

Abstract
Most sudden cardiac deaths in young athletes are caused by previously undetected inherited cardiac diseases. Here, we report a case of a young male athlete in whom a presumptive diagnosis of hypertrophic cardiomyopathy (HCM) was made following a near sudden cardiac death. Although his imaging studies initially suggested HCM, a detailed clinical and genetic evaluation of the patient and his asymptomatic father led to the diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVD) in both. DNA sequencing revealed that each individual was heterozygous for two rare variants in the PKP2 and DSC2 genes, both of which were previously shown to be associated with ARVD and to encode desmosomal proteins, i.e. the previously reported splicing variant c2489 + 1A>G in the PKP2 gene and the novel p.I109M variant in the DSC2 gene. Imaging and electrophysiologic studies further supported a diagnosis of ARVD in the father. This case highlights the importance of detailed clinical evaluation and genetic testing of family members when dealing with sudden cardiac death or unexplained cardiomyopathies in the young.
Key Words: Arrhythmogenic right ventricular dysplasia, Plakophilin-2, Desmocollin-2, Desmosome
Established Facts
-
•
Arrhythmogenic right ventricular dysplasia (ARVD), a common cause of sudden cardiac death in young people, is typically diagnosed based on right ventricular dilation/hypokinesia, characteristic ECG abnormalities, right ventricular fibrofatty infiltrate and characteristic arrhythmia.
-
•
Inherited cardiomyopathies can have variable presentations and can share common characteristics with other inherited cardiomyopathies.
Novel Insights
-
•
We report two cases of ARVD in a father and son with a novel p.I109M variant of DSC2 and a previously reported c2489+1A>G mutation of PKP2, two previously reported ARVD-associated genes with unknown pathogenic mechanisms.
-
•
We highlight the critical role of familial, clinical and genetic evaluation in establishing a diagnosis of ARVD because the heterogeneity of ARVD presentations makes common clinical criteria insufficient.
Introduction
Arrhythmogenic right ventricular dysplasia (ARVD), also known as arrhythmogenic right ventricular cardiomyopathy, is a common cause of sudden cardiac death in young people [1,2]. The pathognomonic hallmark of ARVD is the replacement of cardiomyocytes with fat and fibrous tissues, typically in the right ventricle [1]. ARVD is an autosomal dominant genetic disorder affecting a number of genes encoding the components of the cardiac desmosome complexes that mediate cell-to-cell attachments. The disease prevalence is estimated at 1 in 1,000–5,000 individuals, but it may be higher because of underdiagnosis and misdiagnosis. Complicating efforts to diagnose ARVD is the fact that the disease manifestation varies widely in terms of onset and severity of symptoms, even within an affected family. Furthermore, ARVD can present in an atypical fashion, often mimicking hypertrophic or dilated cardiomyopathy affecting the left ventricle without overt evidence of the pathognomonic fatty fibrous replacement. Here, we present a case of a near sudden cardiac death in a young athlete which was initially attributed to hypertrophic cardiomyopathy (HCM), but further familial and genetic evaluation revealed the ultimate diagnosis of ARVD.
Case Report
A previously healthy 21-year-old male was transferred to our center following aborted sudden cardiac death while playing basketball. As recounted by witnesses, the patient felt suddenly queasy while playing and collapsed while attempting to drink from a water fountain. Bystander CPR was not initiated. Upon arrival of emergency medical services within 5 min of the arrest, the patient was defibrillated 4 times and received intravenous epinephrine and lidocaine. Upon transfer to our center, the patient was unresponsive with evidence of significant anoxic encephalopathy. The patient had no past medical history of hypertension. His only medication was Adderall XR (extended-release mixed amphetamine salts) for treatment of adult attention deficit disorder.
The initial ECG showed sinus rhythm with J point elevation in V1 and inverted and biphasic T waves in V1 and V2 (fig. 1a). A complete metabolic panel, complete blood count and serum and urine toxicology screens were all negative. An urgent coronary angiogram revealed no epicardial coronary artery disease.
Fig. 1.

ECG of the proband and proband's father. a The proband's ECG is notable for J point elevation in V1 and inverted and biphasic T waves in V1 and V2. b The ECG of the proband's father is notable for sinus bradycardia, an incomplete right bundle, an epsilon wave at the end of the QRS complex in V1 and V2, borderline low voltage in the anterior leads and subtle (<1 mm) ST elevation in V2–3 with wave inversions in V1–3.
The patient's provisional diagnosis was nonobstructive HCM based upon the results of cardiac echocardiography and cardiac magnetic resonance (cMR) imaging studies. Cardiac echocardiography showed global, mild depression of left ventricular function with a left ventricular ejection fraction of 40%, a normal left ventricular internal diastolic diameter of 49 mm, mild, concentric left ventricular hypertrophy with a septal thickness of 12 mm and a posterior wall thickness of 11 mm, no valvular abnormalities, no left ventricular outflow gradients, no intracardiac shunts and no diastolic abnormalities (fig. 2). There was mild hypokinesis and dilatation of the right ventricle. cMR showed a symmetrically hypertrophied heart with a septal thickness of 17 mm and a posterior wall thickness of 15 mm (fig. 3). The left ventricular chamber size was normal with an end-diastolic diameter of 45 mm and a left ventricular ejection fraction of 55%. Global right ventricular systolic function was normal. There was no systolic anterior motion of the mitral valve, and no other valvular or great vessel abnormalities were found. Subendocardial delayed gadolinium enhancement was present globally throughout the left ventricle on the short axis view but not on orthogonal two-chamber or four-chamber views due to the patient's inability to hold his breath.
Fig. 2.

Echocardiograph of the proband. The parasternal short axis view (a) and parasternal long axis view (b) demonstrate global, mild depression of left ventricular function with concentric left ventricular hypertrophy and hypokinesis and dilatation of the right ventricle.
Fig. 3.

Short axis cMR of the proband. The cMR was notable for a symmetrically hypertrophied heart and global subendocardial delayed gadolinium enhancement of the left ventricle (arrow).
Over an ensuing 6-week hospitalization, the patient required a gastrostomy tube for feeding and extensive multidisciplinary rehabilitation. He made gradual but substantial recovery from anoxic encephalopathy. Cortical blindness and memory impairment improved, and he regained significant motor control. A Medtronic Sprint Quatro Secure Internal Cardioverter Defibrillator (ICD) was implanted successfully given his improvement. Upon discharge to a rehab facility, the patient was able to follow simple commands, speak a word or two, feed himself, read and walk with assistance, with further sustained improvement in all arrest-related functional deficits. Given the provisional diagnosis of HCM in the patient at the time of the initial presentation, his blood sample was sent for HCM genetic screening (Harvard-Partners Laboratory for Molecular Medicine). Eleven of the genes most commonly associated with HCM were sequenced (ACTC, GLA, LAMP2, MYBPC3, MYH7, MYL2, MYL3, PRKAG2, TNNI3, TNNT2, TMP1), but no pathologic mutations were detected.
Following a prolonged convalescence, the patient (proband), along with the proband's father, was referred to our Inherited Heart Disease Clinic. The proband's 55-year-old father exercised regularly and denied syncope, chest pain, palpitations, light-headedness or dyspnea. The father had no cardiac history and took only aspirin and simvastatin. The father's family history was notable for another 26-year-old son who was alive and well, a 59-year-old brother with S/P coronary artery bypass graft and ICD and a 61-year-old sister who died suddenly after an ICD had been implanted (fig. 4). The remaining family members were alive and well in Iran.
Fig. 4.

Pedigree of the proband and his family. Solid symbols represent individuals with ARVD. Open symbols represent unaffected or undiagnosed relatives. CABG = Coronary artery bypass graft.
The father's ECG showed sinus bradycardia, normal intervals, an incomplete right bundle, an epsilon wave at the end of the QRS complex in V1 and V2, borderline low voltage in the anterior leads and subtle (<1 mm) ST elevation in V2–3 with T wave inversions in leads V1–3 (fig. 1b). The father's ECG was quite similar to the proband's ECG. Since the ECGs of both the proband and his father were atypical for HCM, the proband's father subsequently underwent cMR, which showed normal left ventricle size and function, a dilated right ventricle, moderate right ventricular hypokinesis and delayed enhancement along the entire right ventricular free wall without suggestion of fibrofatty replacement (fig. 5). In aggregate, these ECG and cMR findings were highly suggestive of a clinical diagnosis of ARVD in the father.
Fig. 5.
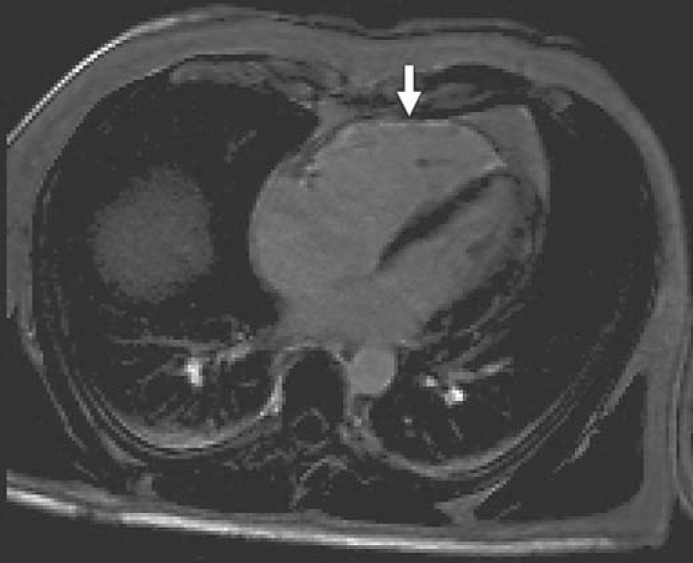
cMR of the proband's father demonstrated a dilated right ventricle, moderate right ventricular hypokinesis and delayed enhancement along the entire right ventricular free wall with some degree of fibrosis but without fibrofatty replacement.
Given the father's ECG and cMR findings suggestive of ARVD and the proband's ECG features that were atypical for HCM, the ARVD genetic screening test was performed on the proband. The ARVD screening panel (Harvard-Partners Laboratory for Molecular Medicine), which examines the coding sequence and splice sites of 5 genes most commonly associated with ARVD (PKP2, DSG2, DSP, DSC2, TMEM43), identified two rare sequence variants. The proband was heterozygous for the previously reported pathologic splice site mutation c.2489 + 1G>A in intron 12 of the desmosomal protein plakophilin-2 gene PKP2 (fig. 6) [3,4,5]. The mutation is predicted to cause abnormal RNA splicing, resulting in abnormal plakophilin-2. In addition, the proband was heterozygous for the c.327A>G (p.I109M) variant in exon 3 of the desmosomal protein desmocollin-2 gene DSC2. Although this variant has not been previously reported in the literature or detected by the reference lab, its pathogenicity could not be determined at this time.
Fig. 6.

The c.2489 + 1G>A variant in intron 12 of the PKP2 gene is predicted to cause abnormal RNA splicing. The variant, found in both the proband and his father, is predicted to disrupt splicing since the substituted guanine (G) adjacent to the splice donor site is absolutely conserved.
The proband's father was then evaluated for risk of sudden cardiac death from possible ARVD. The father's ARVD screen showed the exact same mutations in PKP2 and DSC2. The father was seen in consultation with an electrophysiologist (J.N.R.), who felt that the diagnosis of ARVD was secure in the father. Implantation of an ICD was discussed with the father, who, given considerable financial constraints, was not able to render a final decision.
Discussion
Plakophilin-2
Previous studies have implicated plakophilin-2 and desmocollin-2 in the pathogenesis of ARVD [6,7,8,9,10]. Among patients with ARVD, up to 25–45% harbor mutations in PKP2 [1,3,5,7,8,9,11,12,13]. Plakophilin-2 binds desmosomal transmembrane proteins and desmoplakin, which anchors cytosolic intermediate filaments to the desmosome [1,14]. Plakophilin-2 belongs to the armadillo subfamily of proteins, which consist of an amino-terminal head group and 10 related, short amino acid repeat units (arm units) and a carboxy-terminal domain [14]. The c.2489 + 1G>A mutation in intron 12 of PKP2 detected in both patients (fig. 6) is predicted to cause a splice site mutation that extends the mRNA transcript by 71 base pairs [6,13]. Although mechanistic studies examining the pathogenicity of this mutation in PKP2 have not been reported, the added amino acids probably interfere with the structure of the repeat arm units such that the mutant plakophilin-2 protein has weakened binding to other desmosomal proteins. A weakened cardiac desmosomal unit could predispose an individual to cardiomyopathy, as cardiac myocardium loses the ability to sustain stretch forces.
Desmocollin-2
Several studies of patients with ARVD have revealed multiple pathologic mutations in DSC2 with a frequency ranging from 0 to 7% [8,9,10,11,15,16]. Desmocollin-2 is a desmosomal transmembrane protein and the exclusive desmocollin isoform expressed in cardiac tissue [1,6]. Even though the pathologic mutation in PKP2 was sufficient to support the diagnosis of ARVD, gene sequencing revealed that the patient also harbored a novel missense mutation in DSC2. The c.327A>G mutation observed in the patient is predicted to cause an I109M nonsynonymous amino acid substitution in the propeptide region of desmocollin-2 [6,7]. Although the pathological significance of this novel variant remains unclear, this conservative amino acid substitution in the propeptide region could alter processing of desmocollin-2 to its mature form or inhibit proper targeting of desmocollin-2 to the desmosome. This notion of altered processing is supported by one study in neonatal rat cardiomyocytes demonstrating reduced plasma membrane localization of a desmocollin-2 with a missense mutation in the propeptide region [15]. Furthermore, plakophilin-2 and desmocollin-2 are predicted to bind on the cytosolic side of the desmosome [1,14]. Decreased desmocollin-2 localization to the desmosome could contribute to a dominant negative effect on the integrity of the desmosome, which may explain the observed trend toward exacerbated disease severity in patients with multiple desmosomal mutations [8,9,11]. Thus, the c327A>G (I109M) mutation could function as a modifier that exacerbates the ARVD disease phenotype.
ARVD – A Disease of Junctions
ARVD has been characterized as a disease of cardiac desmosomes because up to 50% of ARVD patients harbor mutations in genes encoding desmosomal proteins [1,6,7,8,9,10,12,16]. The pathogenesis of these mutations remains largely uncharacterized. However, mutations in desmosomal proteins could contribute to weakened cardiac desmosomes through reductions in protein-protein binding affinities or reductions in protein targeting resulting in fewer complete desmosomes. These desmosomal mutations could predispose individuals to reduced cardiac capacity to sustain stretch forces. The mechanism by which these desmosomal abnormalities contribute to clinical characteristics of this disease, such as fibrofatty infiltration in the right ventricle or conduction abnormalities, remains largely uncharacterized. Although the frequency of desmosomal mutations in patients with ARVD is suggestive of a disease of cellular structure, some studies suggest that ARVD could be a disease of cell signaling, because plakophilin-2 has been implicated in Wnt/β-catenin signaling [14,17].
The Critical Importance of ‘Familial Phenotyping’
In the case reported here, the key to the diagnosis of ARVD in this family was the comparative ‘clinical phenotyping’ of the father and son by ECG and MRI. Although the son's ECG was not classic for HCM, his MRI was most suggestive of HCM. MRI in the father was obtained to confirm the provisional diagnosis of HCM in the son. However, the similarity of the ECGs of the son and father, coupled with the father's MRI, which strongly suggested ARVD, and the son's negative HCM genetic screen, prompted further molecular diagnostic evaluation. Had the father not been ‘clinically phenotyped’, the diagnosis of ARVD would likely not have been established.
In the present case, both the proband and his father carried rare variants in PKP2 and DSC2. The c.2489 + 1G>A splice site variant of the PKP2 gene is likely pathogenic on its own. However, the impact of the p.I109M DSC2 variant, either alone or in combination with the PKP2 variant, cannot be determined at this time. Consequently, while familial ARVD is typically inherited in an autosomal dominant manner, the probability of transmitting ARVD to future offspring is unclear. In this regard, it will be very illuminating to examine the genotypes and phenotypes of additional family members.
MRI of the proband did not reveal typical features of ARVD. It is possible that sudden death from the ARVD may have preceded the typical anatomic and functional findings because of synergistic contributions of concurrent medications such as Adderall and physical activity, or because variable expressivity may be present and these changes will not evolve in the proband. This case report raises an important clinical management question, namely whether all young victims of sudden cardiac death should be genetically screened for ARVD in cases where clinical findings are equivocal and the molecular HCM screening studies are normal.
This case also illustrates the lack of predictable phenotype-genotype correlations in families with inherited cardiomyopathies. While variable penetrance of a disease phenotype among family members harboring identical mutations has long been recognized, it is increasingly recognized that distinct phenotypes may result from an identical pathogenic mutation [18]. For example, genotypes classically associated with HCM have been reported in dilated cardiomyopathy phenotypes [19]. Furthermore, a genetic variant classically associated with dilated cardiomyopathy has also been reported in first-degree relatives with dilated cardiomyopathy, HCM and nondilated restrictive cardiomyopathy phenotypes [20]. Given the potential for variable clinical presentations within families, additional diagnostic genetic testing should be pursued if testing for genes traditionally associated with the proband phenotype is negative in the proband.
Due to the variability in the presentation of ARVD, especially within this family, ICD implantation was recommended for the patient's father as a prophylactic measure, but the offsetting factor of prolonged survival without symptoms of arrhythmia was also presented. This clinical recommendation of ICD implantation based on the genetic sequence of the disease genes PKP2 and DSC2 is one of the hallmarks of individualized medicine. Similar decisions may be guided by a patient's genome sequence as more genetic studies identify additional pathologic mutations and clarify genotype-phenotype correlation in disease.
Acknowledgements
The genetic testing program at the Vanderbilt Center for Inherited Heart Disease was funded in part by the generous grant from the Edwards Lifesciences Fund. C.C.H. was funded by the Veterans Affairs Career Development Transition Award, VA Merit Award 101BX000771 and National Institutes of Health grants 5U01HL100398 and 1R01HL104040. B.T.P was supported by NIH T32GM07347 for the Vanderbilt Medical-Scientist Training Program.
References
- 1.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 2.Corrado D, Basso C, Pavei A, Michieli P, Schiavon M, Thiene G. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593–1601. doi: 10.1001/jama.296.13.1593. [DOI] [PubMed] [Google Scholar]
- 3.Dalal D, James C, Devanagondi R, Tichnell C, Tucker A, Prakasa K, Spevak PJ, Bluemke DA, Abraham T, Russell SD, Calkins H, Judge DP. Penetrance of mutations in plakophilin-2 among families with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2006;48:1416–1424. doi: 10.1016/j.jacc.2006.06.045. [DOI] [PubMed] [Google Scholar]
- 4.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 5.van Tintelen JP, Entius MM, Bhuiyan ZA, Jongbloed R, Wiesfeld AC, Wilde AA, van der Smagt J, Boven LG, Mannens MM, van Langen IM, Hofstra RM, Otterspoor LC, Doevendans PA, Rodriguez LM, van Gelder IC, Hauer RN. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–1658. doi: 10.1161/CIRCULATIONAHA.105.609719. [DOI] [PubMed] [Google Scholar]
- 6.Awad MM, Calkins H, Judge DP. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008;5:258–267. doi: 10.1038/ncpcardio1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barahona-Dussault C, Benito B, Campuzano O, Iglesias A, Leung TL, Robb L, Talajic M, Brugada R. Role of genetic testing in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Clin Genet. 2010;77:37–48. doi: 10.1111/j.1399-0004.2009.01282.x. [DOI] [PubMed] [Google Scholar]
- 8.Christensen AH, Benn M, Bundgaard H, Tybjaerg-Hansen A, Haunso S, Svendsen JH. Wide spectrum of desmosomal mutations in Danish patients with arrhythmogenic right ventricular cardiomyopathy. J Med Genet. 2010;47:736–744. doi: 10.1136/jmg.2010.077891. [DOI] [PubMed] [Google Scholar]
- 9.Fressart V, Duthoit G, Donal E, Probst V, Deharo JC, Chevalier P, Klug D, Dubourg O, Delacretaz E, Cosnay P, Scanu P, Extramiana F, Keller D, Hidden-Lucet F, Simon F, Bessirard V, Roux-Buisson N, Hebert JL, Azarine A, Casset-Senon D, Rouzet F, Lecarpentier Y, Fontaine G, Coirault C, Frank R, Hainque B, Charron P. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 2010;12:861–868. doi: 10.1093/europace/euq104. [DOI] [PubMed] [Google Scholar]
- 10.Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;79:978–984. doi: 10.1086/509122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhuiyan ZA, Jongbloed JD, van der Smagt J, Lombardi PM, Wiesfeld AC, Nelen M, Schouten M, Jongbloed R, Cox MG, van Wolferen M, Rodriguez LM, van Gelder IC, Bikker H, Suurmeijer AJ, van den Berg MP, Mannens MM, Hauer RN, Wilde AA, van Tintelen JP. Desmoglein-2 and desmocollin-2 mutations in Dutch arrhythmogenic right ventricular dysplasia/cardiomypathy patients: results from a multicenter study. Circ Cardiovasc Genet. 2009;2:418–427. doi: 10.1161/CIRCGENETICS.108.839829. [DOI] [PubMed] [Google Scholar]
- 12.den Haan AD, Tan BY, Zikusoka MN, Llado LI, Jain R, Daly A, Tichnell C, James C, Amat-Alarcon N, Abraham T, Russell SD, Bluemke DA, Calkins H, Dalal D, Judge DP. Comprehensive desmosome mutation analysis in north Americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Cardiovasc Genet. 2009;2:428–435. doi: 10.1161/CIRCGENETICS.109.858217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasse-Klaassen S, Probst S, Gerull B, Oechslin E, Nurnberg P, Heuser A, Jenni R, Hennies HC, Thierfelder L. Novel gene locus for autosomal dominant left ventricular noncompaction maps to chromosome 11p15. Circulation. 2004;109:2720–2723. doi: 10.1161/01.CIR.0000131865.21260.56. [DOI] [PubMed] [Google Scholar]
- 14.Chen X, Bonne S, Hatzfeld M, van Roy F, Green KJ. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta-catenin signaling. J Biol Chem. 2002;277:10512–10522. doi: 10.1074/jbc.M108765200. [DOI] [PubMed] [Google Scholar]
- 15.Beffagna G, De Bortoli M, Nava A, Salamon M, Lorenzon A, Zaccolo M, Mancuso L, Sigalotti L, Bauce B, Occhi G, Basso C, Lanfranchi G, Towbin JA, Thiene G, Danieli GA, Rampazzo A. Missense mutations in desmocollin-2 N-terminus, associated with arrhythmogenic right ventricular cardiomyopathy, affect intracellular localization of desmocollin-2 in vitro. BMC Med Genet. 2007;8:65. doi: 10.1186/1471-2350-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gehmlich K, Syrris P, Peskett E, Evans A, Ehler E, Asimaki A, Anastasakis A, Tsatsopoulou A, Vouliotis AI, Stefanadis C, Saffitz JE, Protonotarios N, McKenna WJ. Mechanistic insights into arrhythmogenic right ventricular cardiomyopathy caused by desmocollin-2 mutations. Cardiovasc Res. 2011;90:77–87. doi: 10.1093/cvr/cvq353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy – a Heart Failure Society of America practice guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Vasile VC, Will ML, Ommen SR, Edwards WD, Olson TM, Ackerman MJ. Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy. Mol Genet Metab. 2006;87:169–174. doi: 10.1016/j.ymgme.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Menon SC, Michels VV, Pellikka PA, Ballew JD, Karst ML, Herron KJ, Nelson SM, Rodeheffer RJ, Olson TM. Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin Genet. 2008;74:445–454. doi: 10.1111/j.1399-0004.2008.01062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]