

Abstract
Members of the NF-κB family of transcription factors function as dominant regulators of inducible gene expression in virtually all cell types in response to a broad range of stimuli, with particularly important roles in coordinating both innate and adaptive immunity. This review summarizes current knowledge and recent progress toward elucidating the numerous regulatory layers that confer target gene selectivity in response to an NF-κB-inducing stimulus.
The discovery of the NF-κB transcription factor a quarter century ago was a major landmark in the study of eukaryotic transcriptional regulation1. The NF-κB discovery was considered to be of great significance because NF-κB represented the first well-documented example of a transcription factor whose DNA-binding is induced by a post-translational mechanism. Although many other transcription factors induced by post-translational modification or regulation have now been identified, NF-κB has remained at the forefront of the transcription field because of its dominant role in regulating inducible gene transcription in virtually all mammalian cell types in response to a wide range of external, internal, and environmental cues. To make such broad contributions, powerful mechanisms have evolved to ensure that only a limited number of potential target genes are induced when NF-κB's DNA-binding activity is activated in response to a defined stimulus in a specific cell type. A broad framework for understanding NF-κB selectivity and recent progress toward the elucidation of selectivity mechanisms are the focus of this article.
Overview of NF-κB specificity mechanisms
Numerous regulatory layers contribute to the cell-type and stimulus specificity of the NF-κB response (Fig. 1). These layers can be divided into two fundamentally distinct, yet interconnected, categories: those implemented during the development of a responsive cell type, and those that contribute to specificity of the response when a cell encounters a stimulus. Within the first category, recent studies suggest that the cell type-specificity of the NF-κB response is intimately linked to the pathways and factors that control lineage specification and development. For example, transcriptional enhancers for many genes that will be susceptible to NF-κB-induced activation in a differentiated cell become occupied by key developmental regulators in immature cells, perhaps at the time of lineage specification or commitment2,3. Transcription factor occupancy may result in the assembly of a chromatin structure that is permissive to transcriptional activation. Another regulatory strategy within this first category is likely to be the deposition of chromatin barriers at specific sets of NF-κB target genes during development4–7, thereby dictating which target genes will be activated in response to a stimulus; activation of each gene may depend on whether the stimulus can or cannot promote the elimination of its chromatin barriers. A third strategy through which development can contribute to cell-type specificity of the NF-κB response is the developmentally controlled activation or repression of signaling molecules and transcription factors that participate in the response to a stimulus, and thereby help dictate which NF-κB target genes will be activated (or repressed)8. In other words, developmental events can act either in cis, by directly altering NF-κB responsive loci, or in trans, by influencing the expression of factors that may participate in the response following differentiation.
Figure 1.

Contributors to selectivity of the NF-κB response. Selectivity of the NF-κB response is regulated by multiple events that take place during the development of a responsive cell type, and by a broad range of events that act following stimulation.
The second category of regulatory layers, those that contribute to specificity when a differentiated cell encounters a stimulus, is remarkably diverse. As described above, chromatin is one central contributor to specificity, as different sets of potential NF-κB target genes possess different chromatin barriers that must be eliminated to allow efficient transcription. A given stimulus may induce pathways capable of removing some chromatin barriers but not others, limiting the NF-κB response to those genes whose barriers have been successfully eliminated. Some chromatin barriers, such as stable nucleosomes that confer a requirement for nucleosome remodeling, may represent intrinsic properties of a promoter or enhancer and therefore may be largely invariable among cell types for a given gene9. In contrast, other barriers, as suggested above, are likely to be established during development and therefore may differ among cell types. As one example, the promoters for a subset of NF-κB target genes in mouse macrophages possess a repressive histone H3 lysine 9 dimethyl (H3K9me2) mark that, prior to transcriptional activation, must be eliminated through the action of the H3K9me2 demethylase, Aof1 (refs. 4,7); the H3K9me2 mark found in unstimulated macrophages is probably deposited during the development of some, but perhaps not all, cell types. Through this mechanism, development may help dictate which genes in a given cell type will require a particular molecular event (in this example, H3K9me2 demethylation) for transcriptional activation, and this event may be catalyzed by some stimuli but not others.
With respect to the above model linking chromatin barriers established during development to stimulus-dependent pathways capable of eliminating the barriers, it is important to emphasize that the specific DNA sequence within the control regions of each NF-κB target gene is ultimately responsible for dictating how that gene is regulated. To establish a chromatin barrier at only a subset of genes, for example, sequence-specific DNA-binding proteins must recognize DNA motifs in the vicinity of those genes and recruit the machinery that establishes the barrier. Similarly, the removal of a chromatin barrier upon cell stimulation is likely to require recruitment of the necessary machinery by other transcription factors capable of recognizing specific sequences at each target gene. The possible role of non-coding RNAs in sequence-specific recognition at transcriptional control regions also requires further exploration10,11. Thus, although it is important to characterize the chromatin barriers and the chromatin remodeling and modification machinery that contribute to specificity of the response, an equally critical goal for the future is to identify the sequence-specific recognition events responsible for the establishment and elimination of these barriers at key NF-κB target genes. NF-κB may participate in some sequence-specific recognition events linked to chromatin dynamics, but other transcription factors are likely to be involved when chromatin is used to confer specificity to the NF-κB response.
Although chromatin appears to be one central contributor to specificity, there are several additional stimulus-dependent regulatory layers that are of equal importance. One is the frequent, if not universal, need for synergy between NF-κB and other transcription factors that bind DNA motifs within the promoter or enhancers for a given target gene. These additional factors may be inducible, cell type-specific, or expressed ubiquitously. The IFNB enhanceosome complex, which includes multiple transcription factors induced upon viral infection (NF-κB, ATF-2/c-Jun, and IRF3 or IRF7), represents a well-characterized example of transcription factor synergy12–14. Another regulatory layer involves stimulus-dependent post-translational modifications of NF-κB family members, which, at least in some instances, help relieve auto-inhibition of DNA-binding and support interactions with co-regulatory proteins that are needed for the proper induction of defined sets of target genes15–20. Co-regulatory interactions can influence multiple steps in gene transcription, including transcription initiation, transcription elongation and RNA processing21–22.
The specificity of an NF-κB response can be influenced further by the existence of five NF-κB family members, which can assemble into several dimeric species23, raising the possibility that specific dimers are activated by defined signaling pathways and physiological conditions, with each dimer involved in the regulation of a unique set of targets genes. Dimer specificity has been difficult to study because of considerable redundancy between dimeric species, and because each NF-κB family member can participate in multiple different dimers. Nevertheless, the RelB:p52 heterodimer activated by the non-canonical pathway provides one clear example of an NF-κB dimer activated by a unique set of stimuli via a unique pathway, most likely resulting in the activation of a unique set of target genes24,25.
One additional contributor to specificity of the NF-κB transcriptional response is activation kinetics. It is well established that the duration of NF-κB activation and the timing of its subsequent inactivation by nuclear export or degradation vary widely in a stimulus-specific manner. This variability can lead to substantial differences, not only in the kinetics of target gene expression, but also in the sets of target genes induced in response to a stimulus24,26–32.
In addition to the many mechanisms that help dictate which NF-κB target genes will be activated in a particular cell type in response to a given stimulus, powerful mechanisms have evolved to limit the magnitude of induction of NF-κB target genes and to attenuate the response33. Like the basic specificity mechanisms, regulation of induction magnitude and attenuation can be achieved at many levels. For example, well-established regulators of chromatin structure, such as the Mi-2/NuRD nucleosome remodeling complex and the Bcl-6 transcription factor, limit the magnitude of induction of specific subsets of NF-κB target genes in macrophages through mechanisms that have not been fully elucidated34,35. The induction magnitudes of specific target genes are also influenced by intrinsic differences in mRNA stability and by the active regulation of mRNA stability and translation, by miRNAs and proteins that bind 3' untranslated regions36–38. Finally, the duration of the NF-κB response can be regulated by a wide variety of feedback and attenuation mechanisms, ranging from the transcriptional upregulation of the Nfkbia (IκBα) gene and specific miRNA genes to the feedback inhibition of signaling pathways and the direct repression of target gene promoters and enhancers33, 37–38.
The remaining sections of this review highlight in greater depth three areas in which recent advances have led to novel conceptual insights into mechanisms regulating the specificity of the NF-κB response. Other NF-κB specificity mechanisms have been discussed in detail in other recent reviews25,27,30,31,39–43. It is important to emphasize that, although this article focuses primarily on the dynamic events that occur at NF-κB target genes in a chromatin context in the nucleus, the activation of signal transduction pathways at the plasma member and in the cytoplasm - in a stimulus- and cell type-specific manner - often acts upstream of these events to regulate NF-κB specificity20. That is, although the precise DNA sequence at the control regions of each gene ultimately dictates which chromatin events, NF-κB dimers, synergistic transcription factors, post-translational modifications, and co-regulatory interactions participate in proper regulation, all of these events are dependent on and coordinated by the precise set of signaling pathways induced by a given stimulus. Thus, a full understanding of NF-κB specificity will require in depth studies of both signal transduction and transcription, along with the successful merger of the two disciplines.
Establishing competence for an NF-κB response
One of the most exciting recent advances toward understanding the cell-type specificity of an NF-κB response emerged from genome-wide studies that link lineage specification and development to stimulus-dependent gene transcription. It has long been known that, at control regions for genes expressed only in a differentiated cell type, chromatin changes and transcription factor binding can be detected early in development, long before the gene is transcribed44. Studies of the mouse albumin (Alb1) and chicken lysozyme (Lyz1) loci provided early examples of this concept45–47.
Although important insights emerged from studies of individual model genes, recent genome-wide studies have revealed that key regulators of lineage commitment, specification, and development orchestrate cell-type-specific gene transcription and the cell-type specificity of inducible transcription. In the studies of Ghisletti et al.2, a chromatin immunoprecipitation massively parallel sequencing approach (ChIP-Seq) was used to identify genomic sites that associate with the p300 transcriptional co-activator in lipopolysaccharide (LPS)-stimulated mouse macrophages, toward the goal of identifying LPS-induced enhancers. Strikingly, many of the putative enhancers were found to be associated in unstimulated macrophages with PU.1, a key regulator of myeloid and B cell development42,43,48 (Fig. 2). In an independent study, Heinz et al. showed that, in myeloid cell progenitors, PU.1, along with other key regulators of myeloid development, bound thousands of putative enhancers linked to myeloid-specific genes3. In contrast, in B cell progenitors, PU.1 bound with other key regulators of B lymphopoiesis to a distinct set of genomic sites linked to genes expressed in B lineage cells. Other ChIP-chip and ChIP-seq studies have provided additional insights into the orchestration of B cell development at a genome-wide scale49–51.
Figure 2.
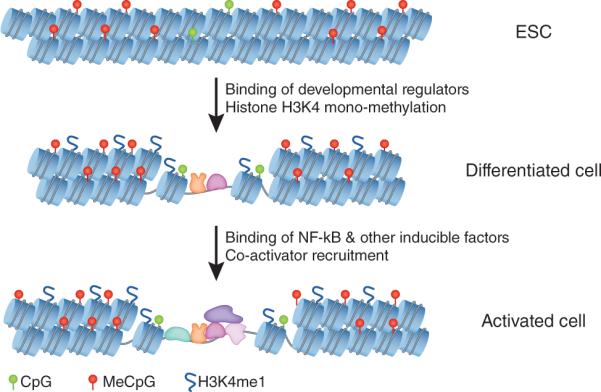
Enhancers for NF-κB target genes may acquire competence for activation at early stages of development. The diagram depicts events that appear to occur at enhancers for NF-κB target genes during development. Enhancers for some inducible genes appear to be associated with transcription factors in pluripotent cells, which may keep CpG dinucleotides in an unmethylated state and serve as placeholders during the earliest stages of development. During early stages of development, key transcription factors involved in lineage commitment, specification, or development, such as PU.1 in myeloid- and B-lineage cells, appear to bind the enhancers and induce local chromatin changes (histone H3K4me1 and nucleosome repositioning) that may confer competence for transcriptional activation in differentiated cells. As the differentiated cell responds to a stimulus, NF-κB and other inducible transcription factors bind the enhancer and recruit essential co-activators, such as p300. The enhancer complex is then thought to interact with the gene's promoter, which also binds constitutive, lineage-specific, and inducible transcription factors, including NF-κB, thereby promoting the cascade of events that culminates in transcription initiation and elongation.
The Ghisletti et al. and Heinz et al. studies both showed that transcription factor occupancy of the putative enhancers was associated with local changes in chromatin structure, as the regions generally exhibited low nucleosome density following PU.1 binding, suggestive of nucleosome eviction or repositioning2,3 (Fig. 2). PU.1 binding (in concert with the binding of other developmental regulatory proteins) also promoted the mono-methylation of histone H3K4; this histone modification is often associated with active enhancers52. The function of the H3K4me1 mark is not known, but the presence of this mark at the genomic regions analyzed in these studies supports the hypothesis that they correspond to enhancers.
These findings suggest that key developmental regulators are directly responsible for dictating which genes can be expressed in a lineage and which genes are permissive to inducible transcription when differentiated cells subsequently encounter a stimulus. One of several unanswered questions is whether the marking of enhancers at an early stage of development induces chromatin changes only locally (i.e. in the vicinity of the enhancer), throughout the locus, or perhaps only at the enhancer and other controls regions for the gene through the formation of an active chromatin hub53. The early enhancer interactions could also help position genes in close proximity to appropriate subnuclear locations that confer competence for transcriptional activation.
Recent studies of one enhancer that appears to conform to the above principles, an enhancer for the mouse Il12b gene, located 10 kb upstream of the Il12b transcription start site54, provide further insight into the mechanisms by which enhancers for NF-κB target genes may be regulated during development and cell activation. The ChIP-Seq datasets of Ghisletti et al.2 and Heinz et al.3 demonstrate that the Il12b enhancer is associated with PU.1 and histone H3K4me1 in both unstimulated and stimulated macrophages, with p300 binding observed only in stimulated cells. However, this enhancer is likely to be associated with transcription factors even at a pluripotent stage, as the enhancer exhibits a window of unmethylated CpG dinucleotides in mouse embryonic stem cells (ESC)55,56 (Fig. 2). This highly specific unmethylated window, which is retained throughout development, probably results from the binding of transcription factors during pluripotency. Thus, the Il12b enhancer, and perhaps others, may first be marked in pluripotent cells by factors that have not yet been identified, possibly providing competence for the binding of PU.1 and other lineage-specifying factors in myeloid progenitors.
Furthermore, although PU.1 and histone H3K4me1 are observed at the Il12b enhancer in unstimulated macrophages, DNase I hypersensitivity, suggestive of substantial chromatin accessibility, is observed only in stimulated cells54. Consistent with the stimulus-dependent DNase I hypersensitivity, SWI/SNF nucleosome remodeling complexes appear to be recruited to the enhancer only after macrophage stimulation34. Thus, although transcription factor binding in early progenitors may alter nucleosome structure at the enhancer to some extent, the dramatic changes in structure associated with DNase I hypersensitivity may occur only in differentiated cells in response to a stimulus. Genome-wide DNase I hypersensitivity experiments and other studies performed with individual enhancers and at a genome-wide scale are needed to further elucidate the mechanisms by which enhancers for NF-κB target genes contribute to induction and the specificity of the response.
Differential contributions of chromatin
The studies of Ghisletti et al.2 and Heintz et al.3 revealed that developmental events can establish competence for transcriptional activation in response to an NF-κB-inducing stimulus. However, development may also be accompanied by the deposition of chromatin barriers, with different barriers deposited at different subsets of genes. As discussed above, transcriptional activation may then be limited to genes that constitutively lack chromatin barriers and genes whose chromatin barriers can be eliminated by the pathways activated by a given stimulus. Among the chromatin barriers that have been suggested to limit activation of specific subsets of NF-κB target genes are histone H3K9 methylation, histone H3K27 methylation, and the simple presence of stable nucleosomes at promoters4–7,9. Many additional barriers to activation are likely to be uncovered in the future.
Importantly, chromatin can contribute to selectivity of the NF-κB response, not just by providing barriers that must be eliminated for transcriptional activation, but also be assisting with molecular events that are needed for efficient gene expression. This concept is exemplified by a recent study that began with the discovery of small-molecule inhibitors of a class of histone binding proteins known as BRD proteins57. These proteins, which include BRD2, BRD3, and BRD4, correspond to a subclass of bromodomain-containing proteins, which bind histones acetylated on specific N-terminal residues58–60. The acetyl-histone binding pocket of BRD proteins was found to be suitable for the design of highly selective small-molecule inhibitors57. Interestingly, the BRD inhibitors potently suppressed transcriptional activation of only a subset of NF-κB target genes in LPS-stimulated macrophages57. The sensitive genes tended to be those that possess non-CpG-island promoters, which appear to assemble into stable nucleosomes, thereby conferring a requirement for SWI/SNF nucleosome remodeling complexes for their activation9. In contrast, genes that lack a nucleosome barrier due to the presence of CpG-island promoter were largely resistant to the BRD inhibitor57.
BRD proteins are thought to serve as a bridge between histone modifications and P-TEFb, a facilitator of transcription elongation and RNA processing22,58–60. This connection raises the question of whether the selective effect of the BRD inhibitor might be because P-TEFb is required at only a subset of NF-κB target genes or, alternatively, because P-TEFb or BRD proteins are recruited to different subsets of NF-κB target genes by different mechanisms. Although this question cannot yet be fully answered, the current results suggest that the selectivity may be due to different BRD and P-TEFb recruitment mechanisms rather than variable requirements for BRD proteins and P-TEFb. BRD knockdown experiments were found to diminish the transcriptional induction of genes that are resistant to the small-molecule BRD inhibitor, suggesting that BRD proteins are important for transcription of both inhibitor-sensitive and inhibitor-resistant genes22,57. Furthermore, an independent study suggested that BRD proteins can be recruited to some NF-κB target genes by interaction with acetylated NF-κB itself rather than by a direct interaction with acetylated histones61.
Together, these results suggest that BRD proteins and P-TEFb may be important for efficient transcription of most or all NF-κB target genes, with different mechanisms of BRD and P-TEFb recruitment. Although acetylation of the histone H4 residues involved in BRD4 recruitment (H4K5, K8, and K12) was found to be potently induced at genes that were resistant to the BRD inhibitor22,57, BRD4 may be recruited to these genes by two redundant mechanisms – recruitment by NF-κB and recruitment by acetylated histone H4 – which would explain the resistance of these genes to the BRD inhibitor.
NF-κB post-translational modifications and co-activator recruitment
It has long been appreciated that NF-κB subunits can be covalently modified in stimulated cells. It also is widely appreciated that transcriptional activation by NF-κB generally requires interactions with transcriptional co-activators, which can influence chromatin structure or other events required for transcriptional activation. Undoubtedly, these two topics are closely linked, as NF-κB modifications will often regulate interactions with co-regulatory proteins. All NF-κB family members appear to be extensively modified under physiological conditions, and the list of modifications and co-regulatory interactions is expanding more rapidly than ever15–19,62–68.
Importantly, post-translational modifications of NF-κB dimers and co-regulatory interactions that may or may not depend on the post-translational modifications are likely to play a major role in specificity of the NF-κB response. Some cell stimuli may activate signal transduction pathways that promote a specific post-translational modification of an NF-κB subunit, whereas other stimuli that efficiently catalyze the nuclear translocation of NF-κB may not promote this post-translational modification. Those NF-κB target genes that require the post-translational modification (and the hypothetical co-regulatory interaction that is dependent on this modification) for their transcriptional activation will be activated only in response to stimuli that promote the modification, whereas target genes that do not require modified NF-κB may be activated by a broader range of stimuli.
A post-translational modification or a co-regulatory interaction may contribute to the activation of only a subset of NF-κB target genes for any of a variety of reasons. First, as with the histone H3K9 demethylase Aof1 or the SWI/SNF nucleosome remodeling complexes discussed above7,9, a co-regulatory interaction may help overcome a chromatin barrier that is present at only a subset of NF-κB target genes. Second, the co-regulatory protein may contribute to a step in the transcriptional activation pathway that is essential at only a subset of NF-κB target genes; activation of other target genes may proceed via fundamentally different pathways that involve, for example, different sets of general transcription factors69. Third, as suggested above for BRD proteins and P-TEFb, the same co-regulatory protein may be recruited to different sets of target genes via different mechanisms, allowing NF-κB target genes to exhibit differential requirements for the post-translational modification. Fourth, a different coregulatory protein recruited via a different mechanism may carry out the same function at a subset of NF-κB target genes.
This selective functions of NF-κB post-translational modifications and co-activator interactions is best exemplified by one of the first NF-κB post-translational modifications to be described: phosphorylation of mouse RelA (also known as p65) on serine 276 (S276). This phosphorylation event, catalyzed by cyclic AMP-dependent protein kinase (PKAc), promotes a conformational change that relieves auto-inhibition of the DNA-binding activity of RelA and allows RelA to interact with the transcriptional co-activators p300 and CBP16,17. In addition to an intriguing epigenetic phenotype, targeted mutagenesis of RelA S276 in the mouse germline revealed a severe defect in the activation of only a subset of NF-κB target genes in tumor necrosis factor (TNF)-stimulated fibroblasts18. Presumably, the RelA:p300/CBP interaction is required at only a subset of NF-κB target genes for one of the reasons listed above, although the precise reason remains to be elucidated. Targeted mutations that disrupt other post-translational modifications will be needed in the future to better understand how the many modifications that have been documented contribute to the complex regulation of NF-κB target genes in various physiological settings.
Although most post-translational modifications of NF-κB family members are thought to contribute to transcriptional induction, Levy et al. recently reported that RelA is monomethylated at lysine 310 (K310) specifically in unstimulated cells, leading to the suggestion that this methylation event contributes to tonic repression of a subset of NF-κB target genes19. The protein methyltransferase SETD6 was found to be responsible for K310 methylation in unstimulated cells, and K310 methylation was observed specifically on the small fraction of nuclear, chromatin-associated RelA found prior to stimulation. RelA that was monomethylated on K310 bound the ankryin-repeat domain of GLP, a partner of the G9a histone H3K9 methyltransferase, leading to evidence that K310-methylated RelA promotes H3K9 methylation and active repression of a subset of NF-κB target genes70. Interestingly, K310 is immediately adjacent to S311, which previously was shown to be phosphorylated by PKC-ζ in TNF-stimulated cells71. S311 phosphorylation was found to block GLP binding to RelA, providing a possible mechanism by which RelA-mediated repression is relieved. In the future, it will be interesting to determine whether this proposed regulatory pathway impacts precisely the same set of NF-κB target genes that require the Aof1 H3K9 demethylase for their activation, as discussed above7.
Concluding Remarks
Since the discovery of NF-κB 25 years ago, much has been learned about the mechanisms that contribute to the cell-type and stimulus specificity of the NF-κB response, as well as the many mechanisms that refine and attenuate the response. When combined with major technological advances, these insights will facilitate further progress over the next quarter century. The availability of genome sequence information and the increasing quality of genomics technologies to take advantage of this information will be especially important. The value of ChIP-Seq for monitoring the binding of NF-κB and other regulators of NF-κB target genes at a genome-wide scale has already been documented. In addition, the quantitative and complete analysis of transcriptomes by RNA-Seq will allow investigators to rigorously evaluate the subsets of target genes influenced by various contributors to selective regulation, and develop hypotheses based on common properties of those sets of genes. Despite the value of studies performed at a genome-wide scale, many mechanistic insights will emerge only from in depth analyses of individual model genes. It is now abundantly clear, however, that an understanding of the selective regulation of model genes will require studies performed largely when the genes and their control regions are assembled into native chromatin structures.
One fundamental issue that remains poorly understood is why NF-κB appears to be employed so frequently as a dominant regulator of inducible transcription. Does it regulate transcription via molecular mechanisms that are fundamentally similar to those used by other transcription factors, as might be suggested by much of the current literature? If so, it may be frequently employed as a dominant regulator of inducible transcription simply because a diverse range of cell stimuli evolved signaling pathways to activate NF-κB dimers. However, an alternative scenario is that NF-κB emerged as a common transcription factor for regulating inducible transcription because it influences gene transcription via unique mechanisms that are fundamentally different from the mechanisms employed by most other transcription factors. Recent high-resolution single-cell imaging studies have provided advances toward understanding the dynamic behavior of NF-κB, which may ultimately reveal key properties that distinguish it from other inducible transcription factors28–38.
A final goal for the future is to take advantage of the accumulated knowledge of NF-κB selectivity mechanisms to develop therapeutic strategies for the treatment of diseases in which NF-κB participates, including cancer and a wide variety of inflammation-related disorders72. Because of NF-κB's broad functions in normal physiology, it may not itself be an ideal therapeutic target. However, a sophisticated understanding of the strategies employed to confer specificity to the NF-κB response may suggest strategies for the therapeutic modulation of individual NF-κB target genes or select subsets of target genes.
REFERENCES
- 1.Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 2.Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A, Wei CL, Ragoussis J, Natoli G. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saccani S, Natoli G. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev. 2002;16:2219–2224. doi: 10.1101/gad.232502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 6.De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, Bucci G, Caganova M, Notarbartolo S, Casola S, Testa G, Sung WK, Wei CL, Natoli G. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. 2009;28:3341–3352. doi: 10.1038/emboj.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Essen D, Zhu Y, Saccani S. A feed-forward circuit controlling inducible NF-kB target gene activation by promoter histone demethylation. Mol. Cell. 2010;39:750–760. doi: 10.1016/j.molcel.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Fan W, Morinaga H, Kim JJ, Bae E, Spann NJ, Heinz S, Glass CK, Olefsky JM. FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J. 2010;29:4223–4236. doi: 10.1038/emboj.2010.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, Smale ST. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. 2009;138:114–128. doi: 10.1016/j.cell.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen LL, Carmichael GG. Decoding the function of nuclear long non-coding RNAs. Curr. Opin. Cell Biol. 2010;22:357–364. doi: 10.1016/j.ceb.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell. 2000;103:667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- 13.Panne D, Maniatis T, Harrison SC. Crystal structure of ATF-2/c-Jun and IRF-3 bound to the interferon-β enhancer. EMBO J. 2004;23:4384–4393. doi: 10.1038/sj.emboj.7600453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-β enhanceosome. Cell. 2007;129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 16.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the co-activator CBP/p300. Mol. Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 17.Zhong H, May MJ, Jimi E, Ghosh S. Phosphorylation of nuclear NF-κB governs its association with either HDAC-1 or CBP/p300: a mechanism for regulating the transcriptional activity of NF-κB. Mol. Cell. 2002;9:625–636. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- 18.Dong J, Jimi E, Zhong H, Hayden MS, Ghosh S. Epigenetic regulation of NF-kB dependent gene expression. Genes Dev. 2008;22:1159–1173. doi: 10.1101/gad.1657408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levy D, et al. Lysine methylation of the NF-kB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-kB signaling. Nat. Immunol. 2011;12:29–36. doi: 10.1038/ni.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cross-reference Sankar Ghosh's review.
- 21.Amir-Zilberstein L, Ainbinder E, Toube L, Yamaguchi Y, Handa H, Dikstein R. Differential regulation of NF-kappaB by elongation factors is determined by core promoter type. Mol. Cell. Biol. 2007;27:5246–5259. doi: 10.1128/MCB.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138:129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2004;25:6706–6716. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 25.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmann A, Baltimore D. Circuitry of NF-kappaB signaling. Immunol. Reviews. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 27.Sen R, Smale ST. Selectivity of the NF-κB response. Cold Spr. Harb. Perspect. Biol. 2010;2:a000257. doi: 10.1101/cshperspect.a000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashall L, et al. Pulsatile stimulation determines timing and specificity of NF-κB-dependent transcription. Science. 2009;324:242–246. doi: 10.1126/science.1164860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tay S, et al. Single-cell NF-kappaB dynamics reveal digital activation and analog information processing. Nature. 466:267–271. doi: 10.1038/nature09145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee TK, Covert MW. High-throughput single-cell NF-κB dynamics. Curr. Opin. Genet. Dev. 2010;20:677–683. doi: 10.1016/j.gde.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paszek P, Jackson DA, White MR. Oscillatory control of signaling molecules. Curr. Opin. Genet. Dev. 2010;20:670–676. doi: 10.1016/j.gde.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, et al. Interactions among oscillatory pathways in NF-kappaB signaling. BMC Syst. Biol. 2011;5:23. doi: 10.1186/1752-0509-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cross-reference Rulan's review.
- 34.Ramirez-Carrozzi VR, et al. Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 2006;20:282–296. doi: 10.1101/gad.1383206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barish GD, Yu RT, Karunasiri M, Ocampo CB, Dixon J, Benner C, Dent AL, Tangirala RK, Evans RM. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010;24:2760–2765. doi: 10.1101/gad.1998010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hao S, Baltimore D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 2009;10:281–288. doi: 10.1038/ni.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 38.Anderson P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat. Rev. Immunol. 2010;10:24–35. doi: 10.1038/nri2685. [DOI] [PubMed] [Google Scholar]
- 39.Foster SL, Medzhitov R. Gene-specific control of the TLR-induced inflammatory response. Clin. Immunol. 2009;130:7–15. doi: 10.1016/j.clim.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 41.Smale ST. Selective transcription in response to an inflammatory stimulus. Cell. 2010;140:833–844. doi: 10.1016/j.cell.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Natoli G. Maintaining cell identity through global control of genomic organization. Immunity. 2010;33:12–24. doi: 10.1016/j.immuni.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Natoli G, Ghisletti S, Barozzi I. The genomic landscapes of inflammation. Genes Dev. 2011;25:101–106. doi: 10.1101/gad.2018811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smale ST. Pioneer factors in embryonic stem cells and differentiation. Curr. Opin. Genet. Dev. 2010;20:519–526. doi: 10.1016/j.gde.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gualdi R, Bossard P, Zheng M, Hamada Y, Coleman JR, Zaret KS. Hepatic specification of the gut endoderm in vitro: cell signaling and transcriptional control. Genes Dev. 1996;10:1670–1682. doi: 10.1101/gad.10.13.1670. [DOI] [PubMed] [Google Scholar]
- 46.Kontaraki J, Chen HH, Riggs A, Bonifer C. Chromatin fine structure profiles for a developmentally regulated gene: reorganization of the lysozyme locus before trans-activator binding and gene expression. Genes Dev. 2000;14:2106–2122. [PMC free article] [PubMed] [Google Scholar]
- 47.Zaret KS, et al. Pioneer factors, genetic competence, and inductive signaling: programming liver and pancreas progenitors from the endoderm. Cold Spr. Harb. Symp. Quant. Biol. 2008;73:119–126. doi: 10.1101/sqb.2008.73.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 49.Schebesta A, et al. Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity. 2007;27:49–63. doi: 10.1016/j.immuni.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 50.Lin YC, et al. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat. Immunol. 2010;11:635–643. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Treiber T, et al. Early B cell factor 1 regulates B cell gene networks by activation, repression, and transcription- independent poising of chromatin. Immunity. 2010;32:714–725. doi: 10.1016/j.immuni.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 52.Heintzman ND, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 53.de Laat W, Grosveld F. Spatial organization of gene expression: the active chromatin hub. Chromosome Res. 2003;11:447–459. doi: 10.1023/a:1024922626726. [DOI] [PubMed] [Google Scholar]
- 54.Zhou L, et al. An inducible enhancer required for Il12b promoter activity in an insulated chromatin environment. Mol. Cell. Biol. 2007;27:2698–2712. doi: 10.1128/MCB.00788-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu J, et al. Pioneer factor interactions and unmethylated CpG dinucleotides mark silent tissue-specific enhancers in embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2007;104:12377–12382. doi: 10.1073/pnas.0704579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu J, et al. Transcriptional competence and the active marking of tissue-specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes Dev. 2009;23:2824–2838. doi: 10.1101/gad.1861209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nicodeme E, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.LeRoy G, Rickards B, Flint SJ. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell. 2008;30:51–60. doi: 10.1016/j.molcel.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jang MK, et al. The bromodomain protein Brd4 is a positive regulatory component of PTEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 60.Yang Z, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 61.Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. Brd4 coactivates transcriptional activation of NF-kB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009;29:1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang XD, et al. Negative regulation of NF-kappaB action by Set9-mediated lysine methylation of the RelA subunit. EMBO J. 2009;28:1055–1066. doi: 10.1038/emboj.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ea CK, Baltimore D. Regulation of NF-κB activity through lysine monomethylation of p65. Proc. Natl. Acad. Sci. 2009;106:18972–18977. doi: 10.1073/pnas.0910439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang XD, Tajkhorshid E, Chen LF. Functional interplay between acetylation and methylation of the RelA subunit of NF-kappaB. Mol. Cell. Biol. 2010;30:2170–2180. doi: 10.1128/MCB.01343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 66.van Essen D, Engist B, Natoli G, Saccani S. Two modes of transcriptional activation at native promoters by NF-kappaB p65. PLoS Biol. 2009;7:e73. doi: 10.1371/journal.pbio.1000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamamoto M, Takeda K. Role of nuclear IkappaB proteins in the regulation of host immune responses. J. Infect. Chemother. 2008;14:265–269. doi: 10.1007/s10156-008-0619-y. [DOI] [PubMed] [Google Scholar]
- 68.Wan F, et al. Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell. 2007;131:927–939. doi: 10.1016/j.cell.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 69.Basehoar AD, Zanton SJ, Pugh BF. Identification and distinct regulation of yeast TATA box-containing genes. Cell. 2004;116:699–709. doi: 10.1016/s0092-8674(04)00205-3. [DOI] [PubMed] [Google Scholar]
- 70.Tachibana M, et al. Histone methyltransferase G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005;19:815–826. doi: 10.1101/gad.1284005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Duran A, Diaz-Meco MT, Moscat J. Essential role of RelA Ser311 phosphorylation by ζPKC in NF-κB transcriptional activation. EMBO J. 2003;22:3910–3918. doi: 10.1093/emboj/cdg370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cross-reference review by Yinon and Karin.