

Abstract
The proteolytic machinery comprising metalloproteases and γ-secretase, an intramembrane aspartyl protease involved in Alzheimer's disease, cleaves several substrates in addition to the extensively studied amyloid precursor protein. Some of these substrates, such as N-cadherin, are synaptic proteins involved in synapse remodeling and maintenance. Here we show, in rats and mice, that metalloproteases and γ-secretase are physiologic regulators of synapses. Both proteases are synaptic, with γ-secretase tethered at the synapse by δ-catenin, a synaptic scaffolding protein that also binds to N-cadherin and, through scaffolds, to AMPA receptor and a metalloprotease. Activity-dependent proteolysis by metalloproteases and γ-secretase takes place at both sides of the synapse, with the metalloprotease cleavage being NMDA receptor-dependent. This proteolysis decreases levels of synaptic proteins and diminishes synaptic transmission. Our results suggest that activity-dependent substrate cleavage by synaptic metalloproteases and γ-secretase modifies synaptic transmission, providing a novel form of synaptic autoregulation.
Introduction
Metalloproteases and γ-secretase act in succession to cleave single-pass transmembrane proteins. Metalloproteases and the closely related a disintegrin and metallopeptidase (ADAM) or, for some substrates, β-secretase 1 (BACE1), initiate the proteolytic pathway by shedding the membrane protein substrate's ectodomain (Thinakaran and Koo, 2008). γ-Secretase, a multisubunit aspartyl protease assembled from four proteins [presenilin 1 (PS1), nicastrin, anterior pharynx-defective 1 (Aph1), and presenilin enhancer 2 (Pen2)], cleaves the resulting C-terminal fragment (CTF) within its transmembrane domain (McCarthy et al., 2009). While this pathway has been studied most extensively for amyloid precursor protein (APP), whose cleavage yields the Aβ peptide, the metalloprotease/γ-secretase proteolytic machinery also cleaves other substrates, especially proteins implicated in synapse remodeling and maintenance, including EphRs, ephrins, and cadherins (Dalva et al., 2007; McCarthy et al., 2009). However, these physiological roles have received very little attention.
The precise location of the metalloprotease/γ-secretase proteolytic machinery is still unclear. ADAM10, ADAM17, and membrane type 5 matrix metalloproteinase (MT5-MMP), the three known metalloproteases that shed the ectodomains of γ-secretase substrates, have been localized at synapses (Monea et al., 2006; Marcello et al., 2007; Malinverno et al., 2010), whereas γ-secretase has been reported mainly in membranes of endosomes, although its presence at the plasma membrane has also been suggested (Lah et al., 1997; Georgakopoulos et al., 2000; Kaether et al., 2006). Interestingly, PS1, the catalytic subunit of γ-secretase, binds to a synaptic protein, δ-catenin, which regulates actin polymerization and cell adhesion (Kosik et al., 2005). We have previously shown that δ-catenin is present in the postsynaptic density (PSD) (Silverman et al., 2007), where it links the N-cadherin intracellular domain via synaptic scaffolds to MT5-MMP, a metalloprotease that cleaves N-cadherin (Monea et al., 2006) and synaptic proteins, particularly the AMPA receptor (AMPAR) (Silverman et al., 2007). Therefore, the proteolytic machinery formed by a metalloprotease and γ-secretase may be present in synaptic complexes with their substrates, where it could modulate synaptic function.
Synaptic dysfunction is a major early event of Alzheimer disease (Knobloch and Mansuy, 2008). Considering the putative role of these proteases in Alzheimer disease (Thinakaran and Koo, 2008), we here investigate possible physiologically relevant synaptic functions of metalloproteases and γ-secretase. We find that the metalloprotease and γ-secretase proteolytic machinery operates on both sides of the synapse and cleaves synaptic N-cadherin. The metalloprotease-mediated cleavage is regulated by NMDA receptor (NMDAR)-dependent synaptic activity, whereas γ-secretase activity is insensitive to the activity of the synapse. This proteolytic machinery downregulates synaptic function. We find a protease-dependent regulation of glutamatergic neurotransmission in the hippocampal neurons in which vesicle recycling and synaptic protein levels are modified. Because the pathway inhibits synaptic transmission in response to synaptic activity, we propose that this metalloprotease/γ-secretase pathway provides a novel form of activity-dependent synaptic autoregulation.
Materials and Methods
Mice and rats.
δ-Catenin knock-out mice (Israely et al., 2004) were kindly provided by Dr X. Liu, University of California, Los Angeles, CA (Israely et al., 2004), and bred in our laboratory. The experiments were done with 12-week-old animals of either sex. Eight-week-old rats of either sex were used for fractionation experiments as indicated.
Antibodies and inhibitors.
Antibodies used were as follows: mouse anti-PSD-95 antibody (1:500; clone K28.43 or clone K28/86.2; NeuroMab); rabbit anti-GluA2/3 antibody (1 μg/ml; Millipore); mouse anti-δ-catenin (1/200; BD Biosciences); mouse anti-N-cadherin (1/100; BD Biosciences); mouse anti-synaptophysin (1/1000; Sigma-Aldrich); mouse anti-synaptotagmin 1 (1/100; Synaptic Systems); rabbit anti-MT5-MMP (1.5 μg/ml; cytoMT5-MMP, catalog #2850) made in our laboratory and described previously (Monea et al., 2006); rabbit anti-EphB2 [1/1000 whole brain (WB), Dr. M. Greenberg, Harvard Medical School, Boston, MA]; rabbit anti-ADAM 10 [1/200 for immunofluorescence (IF), 1/500 for WB, Sigma-Aldrich]; rabbit anti-ADAM 17 (1/200 for IF, 1/500 for WB, Abcam); mouse anti-BACE (1/100, 3D5, Dr. R. Vassar, Northwestern University, Chicago, IL); rabbit anti-presenilin N-terminal (1/500 for IF and 1/1000 for WB, PS1NT, Calbiochem); rabbit anti-presenilin loop domain (1/1000 for WB, Calbiochem); rabbit anti-nicastrin (1/1000, Dr. P. Fraser, University of Toronto, Toronto, Canada); anti-GluR1 (1/1000; Millipore); rabbit anti-tubulin (1/1500; Sigma-Aldrich); mouse anti-APP (clone C1/6.1 and m3.2, 1/1000, and for IF, 1/1000, Millipore).
Secondary antibodies were used at 1/1000, including monoclonal secondary antibodies (Alexa Fluor color; Invitrogen), isotype-specific monoclonal secondary antibodies IgG1 (Alexa Fluor color; Invitrogen), IgG2A (Alexa Fluor color; Invitrogen), and polyclonal secondary antibodies (Alexa Fluor color; Invitrogen).
Inhibitors used were: GM6001 (10 μm, ilomastat; Millipore Bioscience Research Reagents); epoxomicin (1 μm; Peptides International); L-685458 (1–10 μm γ-secretase inhibitor X; Calbiochem); DAPT (10 μm, γ-secretase inhibitor IX; Calbiochem); PP2 (10 μm;Calbiochem); KN-93 [calcium/calmodulin-dependent protein kinase II (CaMKII) inhibitor, 5 μm; Calbiochem]; d-APV (100 μm; Sigma-Aldrich); JNK inhibitor II (50 μm; Calbiochem); bicuculline (40 μm; Tocris Bioscience); NMDA (40 μm; Sigma-Aldrich); glutamate (40 μm; Sigma-Aldrich); 4-aminopyridine (4AP, 100 μm; Tocris Bioscience).
Hippocampal and cortical culture.
Cultures were prepared from embryonic day 19 Sprague Dawley rat embryonic brain tissue. Animals were killed by CO2 in compliance with New York University Medical Center's Institutional Animal Care and Use Committee. Hippocampal and cortical primary neurons were prepared as described previously (Osten et al., 1998). Neurons were plated at a density of 100,000 cells on poly-l-lysine-coated glass coverslips in a six-well plate for immunofluorescence or 3.5 millions on poly-l-lysine-coated 10 cm dishes for biochemical experiments. Neurons were grown in Neurobasal medium with B27 (Invitrogen). At the first change of medium, a one-time dose of the drug AraC (4 μm; Sigma-Aldrich) was added to inhibit growth of dividing cells for immunofluorescence experiments (Macaskill et al., 2009).
Immunofluorescence.
At 18–21 days in vitro (DIV), neurons were fixed with 2% paraformaldehyde (Electron Microscopy Sciences) in 0.12 m sucrose in PBS (15 min at room temperature), permeabilized with 0.2% Triton X-100 (5 min at room temperature), blocked in 10% bovine serum albumin (BSA) in PBS, and then incubated with primary antibodies in 3% BSA. Cells were then washed three times in 1× PBS. Secondary antibodies conjugated to Alexa Fluor probes were incubated in 3% BSA for 1 h and mounted on slides. Images were captured with a Zeiss LSM 510 Meta laser-scanning confocal microscope using a Plan-Apochromat 63× objective and LSM 510 Meta software.
Synaptotagmin reuptake.
Fourteen days in vitro neurons were treated overnight with inhibitors and incubated with synaptotagmin antibody for 5 min. Neurons were washed, fixed, and permeabilized as described in the previous section. Ten neurons per coverslip were imaged, and nine separate experiments were performed. Each experiment was averaged. The nine experiments were then averaged.
Electron microscopy.
Pentobarbital-anesthetized adult Sprague Dawley rats were perfused intracardially with mixed aldehyde fixatives (0.1% glutaraldehyde/4% depolymerized paraformaldehyde or 2% glutaraldehyde/2% paraformaldehyde) dissolved in 0.1 m phosphate buffer (PB; pH 7.4) after a quick rinse with heparinized saline. After overnight postfixation in the same fixative at 4°C, 50 μm brain sections were cut on a Vibratome and collected in PB. Sections containing cortex and hippocampus were incubated 30 min on a shaker at room temperature with 1% sodium borohydride in PBS to block free aldehyde groups, then in 10% normal goat serum to block nonspecific antibody binding. Sections were incubated overnight in primary antibody (rabbit anti-nicastrin; 1:1000–1:2000), rinsed, and then for 2 h in goat anti-rabbit IgG (1:100), conjugated to ultrasmall gold particles (∼1 nm; British BioCell). Sections were then rinsed in 0.01 m acetate buffer, and gold particles were silver intensified using an IntenSE M kit (GE Healthcare). Sections for electron microscopy were postfixed in 0.5–1% osmium tetroxide in PB for 1 h, rinsed in 0.1 m maleate buffer, pH 6, then stained en bloc with 1% uranyl acetate for 1 h. After dehydration in ascending ethanol series and propylene oxide, sections were infiltrated with Epon/Spurr resin, flat mounted between sheets of ACLAR plastic, and polymerized at 60° for 48 h. Seventy nanometer sections from selected areas were cut with a diamond knife, collected on 300 mesh copper grids, stained with uranyl acetate and Sato's lead, and examined with a Tecnai 10 transmission electron microscope (Philips). Images were collected with a 12 bit 1024 × 1024 pixel CCD camera (Gatan); final photomicrographs were prepared using Adobe Photoshop to compose, crop, and adjust contrast.
Treatment and stimulation experiments.
Eighteen days in vitro cortical or hippocampal neurons in culture were stimulated with bicuculline and 4-aminopyridine for 1 h or bicuculline for 24 h, washed three times with PBS, and lysed with 50 mm Tris, pH 7.4, and 2% SDS. Equal amount of total lysate (15–20 μg) were loaded on 8–12% SDS-PAGE gels or precast gradient 4–20% SDS-PAGE gels (Thermo Scientific). Western blotting was further performed with N-cadherin. Tubulin was used as loading control. For most of the experiments otherwise indicated, we quantified the ratio CTF1/full-length protein. For NMDA and glutamate stimulation, cortical neurons were stimulated for the time indicated. Medium was then exchanged for conditioned medium for another hour. Cells were washed and lysed as before. Inhibitors were applied 15 min before stimulation, during stimulation, and for the hour after stimulation.
Postsynaptic density purification.
Synaptosomal and PSD fractions from rat and mouse brain and primary hippocampal cultures were prepared as described previously (Jordan et al., 2004). Equal amount of fractions (10–15 μg) were loaded onto SDS-PAGE gel. Western blots were probed with different antibodies. Synaptophysin was used as loading control for WB, total membrane (P2), and synaptosome (syn) fractions, and PSD-95 was used for PSD fractions.
Presynaptic and postsynaptic fractionation.
Presynaptic and postsynaptic fractions were prepared from synaptosomes by extraction at differential buffer pH as described previously (Phillips et al., 2001). Equal amount of fractions (10–15 μg) were loaded on SDS-PAGE gel. Western blots were probed with different antibodies. Synaptophysin was used as loading control for WB, P2, syn, and presynaptic fractions. PSD-95 was used as loading control for postsynaptic fractions.
Surface biotinylation.
For the biotinylation experiments, cortical neurons in culture were washed and incubated with 2 mg/ml sulfo-NHS-SS-biotin (Pierce) in PBS for 15 min at 4°C. Nonreacted biotin was quenched with 50 mm glycine in PBS, and cells were rinsed twice with cold PBS. Total membrane (P2) and synaptosome fractions were prepared in RIPA buffer (50 mm Tris, pH 7.4,150 mm NaCl, 5 mm EDTA, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate) and complete protease inhibitor cocktail (Roche). Biotinylated surface proteins were purified with UltraLink-immobilized NutrAvidin beads (Pierce) and analyzed by SDS-PAGE and Western blot. Densitometry of the bands and quantification of surface proteins were performed using ImageJ software (NIH), and p values were determined by standard t test and ANOVA analysis when indicated. Equal amounts of proteins were loaded on gels.
Immunoprecipitations.
The synaptosomal fraction was also solubilized in immunoprecipitation buffer [50 mm Tris, pH 7.4,150 mm NaCl, 5 mm EDTA, and 1% 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate (CHAPSO)] for 2 h at 4°C and cleared at 16,000 × g for 10 min at 4°C. Supernatants containing 1 mg of protein were incubated with the antibodies rabbit anti-nicastrin, rabbit anti-PS1NT, and a rabbit IgG negative control (2 μg of antibody) overnight, then 20 μg of protein G or protein A beads (Santa Cruz Biotechnology), as determined by the genotype of the antibody, were added at the reaction for an additional 2 h followed by two washes with immunoprecipitation buffer containing 350 mm NaCl and one wash with immunoprecipitation buffer containing 150 mm NaCl and then eluted with loading buffer.
γ-Secretase assay.
In vitro γ-secretase assays were performed as described previously (Farmery et al., 2003). Briefly, synaptosomal extracts from rat brain were solubilized in CHAPSO buffer and incubated with a fluorogenic peptide probe at 37°C overnight in the presence of inhibitors. The probe contains a consensus aspartic acid cleavage site that, when proteolyzed by γ-secretase, generates a fluorescent peptide whose fluorescence is measured on a plate reader with an excitation wavelength of 340 nm and an emission wavelength of 490 nm. Results were from six independent replicates.
Miniature ΕPSC recording.
Miniature EPSCs (mEPSCs) were assayed in cultured hippocampal neurons at DIV 14. Neurons were voltage clamped with the whole cell ruptured patch technique throughout the experiment. The bath solution consisted of the following (in mm): 119 NaCl, 5 KCl, 20 HEPES, 2 CaCl2, 2 MgCl2, 30 glucose, 0.001 glycine, 0.01 bicuculline, pH 7.3; osmolarity was adjusted to 330 mOsm with sucrose. The solution in the whole cell patch electrode consisted of the following (in mm): 130 K-gluconate, 10 KCl, 5 MgCl2, 0.6 EGTA, 5 HEPES, 0.06 CaCl2, 2 Mg-ATP, 0.2 GTP, 0.2 leupeptin, 20 phosphocreatine, and creatine-phosphokinase at 50 U/ml. Tetrodotoxin at 1 μm was also added to the bath to suppress action potentials. Currents were recorded with a Warner amplifier (model PC-501A) and filtered at 1 kHz. To eliminate artifacts due to variation of the seal properties, the access resistance was monitored for constancy throughout all experiments. The recordings were digitized (Digidata 1440A, Molecular Devices) and analyzed with the Mini Analysis Program, version 4.0, from Synaptosoft.
Results
The proteolytic machinery and its substrates are present at synapses
Although γ-secretase in neurons is found mainly in endosomes (Kaether et al., 2006), synaptic pools of both γ-secretase and metalloproteases have been suggested (Lah et al., 1997; Georgakopoulos et al., 2000; Peiretti et al., 2003; Monea et al., 2006; Marcello et al., 2007). Indeed, the synaptic scaffolding proteins glutamate receptor-interacting protein/AMPA receptor-binding protein (GRIP/ABP) and synapse-associated protein 97 (SAP97) associate, respectively, with MT5-MMP and ADAM10/17, proteases involved in the ectodomain shedding of γ-secretase substrates (Peiretti et al., 2003; Monea et al., 2006; Marcello et al., 2007). Moreover, γ-secretase interacts with δ-catenin, a scaffolding protein that also binds ABP/GRIP (Silverman et al., 2007). These interactions suggest the presence of the proteases in multiprotein complexes at synapses. To test this possibility, we performed immunofluorescence on 21 DIV dissociated hippocampal neurons. We found three metalloproteases that can shed ectodomains of γ-secretase substrates (Reiss et al., 2005; Monea et al., 2006; Postina, 2008), ADAM10, ADAM17, and MT5-MMP (Fig. 1A, arrows), and two components of γ-secretase, nicastrin and PS1 (Fig. 1B, arrows), colocalized with the postsynaptic marker PSD-95.
Figure 1.

The proteolytic machinery and its substrates are present at synapses. A, 21 DIV hippocampal neurons were stained with ADAM10 (i), ADAM17 (ii), or MT5-MMP (iii) and a postsynaptic marker, PSD-95 (FITC staining, middle panel). Top and bottom panels are merged images. Arrows illustrate colocalization with PSD-95. Scale bar, 10 μm. B, 21 DIV hippocampal neurons were stained with PS1 (i) and nicastrin (ii) and PSD-95. Left and bottom panels are merged images. Arrows illustrate colocalization. Scale bar, 10 μm. C, Subcellular fractionation of rat brain into whole brain, labeled WB, and synaptosomes, labeled syn. Synaptosomes were further fractionated into presynaptic (Pre) and postsynaptic (Post) fractions. All the fractions were then blotted for ADAM10, ADAM17, and MT5-MMP. Note enrichment of metalloproteases in the postsynaptic fraction. PSD-95 and synaptophysin (Syp) were used as postsynaptic and presynaptic markers, respectively. D, Subcellular fractionation of rat brain as before. All the fractions were then blotted for PS1NTF, PS1CTF, and nicastrin. All three γ-secretase components were in synaptosomes and in both presynaptic and postsynaptic fractions. Note enrichment of nicastrin in the presynaptic fraction. E, Electron micrographs showing pre-embedded immunogold staining for nicastrin. Scale bar, 200 nm. F, DIV 21 hippocampal neurons were stained with N-cadherin or EphRB (i) and PSD-95 (ii); iii shows merged images. Arrows point to examples of colocalization between substrates and PSD-95. Scale bar, 10 μm. G, Subcellular fractionation of rat brain. Western blotting shows N-cadherin and EphRB in synaptosomes and in both presynaptic and postsynaptic fractions.
Western blotting demonstrated that metalloproteases and two γ-secretase components, nicastrin and PS1 (including the PS1 N- and C-terminal fragments PS1NTF and CTF), were present in synaptosomes fractionated from adult rat brain (Fig. 1C,D). We used differential pH extraction to fractionate synaptosomal presynaptic and postsynaptic components (Phillips et al., 2001). Western blotting revealed the presence of the three sheddases, ADAM10, ADAM17, and MT5-MMP, in presynaptic and postsynaptic fractions with a postsynaptic enrichment (Fig. 1C). PS1-NTF and PS1-CTF were distributed equally between the presynaptic and postsynaptic fractions (Fig. 1D), while nicastrin showed presynaptic enrichment (Fig. 1D). However, the integrity of the γ-secretase complex is highly detergent sensitive (Li et al., 2000); thus, nicastrin may be lost from the postsynaptic fraction because it may detach from PS1 during the fractionation process. We therefore used pre-embedding immunogold electron microscopy to confirm that the γ-secretase complex may be both presynaptic and postsynaptic. We detected nicastrin silver-intensified label postsynaptically at the edge of the PSD (Fig. 1Ei), presynaptically (Fig. 1Eii), and on endomembranes (Fig. 1Eiii). We also detected the γ-secretase substrates N-cadherin and EphRB2 at the synapse by immunofluorescence (Fig. 1F, arrows) (Dalva et al., 2007; McCarthy et al., 2009), and Western blotting revealed their presence in both presynaptic and postsynaptic fractions (Fig. 1G). Thus, we conclude that the proteolytic machinery is present on both side of the synapse. These data raise the possibility that the metalloprotease and γ-secretase are tethered to the PSD through a direct interaction with synaptic scaffolding proteins.
γ-Secretase forms an active complex at synapses
We wondered whether the γ-secretase components, which are assembled in the secretory pathway (Kaether et al., 2006), reach the synapse as an active complex. PS1 coimmunoprecipitated with nicastrin from rat brain synaptosomes that were solubilized in CHAPSO to maintain γ-secretase integrity (Li et al., 2000), demonstrating that PS1 and nicastrin reach the vicinity of synapses as a physical complex (Fig. 2A). Furthermore, we detected γ-secretase enzymatic activity in solubilized synaptosomes by using a γ-secretase fluorogenic substrate (Farmery et al., 2003). This activity was partially but significantly inhibited by two γ-secretase inhibitors, DAPT and L-685458 (Fig. 2B: n = 6, 1.5-fold; p < 0.005), demonstrating the presence of enzymatically active γ-secretase at synapses. Total membrane (P2) was used as positive control (data not shown). Finally, biotinylation of surface proteins in live neurons, followed by synaptosomal fractionation, demonstrated both nicastrin and PS1NT at the synaptic plasma membrane (Fig. 2C). The GluR1 AMPAR subunit (a surface protein used as positive control) was also detected, whereas tubulin (an intracellular protein serving as negative control) was absent (Fig. 2C). These data suggest that γ-secretase components are present at the plasma membrane of synapses in an enzymatically active complex.
Figure 2.

γ-Secretase is active at synapses. A, Coimmunoprecipitation from synaptosomes, showing that nicastrin interacts with PS1. IP, Immunoprecipitation; α-nic, anti-nicastrin; α-PS1, anti-PS1NT; lys, lysate. B, In vitro γ-secretase assay. Incubation of synaptosomes with a fluorogenic γ-secretase substrate probe in the presence of two γ-secretase inhibitors, DAPT (10 μm) or L-685458 (5 μm). Measurement of fluorescence revealed γ-secretase activity, which was significantly blocked with γ-secretase inhibitors in optic density (DO) (**p < 0.005). The remaining activity is due to γ-secretase independent proteolysis. There were six independent replicates. C, Cortical neurons were surface biotinylated and fractionated. Aliquots of total lysate (total) from whole cell (WC), total membrane (P2) and synaptosome (syn) were kept and used as controls for the biotinylated fractions (surface). Nicastrin and PS1 are present at the surface of synaptosomes. The lower PS1 level presumably reflects the limited extent of the PS1 extracellular domain. GluA1 and tubulin were used as positive and negative controls respectively. D, δ-Catenin knock-out (KO) and wild-type (WT) mouse brains were fractionated into WB, P2, syn, and crude postsynaptic density, cPSD, and levels of full-length PS1 (**p < 0.005) (i), N-cadherin (**p < 0.005 and *p < 0.05, respectively) (ii), and GluA2/3 (**p < 0.005 and *p < 0.05, respectively) (iii) were measured by Western blotting. Graph shows normalization of KO to WT.
γ-Secretase is tethered by δ-catenin at synapses
We next analyzed the physical interactions that maintain these proteases at the synapse. PS1 binds to the adherens junction protein δ-catenin (Zhou et al., 1997; Levesque et al., 1999; Tanahashi and Tabira, 1999), which associates with synaptic N-cadherin (Kosik et al., 2005). We had previously shown that δ-catenin is a component of the PSD, where it is linked to the AMPA receptor subunit GluA2 by the scaffolding proteins GRIP/ABP (Silverman et al., 2007). To determine whether δ-catenin is required for tethering PS1 at synapses, we compared PS1 levels in brain subcellular fractions (Jordan et al., 2004) from δ-catenin knock-out and wild-type mice (Israely et al., 2004) (Fig. 2Di). PS1 was decreased in the crude postsynaptic density fraction (cPSD) in the knock-out mice but not in other fractions (cPSD: 0.5, p < 0.005; n = 7), suggesting that for PS1 to associate with the PSD it requires the PS1-δ-catenin interaction, while PS1 outside the PSD does not depend on δ-catenin. This suggests that the PS1-δ-catenin interaction selectively tethers the synaptic fraction of γ-secretase. Unlike PS1, levels of full-length N-cadherin were decreased in all fractions in δ-catenin knock-out mice, especially in synaptosomes and the cPSD, suggesting that δ-catenin is necessary for the stabilization of synaptic N-cadherin (Fig. 2Dii) [WB, 0.79; total membrane (P2), 0.61, p < 0.005; syn, 0.67, p < 0.0005; and cPSD, 0.64, p < 0.05; n = 7]. Interestingly, GluA2 was also decreased in the total membrane (P2), synaptosome, and cPSD fractions (P2, 0.58, p < 0.005; syn, 0.9; and cPSD, 0.72, p < 0.05; n = 7) (Fig. 2Diii), consistent with previous evidence that δ-catenin tethers GluA2-containing AMPA receptors at synapses (Silverman et al., 2007). Together with previous reports of synaptic tethering of metalloproteases by PSD proteins (Peiretti et al., 2003; Monea et al., 2006; Marcello et al., 2007), our data suggest that synaptic scaffolds stabilize the metalloprotease/γ-secretase proteolytic machinery at synapses, positioning the proteases close to their synaptic substrates.
Metalloproteases and γ-secretase cleave N-cadherin at synapses
As shown previously (Marambaud et al., 2003; Reiss et al., 2005; Uemura et al., 2006), N-cadherin ectodomain shedding by a metalloprotease generates a membrane-anchored C-terminal fragment, CTF1, which is further processed by γ-secretase to yield the soluble CTF2 fragment (Fig. 3A). To test whether this processing occurs at synapses, we prepared synaptosomes from cultured neurons treated with a broad metalloprotease inhibitor (ilomastat), γ-secretase inhibitors (L-685458 or DAPT), or with epoxomicin, a proteosome inhibitor. While metalloprotease inhibition decreased the CTF1 levels in synaptosomes, γ-secretase inhibition significantly increased the CTF1 levels, suggesting that CTF1 production by metalloprotease cleavage and its subsequent processing by γ-secretase take place at synapses (n = 8, 1.5-fold, p < 0.05) (Fig. 3B,C). The CTF2 fragment generated by γ-secretase cleavage of CTF1 was only detected in the supernatant of total lysate after treatment with the proteosome inhibitor, epoxomicin, suggesting that normally the cleaved fragment is rapidly degraded by the proteosome (Fig. 3B, bottom panel). Thus, we conclude that N-cadherin is cleaved by metalloproteases and γ-secretase at synapses. Subcellular fractionation showed the presence of the CTF1 fragment in both presynaptic and postsynaptic compartments, suggesting that N-cadherin cleavage occurs at both sides of the synapses (Fig. 3D).
Figure 3.
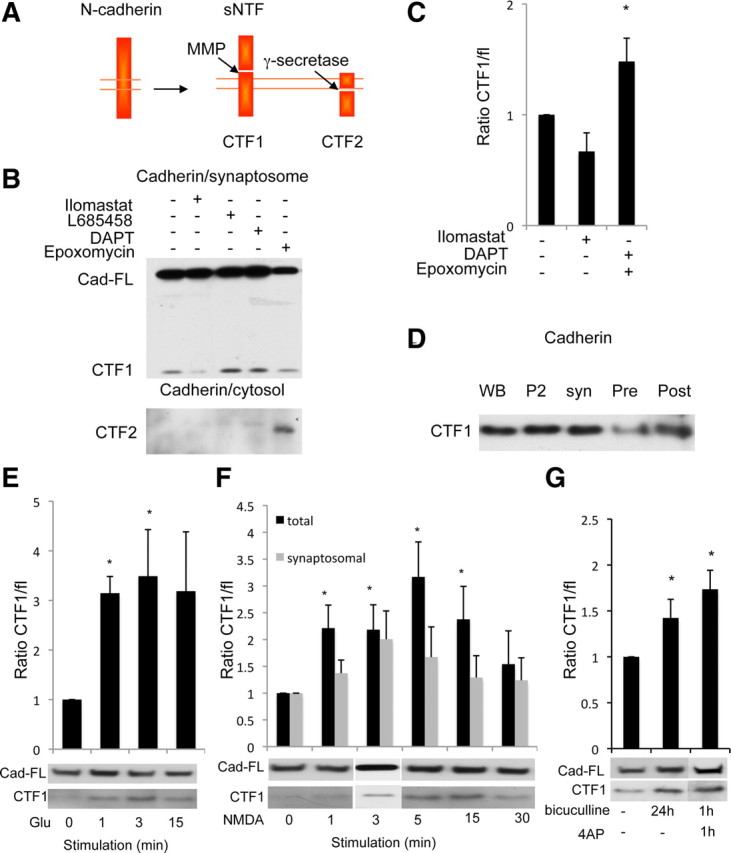
Metalloprotease/γ-secretase complex cleaves N-cadherin at synapses and is influenced by synaptic activity. A, Schematic drawing of N-cadherin cleavage. The cleavage by metalloproteases releases an extracellular (sNTF) and an intracellular (CTF1) fragment. γ-Secretase cleavage of CTF1 yields CTF2. B, Cortical neurons treated overnight with DMSO, 10 μm ilomastat (metalloproteases inhibitor), 1 μm L-685458 or 10 μm DAPT (γ-secretase inhibitors), or 1 μm epoxomicin (proteosome inhibitor) and fractionated. Western blotting detected full-length N-cadherin (Cad-FL) and CTF1 in synaptosomes and CTF2 in cytosol. C, Quantification of the ratio of CTF1/full-length N-cadherin in B, normalized to untreated condition, suggests synaptic γ-secretase cleavage. *p < 0.05. D, Subcellular fractionation of rat brain as described above. Western blotting detected N-cadherin CTF1 both presynaptically (Pre) and postsynaptically (Post). E–G, Neurons were treated with glutamate (E), NMDA (F), or bicuculline (G) for the durations indicated. The ratio of CTF1/full-length N-cadherin was quantified and normalized to the unstimulated condition, after Western blotting. E, N-cadherin CTF1 levels increase significantly with brief glutamate (Glu) (50 μm) stimulation in total lysate. *p < 0.05. Graph shows an average of four experiments. Western blots represent a typical experiment. F, N-cadherin CTF1 levels were increased in total and synaptosomal fractions after a brief NMDA (40 μm) stimulation. *p < 0.05. Graph shows an average of 12 experiments. Western blots represent a typical experiment, with 3 min time point from a separate experiment. G, A significant increase in N-cadherin CTF1 levels was detected after 1 h bicuculline (40 μm) plus 4AP (100 μm), as well as 24 h bicuculline-alone treatment. *p < 0.05. Graph shows an average of 15 experiments. Western blots represent a typical experiment.
N-cadherin cleavage is influenced by activity
Discrepant findings have been reported regarding the effect of synaptic activity on cadherins. Synaptic activity can either stabilize or cleave cadherins (Tanaka et al., 2000; Marambaud et al., 2003; Reiss et al., 2005; Uemura et al., 2006; Tai et al., 2007). To analyze the effects of activity on the metalloprotease and γ-secretase pathway, we first assayed the effects of glutamate and NMDA stimulation on N-cadherin cleavage (Lee et al., 1998; Tai et al., 2007). We detected an increase in N-cadherin CTF1 levels in total lysate following brief glutamate stimulation (1 min, n = 4, 3.2-fold; 3 min, n = 7, 3.8-fold; p < 0.05) (Fig. 3E). Similarly, stimulating cultured neurons with NMDA produced a significant increase in N-cadherin CTF1 level in total lysate (1 min, n = 12, 2.2-fold; 3 min, n = 12, 2.2-fold; 5 min, n = 11, 3.2; 15 min, n = 11, 2.5-fold; p < 0.05) (Fig. 3F). A similar increase was also observed in synaptosomes, although the effect was not significant (1 min, n = 11, 1.4-fold; 3 min, n = 11, 2-fold; and 5 min, n = 8, 1.7-fold) (Fig. 3F). Similar kinetics were observed when the ratio of CTF1/tubulin was evaluated (data not shown).
Addition of glutamate or NMDA to the culture medium stimulates both synaptic and extrasynaptic receptors, which might result in nonspecific effects. To determine the effects of selective synaptic stimulation, we enhanced synaptic transmission by simultaneous application of bicuculline, a GABAA receptor (GABAAR) antagonist, and 4-aminopyridine (4AP), a K+ channel inhibitor, and observed an increase in N-cadherin CTF1 production (n = 15, 1.7-fold and 1.4-fold, p < 0.05) (Fig. 3G) (Ehlers, 2000; Tai et al., 2007). This effect was blocked by inhibition of NMDARs (d-APV, n = 5, p < 0.05) (Fig. 4A). We conclude that synaptic activation stimulates CTF1 production in an NMDA receptor-dependent manner. Likewise, N-cadherin CTF1 production was decreased when action potentials were blocked with the sodium channel blocker tetrodotoxin (TTX, n = 4, 0.5-fold; p < 0.05) (Fig. 4A). Thus, activation of synaptic NMDARs stimulates the cleavage of N-cadherin by proteolytic machinery composed of a metalloprotease and γ-secretase.
Figure 4.

Synaptic activity enhances metalloprotease cleavage, but not γ-secretase cleavage, via NMDA receptors, Src, and JNK. A, Cortical neurons were treated with bicuculline and 4AP for 1 h and then NMDARs were blocked with d-APV. Neuronal activity was also blocked with TTX (*p < 0.05 and **p < 0.005, respectively). B, Cortical neurons were stimulated with glutamate (50 μm, 3 min) in the presence of KN93, PP2, and JNK inhibitor II (CaMKII, Src, and JNK kinases inhibitors, respectively). N-cadherin CTF1 level was significantly decreased by APV, PP2, and JNK inhibitor II but not by KN93 (*p < 0.05 and **p < 0.005, respectively). C, Neurons were stimulated with glutamate (50 μm, 3 min) and treated with a metalloprotease inhibitor (ilomastat, 5 h) as indicated. N-cadherin CTF1 levels in the total lysate fraction were analyzed as before. Addition of ilomastat decreased CTF1 level compared to control condition (c), (*p < 0.05), but no further decrease was observed when neurons were first treated with ilomastat and then stimulated by glutamate (d). D, Effect of γ-secretase inhibition on bicuculline-treated and untreated neurons (40 μm, 24 h). γ-secretase inhibition (L-685458, 1 μm, 24 h) did not affect CTF1 levels in either bicuculline-treated or untreated neurons (*p < 0.05).
Metalloprotease cleavage regulation depends on Src and c-Jun N-terminal kinase
To investigate the pathway by which NMDAR activation regulates N-cadherin cleavage, we stimulated cultured neurons with glutamate, with or without inhibitors of NMDARs or of specific kinases (Fig. 4B). Total lysate fractions were probed in Western blots for the N-cadherin CTF1 fragment. As shown above (Fig. 3E), CTF1 fragment production increased after glutamate stimulation. The glutamate-induced increase in CTF1 fragment production was blocked by an NMDAR inhibitor (d-APV, n = 4, 0.9-fold; p < 0.05) (Fig. 4B). The addition of a Src or JNK inhibitor (PP2 or JNK inhibitor II, respectively) also blocked the glutamate-induced increase in CTF1 fragment (n = 6, respectively, 0.6-fold, p < 0.005; 0.75-fold, p < 0.05) (Fig. 4B). In contrast, KN93, an inhibitor of CaMKII, had no effect. These data suggest that glutamate stimulation of N-cadherin ectodomain shedding by a metalloprotease depends on the activation of the NMDA receptor and Src and JNK kinases.
γ-secretase cleavage is not influenced by activity
We next examined whether glutamate stimulation differentially regulates the two N-cadherin cleavage steps. As shown above (Fig. 3F) NMDAR activation stimulates CTF1 production. A direct assay of CTF2 production that would reflect γ-secretase cleavage of CTF1 was unfeasible, because CTF2 was detected at very low levels only in the supernatants of total neuron lysates (Fig. 3B). As an alternative, we treated neurons with the metalloprotease inhibitor ilomastat to inhibit production of CTF1. In agreement with Figure 3E, glutamate stimulation in the absence of any protease inhibitor increased the level of the CTF1 fragment (Fig. 4Cb: n = 16, 4-fold, p < 0.05). This increase was inhibited by ilomastat (Fig. 4, compare Cb, Cd, p < 0.05), suggesting that the metalloprotease is activated by glutamate stimulation. We reasoned that since ilomastat blocks the production of new CTF1, a decrease in CTF1 levels caused by glutamate stimulation in the presence of ilomastat would imply an activation of the γ-secretase cleavage step. We found that addition of ilomastat decreased the level of CTF1 compared to the control condition, which is as expected if γ-secretase cleaves CTF1 fragments that existed before glutamate stimulation (Fig. 4Cc: n = 6, 0.56-fold). However we did not observe a further decrease in CTF1 levels when neurons were first treated with ilomastat (to block CTF1 production) and then stimulated with glutamate (Fig. 4, compare Cd, Cc), suggesting that glutamate stimulation does not increase γ-secretase cleavage of CTF1. We conclude that glutamate stimulation regulates the metalloprotease cleavage of N-cadherin, but not the cleavage by γ-secretase.
To confirm this result, we assessed γ-secretase regulation by synaptic activity. By comparing the effects of a γ-secretase inhibitor (L-685458) on bicuculline-treated and untreated neurons, we could determine whether the bicuculline-dependent increase in synaptic transmission influences the rate of γ-secretase cleavage of CTF1. We analyzed two pairs of dishes of cultured neurons: one pair of unstimulated control dishes and a pair of bicuculline-treated dishes. In one dish of each pair, we inhibited γ-secretase for the duration of the bicuculline exposure. The N-cadherin CTF1 was quantified in the total lysates of the four dishes by Western blots. In both the control and the L-685458-treated cultures, CTF1 fragment production increased with bicuculline treatment, as expected (Fig. 4Dc, 4.96-fold, p < 0.05; Fig. 4Dd, 4.84-fold; n = 15). Inhibition of γ-secretase increased CTF1 fragment levels in both untreated and bicuculline-treated cultures (Fig. 4Db, control: n = 15, 1.4-fold; Fig. 4Dd, stimulated: n = 9, 4.84-fold). However, when we compared the effect of γ-secretase inhibition on bicuculline-treated and untreated control neurons, no significant difference was observed (Fig. 4D: ratio b/a = 4.96 compared to d/c = 4.03, p = 0.53), confirming that synaptic activation did not regulate γ-secretase cleavage of CTF1.
We conclude that synaptic activation following bicuculline treatment or glutamate stimulation has little or no influence on the rate of γ-secretase cleavage of CTF1, suggesting that the increased rate of cleavage of N-cadherin induced by neuronal activity results specifically from activity-dependent stimulation of ectodomain shedding by a metalloprotease.
Inhibition of metalloprotease or γ-secretase modifies mEPSCs
Cleavage of synaptic substrates by metalloproteases and γ-secretase might alter synaptic architecture or modify components of the transmission machinery; either effect could lead to functional changes. To determine whether metalloproteases or γ-secretase have an effect on synaptic function, we recorded mEPSCs from cultured hippocampal neurons treated overnight with the γ-secretase inhibitor, L-685458 (1 μm), or the metalloprotease inhibitor, ilomastat (10 μm) (Fig. 5). Changes in mEPSC frequency and amplitude are usually attributed to changes in presynaptic and postsynaptic functions, respectively (Wierenga et al., 2006). While we observed significant increases in both the frequency and the amplitude of mEPSCs in L-685458-treated neurons compared to vehicle-treated neurons (p < 0.05), ilomastat-treated neurons showed a selective enhancement of mEPSC frequency (p < 0.05) with no effect on mEPSC amplitude (p > 0.05) [frequency, 2.3 ± 0.2 (vehicle), 6.2 ± 0.8 (ilomastat), 3.5 ± 0.4 (L-685458); amplitude: 13.9 ± 0.4 (vehicle), 14 ± 0.3 (ilomastat), 18.5 ± 0.6 (L685458); N = 17 (vehicle), 15 (ilomastat), and 15 (L-685458)] (Fig. 5). These results suggest that the activities of both γ-secretase and metalloproteases negatively regulate glutamatergic transmission in cultured hippocampal neurons; furthermore, γ-secretase activity appears to affect both presynaptic and postsynaptic functions, whereas metalloprotease activity selectively reduces presynaptic glutamate release. The decrease in mEPSC frequency could possibly also reflect a postsynaptic effect, for example one resulting from a reduction of spine or receptor numbers.
Figure 5.

Effect of metalloprotease or γ-secretase inhibition on synaptic transmission. Inhibition of metalloproteases or γ-secretase enhanced glutamatergic neurotransmission in cultured hippocampal neurons. A, Examples of mEPSC recordings in vehicle (Control)-, ilomastat-, and L-685458-treated neurons. B, C, Cumulative probability distribution of mEPSC inter-event interval (B) and amplitude (C) in vehicle-, ilomastat (Ilo)-, and L-685458-treated neurons. D, E, Average mEPSC frequency (D) and amplitude (E) in vehicle-, ilomastat-, and L-685458-treated neurons. While ilomastat selectively increased mEPSC frequency, L-685458 increased both the frequency and amplitude of mEPSCs (*p < 0.05).
Inhibition of metalloprotease or γ-secretase modifies synaptic proteins
To explore the mechanistic basis for these electrophysiological changes, we assayed whether inhibition of metalloproteases or γ-secretase affects synaptic protein levels or functions. Inhibition of metalloprotease (ilomastat) or γ-secretase (L685458) activity did not change the level of the presynaptic marker synaptophysin as assayed by immunofluorescence (total intensity: 1.08 and 0.99, respectively; n = 9) (Fig. 6Ai,Aii). However, an increase in the rate of synaptic vesicle cycling, as reflected in the level of synaptotagmin reuptake measured by an antibody-feeding assay (Matteoli et al., 1992), could be detected after metalloprotease but not γ-secretase inhibition (total intensity: 1.2 and 0.97, respectively; n = 5) (Fig. 6Bi,Bii). This effect is consistent with the increase in mEPSC frequency recorded after metalloprotease inhibition (Fig. 5), suggesting that a metalloprotease decreases the rate of synaptic vesicle recycling.
Figure 6.
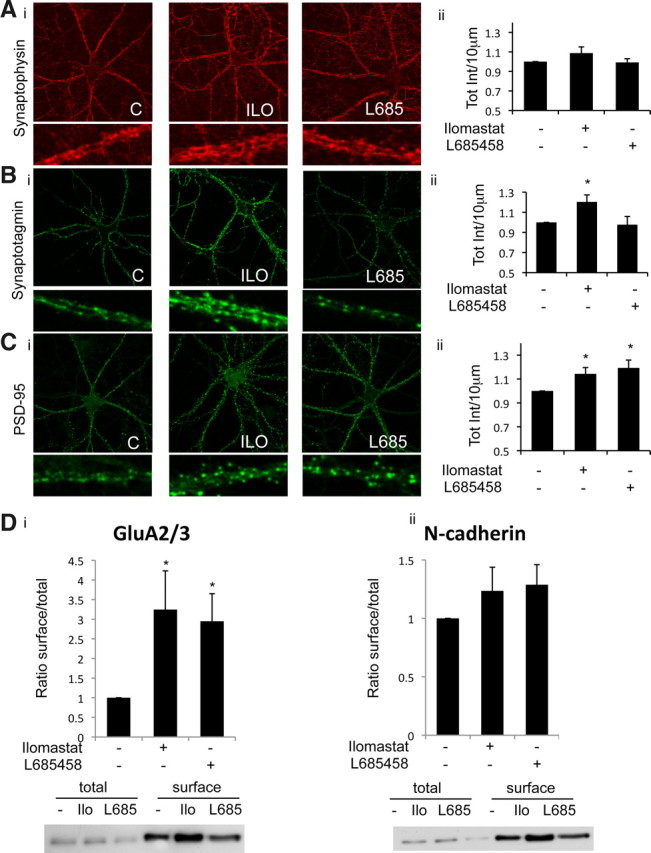
Effect of metalloproteases or γ-secretase inhibition on synaptic proteins. Ai, 14 DIV hippocampal neurons in culture stained with synaptophysin after inhibition of metalloprotease (ilomastat) or γ-secretase (L685458). Aii, Total intensity of fluorescence per 10 μm was measured and normalized to control. Average fluorescence intensity is shown for control (C), ilomastat (ILO), and L685458 (L685) groups. Bi, Synaptotagmin reuptake in 14 DIV hippocampal neurons exposed to ILO and L685 overnight. Bii, Total intensity of fluorescence per 10 μm was measured and normalized to control. Average fluorescence intensity is shown for control, ilomastat, and L685458 groups (*p < 0.05). Ci, Same experiment as in A stained for PSD-95. Cii, Total intensity of fluorescence per 10 μm was measured and normalized to control. Average fluorescence intensity is shown for control, ilomastat, and L685458 groups (*p < 0.05). D, 14 DIV cortical neurons in culture were biotinylated and total cell fraction was prepared. The ilomastat and L685458 conditions were normalized to untreated condition for each experiment. Western blotting of GluA2/3 (i) and N-cadherin (ii) shows a significant increase of surface GluA2 after metalloprotease (Ilo) or γ-secretase (L685) inhibition, but no effect on surface N-cadherin (*p < 0.05).
We next assayed changes in postsynaptic proteins after inhibition of the proteolytic machinery. Inhibition of metalloproteases or γ-secretase increased the level of PSD-95 as measured by immunofluorescence (total intensity: 1.14 and 1.19, respectively; p < 0.05, n = 9) (Fig. 6Ci,Cii) in agreement with previous reports (Parent et al., 2005). Moreover, a significant increase of surface GluA2, but not N-cadherin, was detected by a live neuron biotinylation assay after inhibition of metalloproteases or γ-secretase (GluA2: 3.24 and 2.94, respectively, p < 0.05; N-cadherin: 1.25 and 1.28, respectively) (Fig. 6Di,Dii). We conclude that the activity of the proteolytic machinery decreases levels of a postsynaptic scaffold and an AMPA receptor subunit, consistent with a role for these enzymes in attenuating synaptic transmission.
Discussion
Proteolytic enzymes can regulate synaptic efficacy (Cartier et al., 2009; Mabb and Ehlers, 2010; Malinverno et al., 2010), but this regulatory mechanism is still largely uncharacterized. Here we have identified synaptic functions of metalloproteases and γ-secretase, two proteases that act in succession to cleave single-pass transmembrane proteins. Our work indicates that the proteolytic machinery might be part of multiprotein complexes containing the proteases and their substrates and tethered to the PSD by synaptic scaffold proteins. Synaptic activity stimulates proteolysis through enhancement of the metalloprotease cleavage, leading to protease-dependent decreases in synaptic efficacy through both presynaptic and postsynaptic mechanisms, providing a novel form of synaptic autoregulation.
The cleavage machinery and its substrates
We find that the proteolytic machinery processes N-cadherin on both sides of the synapse, consistent with the homophilic nature of the N-cadherin interaction and with the observed distribution of the metalloproteases ADAM10, ADAM17, and MT5-MMP and the presence of the γ-secretase in both presynaptic and postsynaptic compartments. The postsynaptic localization of the metalloproteases may be a consequence of the tethering of ADAM10, ADAM17, and MT5-MMP by the postsynaptic scaffolds SAP97 (Peiretti et al., 2003; Marcello et al., 2007) and ABP/GRIP (Monea et al., 2006). Likewise, the association of the γ-secretase catalytic subunit PS1 and of the ADAMs with the presynaptic scaffold X11 and with the postsynaptic protein δ-catenin (Leonoudakis et al., 2004; Kosik et al., 2005; Miller et al., 2006; McCarthy et al., 2009) may enable γ-secretase to function at both sides of the synapse.
Several metalloproteases can shed γ-secretase substrates (Reiss et al., 2005; Monea et al., 2006; Uemura et al., 2006; Postina, 2008). A number of them are tethered to synaptic scaffold proteins and could thus cleave synaptic substrates (Peiretti et al., 2003; Monea et al., 2006; Marcello et al., 2007). Indeed, in this study we identified three metalloproteases, MT5-MMP, ADAM 10, and ADAM 17, at synapses. We have then focused our study on understanding the general mechanism of metalloprotease cleavage regulation. Each metalloprotease might have a substrate or location preference, but little is yet known regarding possible differences (Postina, 2008).
The role of δ-catenin, which anchors PS1 (Zhou et al., 1997; Levesque et al., 1999; Tanahashi and Tabira, 1999), is of particular interest. δ-Catenin can also bind to N-cadherin (Kosik et al., 2005), PSD-95, and ABP/GRIP (Silverman et al., 2007). The ABP/GRIP scaffolds, in turn, can bind to the metalloprotease MT5-MMP, which cleaves N-cadherin to release a CTF that is a substrate of γ-secretase (Monea et al., 2006). The ABP/GRIP scaffolds also bind to Eph receptors, ephrins, and LAR (Brückner et al., 1999; Dunah et al., 2005; McCarthy et al., 2009), all potential synaptic substrates of the proteolytic pathway (Georgakopoulos et al., 2006; Haapasalo et al., 2007). This suggests the existence of large, supramolecular synaptic complexes containing metalloproteases and γ-secretase and lying close to cadherins and other synaptic substrates of these proteases. The synaptic tethering of AMPAR complexes by ABP/GRIP and PSD-95 (Silverman et al., 2007) raises the possibility that disruption of these complexes could release AMPARs from the synapse. δ-Catenin may serve as a central organizer of these supramolecular complexes, as has been described in non-neuronal cells (Kouchi et al., 2009). In support of this model, we observed that synaptic levels of PS1, N-cadherin, and GluA2 are markedly decreased in δ-catenin knock-out mice.
Synaptic protease cleavage as an activity-dependent regulatory mechanism
Application of glutamate or NMDA increased the cleavage of N-cadherin by metalloproteases, as did enhancement of synaptic activity by bicuculline. This action on N-cadherin was NMDAR dependent and enhanced the metalloprotease cleavage, but not the γ-secretase cleavage. Different members of the metalloprotease family may be localized or regulated differently with respect to particular substrates. We suggest that the presence of the NMDA receptor adjacent to the postsynaptic δ-catenin-cadherin complexes may account for rapid N-cadherin cleavage. Previous work showing that metalloproteases can be activated by kinases via G protein-coupled receptors (Postina, 2008) and by Src phosphorylation (Zhang et al., 2006) is consistent with the present observation that NMDAR stimulation of the metalloprotease cleavage of N-cadherin depended on Src and JNK kinases.
The metalloprotease/γ-secretase proteolytic machinery regulates synaptic function
Our work demonstrates that both metalloprotease and γ-secretase activities reduced the efficacy of synaptic transmission. Consistent with an action on both sides of the synapse, inhibition of protease activity increased both mEPSC frequency and mEPSC amplitude. Furthermore, protease activity reduced the rate of synaptic vesicle reuptake (consistent with a decrease in mEPSC frequency) while also reducing levels of PSD-95 and surface expression of the AMPA receptor subunit GluA2 (consistent with a decrease in mEPSC amplitude). This latter effect suggests that postsynaptic cleavage can disrupt a supramolecular complex that tethers AMPA receptors at synapses, as implied in previous studies of PS1 knock-out mice (Parent et al., 2005; Pratt et al., 2011).
The level of full-length N-cadherin showed little change with synaptic activity, and only a small fraction of full-length N-cadherin underwent metalloprotease/γ-secretase cleavage. Although the subpopulation of N-cadherin that is degraded remains to be identified, it is likely to include the synaptic pool, since substantial levels of N-cadherin CTF1 were found at synapses. Cleavage of the CTF1 pool by γ-secretase might play a structural role, but of special interest is its possible role in synaptic regulation. Besides releasing δ-catenin and associated components, cleavage of CTF1 releases an intracellular signaling peptide, like other peptides released by γ-secretase from adhesion molecules and related proteins (Marambaud et al., 2003; Bao et al., 2004; Hass et al., 2009; McCarthy et al., 2009). Indeed, cleavage of N-cadherin CTF1 by γ-secretase releases the CTF2 fragment, which represses CBP/CREB-dependent transcription (Marambaud et al., 2003). This dual effect might explain discrepancies between previous reports suggesting that synaptic activity can either stabilize or degrade cadherins at synapses (Tanaka et al., 2000; Marambaud et al., 2003).
Our work demonstrates a synaptic autoregulatory loop involving proteases. The finding that metalloproteases are induced by synaptic activity and that the cleavage machinery in turn negatively regulates synaptic function suggests a negative autoregulatory loop at the synapse reminiscent of homeostatic plasticity (Pozo and Goda, 2010). This specialized proteolytic machinery may provide novel pathways for modification of the number, shape, or function of synapses, modifications that may contribute to the cellular basis of memory (Segal, 2005). Indeed, changes in synaptic plasticity and spine morphology have been observed in PS1 knock-out and familial Alzheimer's disease mutant mice (Saura et al., 2004; Auffret et al., 2009). Pathological disruption of synapses through deregulation of the metalloprotease and γ-secretase cleavage pathways could play a role in early synaptic deficits which subsequently lead to cognitive dysfunction and neurodegenerative changes in Alzheimer's disease (Knobloch and Mansuy, 2008).
Footnotes
SR was supported by a Levine Fellowship and the work was supported by Alzheimer Association Grants NIRG-06-25401 (to S.R.) and NIRG-08-90001 (to I.N.) and National Institutes of Health Grants R01 AG13620 (to E.B.Z.), R01 NS35527 (to R.J.W.), and P01 AG017617 (to P.M.M.). We thank Sebastien Thuault, Nicholas J Cowan, Randy Nixon, and Gordon Fishell for critical readings of the manuscript and discussions, Pierre Trifilieff for help with statistical analysis, and Yan Deng for help with image analysis. We thank Bryen Jordan, Vivien Chevaleyre, and Pierre Trifilieff for discussion, and Kristen Phend for the electron microscopy experiments.
References
- Auffret A, Gautheron V, Repici M, Kraftsik R, Mount HT, Mariani J, Rovira C. Age-dependent impairment of spine morphology and synaptic plasticity in hippocampal CA1 neurons of a presenilin 1 transgenic mouse model of Alzheimer's disease. J Neurosci. 2009;29:10144–10152. doi: 10.1523/JNEUROSCI.1856-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao J, Lin H, Ouyang Y, Lei D, Osman A, Kim TW, Mei L, Dai P, Ohlemiller KK, Ambron RT. Activity-dependent transcription regulation of PSD-95 by neuregulin-1 and Eos. Nat Neurosci. 2004;7:1250–1258. doi: 10.1038/nn1342. [DOI] [PubMed] [Google Scholar]
- Brückner K, Pablo Labrador J, Scheiffele P, Herb A, Seeburg PH, Klein R. EphrinB ligands recruit GRIP family PDZ adaptor proteins into raft membrane microdomains. Neuron. 1999;22:511–524. doi: 10.1016/s0896-6273(00)80706-0. [DOI] [PubMed] [Google Scholar]
- Cartier AE, Djakovic SN, Salehi A, Wilson SM, Masliah E, Patrick GN. Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J Neurosci. 2009;29:7857–7868. doi: 10.1523/JNEUROSCI.1817-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signalling functions at the synapse. Nat Rev Neurosci. 2007;8:206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Hueske E, Wyszynski M, Hoogenraad CC, Jaworski J, Pak DT, Simonetta A, Liu G, Sheng M. LAR receptor protein tyrosine phosphatases in the development and maintenance of excitatory synapses. Nat Neurosci. 2005;8:458–467. doi: 10.1038/nn1416. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Farmery MR, Tjernberg LO, Pursglove SE, Bergman A, Winblad B, Näslund J. Partial purification and characterization of gamma-secretase from post-mortem human brain. J Biol Chem. 2003;278:24277–24284. doi: 10.1074/jbc.M211992200. [DOI] [PubMed] [Google Scholar]
- Georgakopoulos A, Marambaud P, Friedrich VL, Jr, Shioi J, Efthimiopoulos S, Robakis NK. Presenilin-1: a component of synaptic and endothelial adherens junctions. Ann NY Acad Sci. 2000;920:209–214. doi: 10.1111/j.1749-6632.2000.tb06924.x. [DOI] [PubMed] [Google Scholar]
- Georgakopoulos A, Litterst C, Ghersi E, Baki L, Xu C, Serban G, Robakis NK. Metalloproteinase/Presenilin1 processing of ephrinB regulates EphB-induced Src phosphorylation and signaling. EMBO J. 2006;25:1242–1252. doi: 10.1038/sj.emboj.7601031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapasalo A, Kim DY, Carey BW, Turunen MK, Pettingell WH, Kovacs DM. Presenilin/gamma-secretase-mediated cleavage regulates association of leukocyte-common antigen-related (LAR) receptor tyrosine phosphatase with beta-catenin. J Biol Chem. 2007;282:9063–9072. doi: 10.1074/jbc.M611324200. [DOI] [PubMed] [Google Scholar]
- Hass MR, Sato C, Kopan R, Zhao G. Presenilin: RIP and beyond. Semin Cell Dev Biol. 2009;20:201–210. doi: 10.1016/j.semcdb.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israely I, Costa RM, Xie CW, Silva AJ, Kosik KS, Liu X. Deletion of the neuron-specific protein delta-catenin leads to severe cognitive and synaptic dysfunction. Curr Biol. 2004;14:1657–1663. doi: 10.1016/j.cub.2004.08.065. [DOI] [PubMed] [Google Scholar]
- Jordan BA, Fernholz BD, Boussac M, Xu C, Grigorean G, Ziff EB, Neubert TA. Identification and verification of novel rodent postsynaptic density proteins. Mol Cell Proteomics. 2004;3:857–871. doi: 10.1074/mcp.M400045-MCP200. [DOI] [PubMed] [Google Scholar]
- Kaether C, Haass C, Steiner H. Assembly, trafficking and function of gamma-secretase. Neurodegener Dis. 2006;3:275–283. doi: 10.1159/000095267. [DOI] [PubMed] [Google Scholar]
- Knobloch M, Mansuy IM. Dendritic spine loss and synaptic alterations in Alzheimer's disease. Mol Neurobiol. 2008;37:73–82. doi: 10.1007/s12035-008-8018-z. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Donahue CP, Israely I, Liu X, Ochiishi T. Delta-catenin at the synaptic-adherens junction. Trends Cell Biol. 2005;15:172–178. doi: 10.1016/j.tcb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Kouchi Z, Barthet G, Serban G, Georgakopoulos A, Shioi J, Robakis NK. p120 catenin recruits cadherins to γ-secretase and inhibits production of Aβ peptide. J Biol Chem. 2009;284:1954–1961. doi: 10.1074/jbc.M806250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Levey AI. Light and electron microscopic localization of presenilin-1 in primate brain. J Neurosci. 1997;17:1971–1980. doi: 10.1523/JNEUROSCI.17-06-01971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Leonoudakis D, Conti LR, Radeke CM, McGuire LM, Vandenberg CA. A multiprotein trafficking complex composed of SAP97, CASK, Veli, and Mint1 is associated with inward rectifier Kir2 potassium channels. J Biol Chem. 2004;279:19051–19063. doi: 10.1074/jbc.M400284200. [DOI] [PubMed] [Google Scholar]
- Levesque G, Yu G, Nishimura M, Zhang DM, Levesque L, Yu H, Xu D, Liang Y, Rogaeva E, Ikeda M, Duthie M, Murgolo N, Wang L, VanderVere P, Bayne ML, Strader CD, Rommens JM, Fraser PE, St George-Hyslop P. Presenilins interact with armadillo proteins including neural-specific plakophilin-related protein and beta-catenin. J Neurochem. 1999;72:999–1008. doi: 10.1046/j.1471-4159.1999.0720999.x. [DOI] [PubMed] [Google Scholar]
- Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci U S A. 2000;97:6138–6143. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Ehlers MD. Ubiquitination in postsynaptic function and plasticity. Annu Rev Cell Dev Biol. 2010;26:179–210. doi: 10.1146/annurev-cellbio-100109-104129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinverno M, Carta M, Epis R, Marcello E, Verpelli C, Cattabeni F, Sala C, Mulle C, Di Luca M, Gardoni F. Synaptic localization and activity of ADAM10 regulate excitatory synapses through N-cadherin cleavage. J Neurosci. 2010;30:16343–16355. doi: 10.1523/JNEUROSCI.1984-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–645. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Marcello E, Gardoni F, Mauceri D, Romorini S, Jeromin A, Epis R, Borroni B, Cattabeni F, Sala C, Padovani A, Di Luca M. Synapse-associated protein-97 mediates alpha-secretase ADAM10 trafficking and promotes its activity. J Neurosci. 2007;27:1682–1691. doi: 10.1523/JNEUROSCI.3439-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteoli M, Takei K, Perin MS, Südhof TC, De Camilli P. Exo-endocytotic recycling of synaptic vesicles in developing processes of cultured hippocampal neurons. J Cell Biol. 1992;117:849–861. doi: 10.1083/jcb.117.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JV, Twomey C, Wujek P. Presenilin-dependent regulated intramembrane proteolysis and gamma-secretase activity. Cell Mol Life Sci. 2009;66:1534–1555. doi: 10.1007/s00018-009-8435-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CC, McLoughlin DM, Lau KF, Tennant ME, Rogelj B. The X11 proteins, Abeta production and Alzheimer's disease. Trends Neurosci. 2006;29:280–285. doi: 10.1016/j.tins.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Monea S, Jordan BA, Srivastava S, DeSouza S, Ziff EB. Membrane localization of membrane type 5 matrix metalloproteinase by AMPA receptor binding protein and cleavage of cadherins. J Neurosci. 2006;26:2300–2312. doi: 10.1523/JNEUROSCI.3521-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB. The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and alpha- and beta-SNAPs. Neuron. 1998;21:99–110. doi: 10.1016/s0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- Parent AT, Barnes NY, Taniguchi Y, Thinakaran G, Sisodia SS. Presenilin attenuates receptor-mediated signaling and synaptic function. J Neurosci. 2005;25:1540–1549. doi: 10.1523/JNEUROSCI.3850-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiretti F, Deprez-Beauclair P, Bonardo B, Aubert H, Juhan-Vague I, Nalbone G. Identification of SAP97 as an intracellular binding partner of TACE. J Cell Sci. 2003;116:1949–1957. doi: 10.1242/jcs.00415. [DOI] [PubMed] [Google Scholar]
- Phillips GR, Huang JK, Wang Y, Tanaka H, Shapiro L, Zhang W, Shan WS, Arndt K, Frank M, Gordon RE, Gawinowicz MA, Zhao Y, Colman DR. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron. 2001;32:63–77. doi: 10.1016/s0896-6273(01)00450-0. [DOI] [PubMed] [Google Scholar]
- Postina R. A closer look at alpha-secretase. Current Alzheimer research. 2008;5:179–186. doi: 10.2174/156720508783954668. [DOI] [PubMed] [Google Scholar]
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–351. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, Zhu P, Watari H, Cook DG, Sullivan JM. A novel role for {gamma}-secretase: selective regulation of spontaneous neurotransmitter release from hippocampal neurons. J Neurosci. 2011;31:899–906. doi: 10.1523/JNEUROSCI.4625-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss K, Maretzky T, Ludwig A, Tousseyn T, de Strooper B, Hartmann D, Saftig P. ADAM10 cleavage of N-cadherin and regulation of cell–celladhesion and beta-catenin nuclear signalling. EMBO J. 2005;24:742–752. doi: 10.1038/sj.emboj.7600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Silverman JB, Restituito S, Lu W, Lee-Edwards L, Khatri L, Ziff EB. Synaptic anchorage of AMPA receptors by cadherins through neural plakophilin-related arm protein AMPA receptor-binding protein complexes. J Neurosci. 2007;27:8505–8516. doi: 10.1523/JNEUROSCI.1395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai CY, Mysore SP, Chiu C, Schuman EM. Activity-regulated N-cadherin endocytosis. Neuron. 2007;54:771–785. doi: 10.1016/j.neuron.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Tanahashi H, Tabira T. Isolation of human delta-catenin and its binding specificity with presenilin 1. Neuroreport. 1999;10:563–568. doi: 10.1097/00001756-199902250-00022. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Shan W, Phillips GR, Arndt K, Bozdagi O, Shapiro L, Huntley GW, Benson DL, Colman DR. Molecular modification of N-cadherin in response to synaptic activity. Neuron. 2000;25:93–107. doi: 10.1016/s0896-6273(00)80874-0. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura K, Kihara T, Kuzuya A, Okawa K, Nishimoto T, Ninomiya H, Sugimoto H, Kinoshita A, Shimohama S. Characterization of sequential N-cadherin cleavage by ADAM10 and PS1. Neurosci Lett. 2006;402:278–283. doi: 10.1016/j.neulet.2006.04.018. [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Walsh MF, Turrigiano GG. Temporal regulation of the expression locus of homeostatic plasticity. J Neurophysiol. 2006;96:2127–2133. doi: 10.1152/jn.00107.2006. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Thomas SM, Lui VW, Xi S, Siegfried JM, Fan H, Smithgall TE, Mills GB, Grandis JR. Phosphorylation of TNF-alpha converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc Natl Acad Sci U S A. 2006;103:6901–6906. doi: 10.1073/pnas.0509719103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Liyanage U, Medina M, Ho C, Simmons AD, Lovett M, Kosik KS. Presenilin 1 interaction in the brain with a novel member of the Armadillo family. Neuroreport. 1997;8:2085–2090. doi: 10.1097/00001756-199705260-00054. [DOI] [PubMed] [Google Scholar]