUROSCI.0854-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastock R, St Johnston D. Drosophila oogenesis. Curr Biol. 2008;18:R1082–R1087. doi: 10.1016/j.cub.2008.09.011. [DOI] [PubMed] [Google Scholar]
- Burns DM, Richter JD. CPEB regulation of human cellular senescence, energy metabolism, and p53 mRNA translation. Genes Dev. 2008;22:3449–3460. doi: 10.1101/gad.1697808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Cooper S. Reappraisal of serum starvation, the restriction point, G0, and G1 phase arrest points. FASEB J. 2003;17:333–340. doi: 10.1096/fj.02-0352rev. [DOI] [PubMed] [Google Scholar]
- Crasta K, Huang P, Morgan G, Winey M, Surana U. Cdk1 regulates centrosome separation by restraining proteolysis of microtubule-associated proteins. EMBO J. 2006;25:2551–2563. doi: 10.1038/sj.emboj.7601136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moor CH, Richter JD. Cytoplasmic polyadenylation elements mediate masking and unmasking of cyclin B1 mRNA. EMBO J. 1999;18:2294–2303. doi: 10.1093/emboj/18.8.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxsey S, Zimmerman W, Mikule K. Centrosome control of the cell cycle. Trends Cell Biol. 2005;15:303–311. doi: 10.1016/j.tcb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman I, Huang YS, Mendez R, Cao Q, Theurkauf W, Richter JD. CPEB, maskin, and cyclin B1 mRNA at the mitotic apparatus: implications for local translational control of cell division. Cell. 2000;103:435–447. doi: 10.1016/s0092-8674(00)00135-5. [DOI] [PubMed] [Google Scholar]
- Groppo R, Richter JD. Translational control from head to tail. Curr Opin Cell Biol. 2009;21:444–451. doi: 10.1016/j.ceb.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchcliffe EH, Sluder G. “It takes two to tango”: understanding how centrosome duplication is regulated throughout the cell cycle. Genes Dev. 2001;15:1167–1181. doi: 10.1101/gad.894001. [DOI] [PubMed] [Google Scholar]
- Hinchcliffe EH, Miller FJ, Cham M, Khodjakov A, Sluder G. Requirement of a centrosomal activity for cell cycle progression through G1 into S phase. Science. 2001;291:1547–1550. doi: 10.1126/science.1056866. [DOI] [PubMed] [Google Scholar]
- Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, Hatakeyama K, Saya H. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585–598. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- Jackman M, Lindon C, Nigg EA, Pines J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol. 2003;5:143–148. doi: 10.1038/ncb918. [DOI] [PubMed] [Google Scholar]
- Jones KJ, Korb E, Kundel MA, Kochanek AR, Kabraji S, McEvoy M, Shin CY, Wells DG. CPEB1 regulates beta-catenin mRNA translation and cell migration in astrocytes. Glia. 2008;56:1401–1413. doi: 10.1002/glia.20707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanu OO, Mehta A, Di C, Lin N, Bortoff K, Bigner DD, Yan H, Adamson DC. Glioblastoma multiforme: a review of therapeutic targets. Expert Opin Ther Targets. 2009;13:701–718. doi: 10.1517/14728220902942348. [DOI] [PubMed] [Google Scholar]
- Khodjakov A, Rieder CL. The sudden recruitment of gamma-tubulin to the centrosome at the onset of mitosis and its dynamic exchange throughout the cell cycle, do not require microtubules. J Cell Biol. 1999;146:585–596. doi: 10.1083/jcb.146.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodjakov A, Rieder CL. Centrosomes enhance the fidelity of cytokinesis in vertebrates and are required for cell cycle progression. J Cell Biol. 2001;153:237–242. doi: 10.1083/jcb.153.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebler MA, Bassell GJ. Neuronal RNA granules: movers and makers. Neuron. 2006;51:685–690. doi: 10.1016/j.neuron.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Kundel M, Jones KJ, Shin CY, Wells DG. Cytoplasmic polyadenylation element-binding protein regulates neurotrophin-3-dependent beta-catenin mRNA translation in developing hippocampal neurons. J Neurosci. 2009;29:13630–13639. doi: 10.1523/JNEUROSCI.2910-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist A, Källström H, Lundgren A, Barsoum E, Rosenthal CK. Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1-Cdk1 at the centrosome. J Cell Biol. 2005;171:35–45. doi: 10.1083/jcb.200503066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy M, Cao G, Montero Llopis P, Kundel M, Jones K, Hofler C, Shin C, Wells DG. Cytoplasmic polyadenylation element binding protein 1-mediated mRNA translation in Purkinje neurons is required for cerebellar long-term depression and motor coordination. J Neurosci. 2007;27:6400–6411. doi: 10.1523/JNEUROSCI.5211-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez R, Hake LE, Andresson T, Littlepage LE, Ruderman JV, Richter JD. Phosphorylation of CPE binding factor by Eg2 regulates translation of c-mos mRNA. Nature. 2000a;404:302–307. doi: 10.1038/35005126. [DOI] [PubMed] [Google Scholar]
- Mendez R, Murthy KG, Ryan K, Manley JL, Richter JD. Phosphorylation of CPEB by Eg2 mediates the recruitment of CPSF into an active cytoplasmic polyadenylation complex. Mol Cell. 2000b;6:1253–1259. doi: 10.1016/s1097-2765(00)00121-0. [DOI] [PubMed] [Google Scholar]
- Mendez R, Barnard D, Richter JD. Differential mRNA translation and meiotic progression require Cdc2-mediated CPEB destruction. EMBO J. 2002;21:1833–1844. doi: 10.1093/emboj/21.7.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menet V, Prieto M, Privat A, Giménez y Ribotta M. Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin genes. Proc Natl Acad Sci U S A. 2003;100:8999–9004. doi: 10.1073/pnas.1533187100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshull J, Blow JJ, Hunt T. Translation of cyclin mRNA is necessary for extracts of activated Xenopus eggs to enter mitosis. Cell. 1989;56:947–956. doi: 10.1016/0092-8674(89)90628-4. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ, Kirschner MW. Isolation of mammalian centrosomes. Methods Enzymol. 1986;134:261–268. doi: 10.1016/0076-6879(86)34094-1. [DOI] [PubMed] [Google Scholar]
- Murray AW, Kirschner MW. Cyclin synthesis drives the early embryonic cell cycle. Nature. 1989;339:275–280. doi: 10.1038/339275a0. [DOI] [PubMed] [Google Scholar]
- Novoa I, Gallego J, Ferreira PG, Mendez R. Mitotic cell-cycle progression is regulated by CPEB1 and CPEB4-dependent translational control. Nat Cell Biol. 2010;12:447–456. doi: 10.1038/ncb2046. [DOI] [PubMed] [Google Scholar]
- Nurse P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344:503–508. doi: 10.1038/344503a0. [DOI] [PubMed] [Google Scholar]
- Piel M, Nordberg J, Euteneuer U, Bornens M. Centrosome-dependent exit of cytokinesis in animal cells. Science. 2001;291:1550–1553. doi: 10.1126/science.1057330. [DOI] [PubMed] [Google Scholar]
- Piqué M, López JM, Foissac S, Guigó R, Méndez R. A combinatorial code for CPE-mediated translational control. Cell. 2008;132:434–448. doi: 10.1016/j.cell.2007.12.038. [DOI] [PubMed] [Google Scholar]
- Rassa JC, Wilson GM, Brewer GA, Parks GD. Spacing constraints on reinitiation of paramyxovirus transcription: the gene end U tract acts as a spacer to separate gene end from gene start sites. Virology. 2000;274:438–449. doi: 10.1006/viro.2000.0494. [DOI] [PubMed] [Google Scholar]
- Saeki T, Ouchi M, Ouchi T. Physiological and oncogenic Aurora-A pathway. Int J Biol Sci. 2009;5:758–762. doi: 10.7150/ijbs.5.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin CY, Choi JW, Ryu JR, Ryu JH, Kim W, Kim H, Ko KH. Immunostimulation of rat primary astrocytes decreases intracellular ATP level. Brain Res. 2001;902:198–204. doi: 10.1016/s0006-8993(01)02385-x. [DOI] [PubMed] [Google Scholar]
- Shin CY, Kundel M, Wells DG. Rapid, activity-induced increase in tissue plasminogen activator is mediated by metabotropic glutamate receptor-dependent mRNA translation. J Neurosci. 2004;24:9425–9433. doi: 10.1523/JNEUROSCI.2457-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins-Boaz B, Hake LE, Richter JD. CPEB controls the cytoplasmic polyadenylation of cyclin, Cdk2 and c-mos mRNAs and is necessary for oocyte maturation in Xenopus. EMBO J. 1996;15:2582–2592. [PMC free article] [PubMed] [Google Scholar]
- Tay J, Richter JD. Germ cell differentiation and synaptonemal complex formation are disrupted in CPEB knockout mice. Dev Cell. 2001;1:201–213. doi: 10.1016/s1534-5807(01)00025-9. [DOI] [PubMed] [Google Scholar]
- Tay J, Hodgman R, Richter JD. The control of cyclin B1 mRNA translation during mouse oocyte maturation. Dev Biol. 2000;221:1–9. doi: 10.1006/dbio.2000.9669. [DOI] [PubMed] [Google Scholar]
- van de Loosdrecht AA, Beelen RH, Ossenkoppele GJ, Broekhoven MG, Langenhuijsen MM. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J Immunol Methods. 1994;174:311–320. doi: 10.1016/0022-1759(94)90034-5. [DOI] [PubMed] [Google Scholar]
- Wells DG. RNA-binding proteins: a lesson in repression. J Neurosci. 2006;26:7135–7138. doi: 10.1523/JNEUROSCI.1795-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Wells D, Tay J, Mendis D, Abbott MA, Barnitt A, Quinlan E, Heynen A, Fallon JR, Richter JD. CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of alpha-CaMKII mRNA at synapses. Neuron. 1998;21:1129–1139. doi: 10.1016/s0896-6273(00)80630-3. [DOI] [PubMed] [Google Scholar]