

B. henselae is the etiological agent responsible for cat scratch fever (bartonellosis). The crystal structure of the smaller of the two Nudix hydrolases encoded in the genome of B. henselae, Bh-MutT, was determined to 2.1 Å resolution.
Keywords: Nudix hydrolases, Bartonella henselae, MutT, cat scratch fever
Abstract
Cat scratch fever (also known as cat scratch disease and bartonellosis) is an infectious disease caused by the proteobacterium Bartonella henselae following a cat scratch. Although the infection usually resolves spontaneously without treatment in healthy adults, bartonellosis may lead to severe complications in young children and immunocompromised patients, and there is new evidence suggesting that B. henselae may be associated with a broader range of clinical symptoms then previously believed. The genome of B. henselae contains genes for two putative Nudix hydrolases, BH02020 and BH01640 (KEGG). Nudix proteins play an important role in regulating the intracellular concentration of nucleotide cofactors and signaling molecules. The amino-acid sequence of BH02020 is similar to that of the prototypical member of the Nudix superfamily, Escherichia coli MutT, a protein that is best known for its ability to neutralize the promutagenic compound 7,8-dihydro-8-oxoguanosine triphosphate. Here, the crystal structure of BH02020 (Bh-MutT) in the Mg2+-bound state was determined at 2.1 Å resolution (PDB entry 3hhj). As observed in all Nudix hydrolase structures, the α-helix of the highly conserved ‘Nudix box’ in Bh-MutT is one of two helices that sandwich a four-stranded mixed β-sheet with the central two β-strands parallel to each other. The catalytically essential divalent cation observed in the Bh-MutT structure, Mg2+, is coordinated to the side chains of Glu57 and Glu61. The structure is not especially robust; a temperature melt obtained using circular dichroism spectroscopy shows that Bh-MutT irreversibly unfolds and precipitates out of solution upon heating, with a T m of 333 K.
1. Introduction
Cat scratch fever (also known as cat scratch disease and bartonellosis) is a bacterial infection characterized by a bump or blister at the original site of entry, swollen lymph nodes, headaches and overall general discomfort. The etiological agents responsible for the disease are bacteria from the fastidious hemotropic Gram-negative genus Bartonella (Chomel & Kasten, 2010 ▶). This genus contains 19 species, of which six have been associated with the human disease (Florin et al., 2008 ▶). Of these six species, B. henselae is the primary agent responsible for cat scratch fever. As the name of the disease suggests, the chief reservoir for B. henselae is the domestic cat, with the bacteria being transmitted to humans primarily via scratches (Florin et al., 2008 ▶). It is debatable whether cats themselves become ill owing to B. henselae infection (Chomel et al., 1996 ▶). There is no horizontal transfer of B. henselae between cats; instead, inter-feline spread of B. henselae occurs through a vector, Ctenocephalides felix, the cat flea (Chomel et al., 1996 ▶). It was generally believed that the infection did not require medical treatment, except in immunocompromised individuals, including those with human immunodeficiency virus infections. However, as techniques to diagnosis the presence of B. henselae have improved, there is a new concern that it may be responsible for a broader range of clinical symptoms, including prolonged fever of unknown origin (Florin et al., 2008 ▶). Owing to the widening spectrum of B. henselae infections and the widespread presence of B. henselae in the domestic cat, there is a renewed interest in understanding the biochemistry of B. henselae and in developing new therapies to combat this organism.
The genome of B. henselae contains genes for two putative Nudix hydrolases, BH02020 and BH01640 (KEGG). The Nudix superfamily of proteins (Pfam PF00293) is large, with over 16 000 members as of the end of 2010, and widely distributed, with members identified in organisms as diverse as viruses and humans (McLennan, 2006 ▶). Associated with their wide distribution is an ability to act, primarily as polyphosphate pyrophosphohydrolases (Bessman et al., 1996 ▶), on a wide range of substrates of the general structure shown in Fig. 1 ▶. It is from this general structure, Nucleoside diphosphates linked to some other moiety x, that the acronym ‘Nudix’ arose (Abeygunawardana et al., 1995 ▶; Bessman et al., 1996 ▶). Identified Nudix substrates include capped mRNA (Wang, Jiao et al., 2002 ▶), dinucleotide coenzymes, nucleotide sugars, nucleotide alcohols, dinucleotide polyphosphates, both canonical and oxidized (deoxy)ribonucleoside triphosphates (NTPs) (Dunn et al., 1999 ▶) and canonical (deoxy)ribonucleoside diphosphates (NDPs) (Fisher et al., 2004 ▶; Buchko et al., 2008 ▶). Some Nudix substrates have been reported without a nucleoside, such as thiamine pyrophosphate (Lawhorn et al., 2004 ▶), diphosphoinositol polyphosphates (Safrany et al., 1998 ▶) and dihydroneopterin triphosphate (Klaus et al., 2005 ▶). With a few exceptions, catalysis depends on a highly conserved sequence of amino acids, GX 5EX 7REU XEEXGU (where U is an aliphatic and hydrophobic amino acid and X is any amino acid), called the ‘Nudix box’ (Abeygunawardana et al., 1995 ▶; Bessman et al., 1996 ▶). This box forms a loop–helix–loop structural motif that is part of the larger substrate-binding Nudix fold. Glutamic acid residues in the core of the Nudix box, REUXEE, bind one to three divalent cations, and at least one cation is required for hydrolase activity. While Nudix hydrolases can bind a number of different divalent cations, including Mn2+ and Co2+ (Lin et al., 1997 ▶; Buchko et al., 2008 ▶), the highest activity observed in vitro is usually with Mg2+, the cation that is most likely to be relevant under physiological conditions (Bessman et al., 1996 ▶). Detailed features of the Nudix fold, a mixed β-sheet sandwiched between the catalytic α-helix of the Nudix box and a second α-helix, confer the substrate specificity of Nudix hydrolases (Mildvan et al., 2005 ▶; McLennan, 2006 ▶). By hydrolyzing their substrates, Nudix proteins effect the elimination of potentially toxic endogenous nucleotides from the cell and regulate the concentration of nucleotide cofactors and signaling molecules inside the cell (Bessman et al., 1996 ▶; Galperin et al., 2006 ▶). For example, Escherichia coli MutT protein, the prototypical member of the Nudix superfamily, preferably hydrolyzes 7,8-dihydro-8-oxoguanosine triphosphate, a promutagenic compound that is generated during normal cellular metabolism and upon exposure to oxidative stress (Grollman & Moriya, 1993 ▶), into nonmutagenic nucleoside monophosphate and inorganic phosphate (Maki & Sekiguchi, 1992 ▶; Weber et al., 1992 ▶). Given the important housekeeping duties performed by Nudix hydrolases, the crystal structure of one of the two Nudix hydrolases present in the genome of B. henselae, BH02020 (Bh-MutT), was solved and its structure described in relation to other Nudix protein structures.
Figure 1.
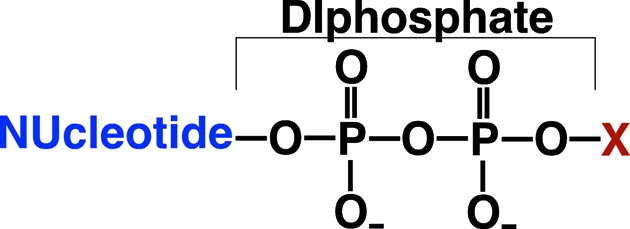
General chemical structure of a Nudix substrate: a Nucleotide diphosphate linked to a moiety ‘x’. The moiety ‘x’ may be as small as a hydrogen atom or metal ion.
2. Materials and methods
2.1. Cloning, expression and purification
The Bh-MutT gene (BH02020/YP_033055.1; NCBI) was amplified using the genomic DNA of B. henselae strain Houston-1 and the oligonucleotide primers 5′-GGGTCCTGGTTCGATGCCTATAAAAAGTTCACTTCTTATT-3′ (forward) and 5′-CTTGTTCGTGCTGTTTATTAAATAAAGAAATTTTTTAATACCTGAACTA-3′ (reverse) (Invitrogen, Carlsbad, California, USA). The amplified Bh-MutT gene was then inserted into NruI/PmeI-digested expression vector AVA0421 by ligation-independent cloning at a site that provided a 21-residue tag (MAHHHHHHMGTLEAQTQGPGS–) at the N-terminus of the expressed protein (Choi et al., 2011 ▶). The recombinant plasmid was transformed into E. coli BL21(DE3)-R3-pRARE2 cells (a gift from SGC-Toronto, Toronto, Ontario, Canada) via a heat-shock method and the resulting clone was used to inoculate 2 l of autoinduction medium (Studier, 2005 ▶) in a LEX bioreactor (Harbinger Biotechnology and Engineering Corporation, Markam, Ontario, Canada). After 72 h of growth at 293 K, the cells were harvested, flash-frozen in liquid nitrogen and stored at 193 K. The cell pellet was subsequently solubilized in 200 ml lysis buffer (25 mM HEPES, 500 mM NaCl, 5% glycerol, 30 mM imidazole, 0.5% CHAPS, 10 mM MgCl2, 1 mM TCEP, 1 mM AEBSF, pH 7.0) containing 10 mg lysozyme and sonicated for 30 min. After sonication, samples were treated with Benzonase (500 units) and centrifuged for 1 h (14 000 rev min−1 in a Sorvall SLA-1500 rotor) to remove cell debris. The protein was purified by immobilized metal-ion affinity chromatography on a 5 ml HisTrap FF Nickel Sepharose column (GE Healthcare, Piscataway, New Jersey, USA) using an ÄTKAexplorer (GE Healthcare, Piscataway, New Jersey, USA). Fractions containing Bh-MutT were pooled, concentrated and further purified by size-exclusion chromatography on a Superdex75 HiLoad 26/60 column (GE Healthcare, Piscataway, New Jersey) that also exchanged the protein into crystallization buffer (25 mM HEPES, 500 mM NaCl, 5% glycerol, 2 mM dithiothreitol, 0.025% sodium azide pH 7.5). Fractions containing Bh-MutT were pooled and concentrated (Amicon Ultra-15 with a 3000 Da MWCO) to 27 mg ml−1. Protein samples were aliquoted in 100 µl volumes into PCR tubes, flash-frozen in liquid nitrogen and stored at 193 K (Deng et al., 2004 ▶). For the circular dichroism and nuclear magnetic resonance spectroscopy experiments, 15N-labeled uncleaved Bh-MutT was prepared from 1 l of autoinduction medium (Studier, 2005 ▶) and purified by a conventional two-step protocol involving Ni- NTA affinity chromatography followed by size-exclusion chromatography using CD buffer (100 mM NaCl, 20 mM Tris, 1.0 mM dithiothreitol, pH 7.1).
2.2. Nuclear magnetic resonance spectroscopy
A Varian 600-Inova spectrometer equipped with a 1H{13C,15N} triple-resonance probe and pulse-field gradients was used to collect a 2D 1H–15N HSQC spectrum from 15N-labeled Bh-MutT in CD buffer (293 K).
2.3. Circular dichroism spectroscopy
A calibrated Aviv Model 410 spectropolarimeter (Lakewood, New Jersey, USA) was used to collect circular dichroism data from a 0.05 mM sample of Bh-MutT in CD buffer in a quartz cell of 0.1 cm path length. Using the same sample, a thermal denaturation curve was obtained by recording the ellipticity at 220 nm in 2.0 K intervals from 283 to 353 K. A quantitative estimation of the melting temperature, T m, was obtained by taking the first derivative of the thermal denaturation curve (Greenfield, 2006 ▶) using the Aviv software. Steady-state wavelength spectra for Bh-MutT were recorded in 0.5 nm increments between 200 and 260 nm at 298 K. The reported steady-state wavelength spectrum was the result of averaging two consecutive scans with a bandwidth of 1.0 nm and a time constant of 1.0 s. The spectrum was processed by subtracting a blank spectrum from the protein spectrum and then automatically line-smoothing the data using the Aviv software.
2.4. Crystallization
Sitting-drop vapor-diffusion crystallization trials were set up at 289 K using the Wizard full matrix (Emerald BioSystems, Bainbridge, Washington, USA) or ProPlex sparse-matrix (Molecular Dimensions, Newmarket, England) screens with or without in situ proteolysis with chymotrypsin (0.01 mg ml−1; Wernimont & Edwards, 2009 ▶). Bh-MutT stock solutions (0.4 µl) were mixed with reservoir solution (0.4 µl) and equilibrated against reservoir solution (100 µl) using 96-well Compact Jr plates (Emerald BioSystems, Bainbridge, Washington, USA). Crystals grew in several conditions, but those used in X-ray data collection and structure determination were obtained from ProPlex condition B5 without in situ proteolysis (27 mg ml−1 protein against 0.1 M HEPES, pH 7.5, 10% PEG 4000, 0.1 M MgCl2).
2.5. Data collection and structure refinement
A crystal of Bh-MutT was cryoprotected with reservoir solution supplemented with 20% glycerol, flash-frozen in liquid nitrogen, stored under liquid nitrogen, and shipped to the Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory for X-ray data collection. A data set was collected on ALS beamline 5.0.3 with an ADSC Quantum 315 detector (Table 1 ▶) and reduced with XDS/XSCALE (Kabsch, 2010 ▶). The structure was solved by molecular replacement using Phaser (McCoy et al., 2007 ▶) from the CCP4 suite (Winn et al., 2011 ▶) with the NMR solution structure of E. coli MutT (PDB entry 1tum, first model; Lin et al., 1997 ▶) as the search model after alignment modification in CHAINSAW (Stein, 2008 ▶). The structure was initially rebuilt with Buccaneer (Cowtan, 2006 ▶) followed by additional model building in ARP/wARP (Langer et al., 2008 ▶). The final model was produced after numerous iterative rounds of refinement in REFMAC (Murshudov et al., 1997 ▶) and manual building in Coot (Emsley & Cowtan, 2004 ▶). The final model showed good geometry and fitness (Table 1 ▶) according to analysis with MolProbity (Chen et al., 2010 ▶).
Table 1. Summary of the diffraction data-collection and refinement statistics for Bh-MutT.
Value in parentheses are for the highest resolution shell.
| Data collection | |
| X-ray source | ALS beamline 5.0.3 |
| Detector | ADSC Q315 CCD |
| X-ray wavelength (Å) | 0.97946 |
| Temperature (K) | 100 |
| Data set | Native |
| Space group | C2 |
| Unit-cell parameters | |
| a (Å) | 96.50 |
| b (Å) | 93.49 |
| c (Å) | 43.88 |
| α = γ (°) | 90 |
| β (°) | 109.24 |
| Matthews coefficient (Å3 Da−1) | 2.62 |
| Solvent content (%) | 53.12 |
| Resolution range (Å) | 50–2.1 (2.15–2.10) |
| Mean I/σ(I) | 12.71 (2.06) |
| No. of observed reflections† | 21194 (1406) |
| Completeness (%) | 98.4 (98.2) |
| Multiplicity | 3.6 (2.6) |
| Rmerge‡ (%) | 0.073 (0.488) |
| Rmeas§ (%) | 0.086 (0.606) |
| Phasing | |
| Molecular replacement model | 1tum |
| Refinement | |
| No. of reflections used | 20108 |
| Rwork¶ (%) | 20.2 (27.7) |
| Rfree†† (%) | 23.4 (31.5) |
| Mean B factor (Å2) | 30.5 |
| R.m.s.d. bonds (Å) | 0.014 |
| R.m.s.d. angles (°) | 1.391 |
| Model validation | |
| MolProbity Ramachandran analysis | |
| Most favored (%) | 98 |
| Additionally allowed (%) | 2 |
| MolProbity | |
| Clash score, all atoms [percentile] | 7.59 [94th] |
| MolProbity score [percentile] | 1.82 [90th] |
All reflections were used in data reduction.
R
merge = 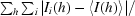
 .
.
R
meas = 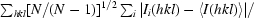
 (Diederichs & Karplus, 1997 ▶).
(Diederichs & Karplus, 1997 ▶).
R
work = 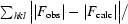
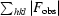 , where F
obs and F
calc are the observed and calculated structure factors, respectively, calculated with the 95% of the reflections remaining after setting aside 5% for R
free.
, where F
obs and F
calc are the observed and calculated structure factors, respectively, calculated with the 95% of the reflections remaining after setting aside 5% for R
free.
R
free =
. The free R factor was calculated using 5% (1086) of the reflections, which were omitted from the refinement.
The amino-acid sequence of Bh-MutT deposited in the PDB is numbered sequentially, Met1–Ile158, starting with the 21 non-native residues that were part of the N-terminal polyhistidine tag. Here, the first native residue, Met22 in the PDB deposition, is labeled Met1.
3. Results and discussion
3.1. Structure of Bh-MutT
The crystal structure includes two molecules of Bh-MutT, A and B, packed together in the asymmetric unit. Electron density was missing at the N-terminus for the 21-residue tag plus the first six native residues in both subunits. At the C-terminus, electron density was missing for the last two residues of subunit A and the last residue of subunit B. The structure of each molecule in the asymmetric unit was similar; the Cα atoms of residues Ser6–Phe135 of the two subunits superposed with a root-mean-square deviation (r.m.s.d.) of 0.61 Å (Winn et al., 2011 ▶). With a buried surface area of 11.4%, the PDBe PISA server (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html) predicts the biological unit of the protein to be a dimer as shown in Fig. 2 ▶. This observation in the crystalline state was also corroborated in solution. Bh-MutT eluted from a Superdex75 size-exclusion column with a retention time of 68 min, a value that is more consistent with a 36 kDa dimer than an 18 kDa monomer (21-residue N-terminal tag present). Furthermore, the 1H–15N HSQC spectrum of uncleaved 15N-labeled Bh-MutT featured broad cross-peaks characteristic of a protein much larger than 18 kDa (data not shown). Dimeric Nudix hydrolases have previously been observed both in solution and in the crystalline state and are sometimes required for the Nudix hydrolase to function optimally (Wang, Mura et al., 2002 ▶).
Figure 2.

A transparent surface representation of the two subunits in the asymmetric unit of the crystal structure of Bh-MutT (PDB entry 3hhj). A cartoon representation of the structure is illustrated underneath the surface. The two subunits are colored cyan and purple and the Mg2+ ions are colored yellow.
Fig. 3 ▶(a) shows a cartoon representation of the structure of Bh-MutT and Fig. 3 ▶(b) shows a two-dimensional schematic of the individual secondary structure components. The molecule is composed of six β-strands organized into two β-sheets, two α-helices and two 310-helices. Four of the β-strands form the core of the Nudix fold, a mixed β-sheet with the central two β-strands parallel to each other, sandwiched between the catalytic helix (α1) and an orthogonally orientated C-terminal helix (α2). Such an arrangement is observed in Nudix proteins of known structure (Mildvan et al., 2005 ▶; McLennan, 2006 ▶).
Figure 3.

(a) Cartoon representation of the crystal structure of Bh-MutT in the Mg2+-bound state (PDB entry 3hhj). The β-strands are colored blue, α-helices red and 310-helices magenta. The 23-residue ‘Nudix box’ consists of residues Gly42–Val64. (b) Secondary-structure diagram of Bh-MutT. The α-helices are drawn as red ovals and 310-helices as magenta ovals, with the residue number of the beginning and the end of each element shown. The β-strands are drawn as solid blue arrows with each residue indicated within a box. The β-sheets contain a bulge at residue Pro73. A solid pink line between β-strand residues indicates dual hydrogen bonds between two residues in an antiparallel β-sheet. The regions between secondary-structure elements (black lines) are not drawn to scale.
Nudix proteins are all characterized by the presence of a highly conserved 23-residue ‘Nudix box’, GX 5EX 7REUXEEXGU, where U is a bulky hydrophobic group and X is any residue (Abeygunawardana et al., 1995 ▶; Bessman et al., 1996 ▶). The Nudix sequence adopts a loop–helix–loop motif (Koonin, 1993 ▶) that serves to coordinate the catalytically essential divalent cation (usually Mg2+) to the protein (Lin et al., 1997 ▶; Bailey et al., 2002 ▶; Gabelli et al., 2001 ▶). Nine of the 23 residues in the Nudix box are highly conserved, G 42 X 5 E 48 X 7 R 56 E 57 L 58 X E 60 E 61 X G 63 V 64 (bold, with the superscripts referring to the residue numbers in the Bh-MutT sequence), and Fig. 4 ▶(a) illustrates the relative orientation of these highly conserved residues in the loop–helix–loop Nudix motif of Bh-MutT. As observed in other Nudix structures, all of the polar conserved residues in the α-helix of Bh-MutT are orientated on the same side and are opposite the central mixed β-sheet. A small hydrophobic pocket formed by the side chains of Leu58 and Val64 is likely to play a conserved role in stabilizing the loop–helix–loop Nudix motif. The similar three-dimensional orientations of the conserved residues in the Nudix box of all known Nudix protein structures suggest that these residues play similar roles in all Nudix hydrolases, binding one or more divalent cations (Wang, Mura et al., 2002 ▶). This is true for Bh-MutT, as shown in the electron-density map in Fig. 4(b), in which the hydroxyl groups of Glu57 and Glu61 are near the Mg2+ ion at distances of 2.10 and 2.11 Å, respectively, which are within the expected Mg2+-coordination distance range (Zheng et al., 2008 ▶). It has been established that binding of the catalytic divalent metal ion occurs through the side chains of the glutamic acid residues in the loop–helix–loop Nudix motif (Lin et al., 1997 ▶; Bailey et al., 2002 ▶; Gabelli et al., 2001 ▶; Buchko et al., 2008 ▶). The bound metal in turn complexes with the diphosphate part of the substrate (Fig. 1 ▶) and forms part of the enzyme’s active site (Lin et al., 1997 ▶). Diphosphate interactions with both the loop–helix–loop Nudix motif and the divalent metal cation are features that are common to all Nudix proteins. The other parts of the Nudix substrate, the nucleoside and the moiety ‘x’, interact with the side chains and motifs elsewhere in the protein (Lin et al., 1997 ▶; Gabelli et al., 2001 ▶; Bailey et al., 2002 ▶; Massiah et al., 2003 ▶; Mildvan et al., 2005 ▶). Differences in the shape and nature of the environment surrounding the Nudix box are responsible for the different substrate specificities of different Nudix hydrolases and some of the structural differences between Nudix hydrolases (Ranatunga et al., 2004 ▶).
Figure 4.

(a) Cartoon representation of the crystal structure of Bh-MutT with the 23-residue Nudix box highlighted in wheat. The side chains of the residues that form the Nudix box are shown (O, red; N, blue) along with the Mg2+ ion (black sphere). (b) The electron density surrounding the Mg2+ ion in the crystal structure of Bh-MutT illustrated at 1.2σ. The O atoms from the side chains of Glu61 and Glu57 are coordinated to the ion.
3.2. Circular dichroism profile and thermal stability of Bh-MutT
Circular dichroism spectroscopy is sensitive to small changes in a protein’s backbone and consequently it is a useful tool to quickly probe the conformation of proteins in solution and assess the effect of variables on a protein’s structure (Woody, 1974 ▶). Fig. 5 ▶(a) shows the steady-state CD spectrum for Bh-MutT collected at 298 K. The dominant feature of the spectrum is characteristic of an α-helical secondary structure: a double minimum at approximately 222 and 208 nm and an extrapolated maximum around 195 nm (Holzwarth & Doty, 1965 ▶; Greenfield, 2006 ▶). Note that the double minimum is skewed and more intense around 220 nm, which is likely to be a consequence of the contribution of other secondary-structure elements to the CD steady-state spectrum.
Figure 5.

(a) Circular dichroism steady-state wavelength spectrum for Bh-MutT (0.05 mM) in CD buffer collected at 298 K. (b) The CD thermal melt for Bh-MutT obtained by measuring the ellipticity at 220 nm in 2.0 K intervals between 283 and 353 K. (c) The first derivative of the thermal melt curve shows that the protein has a melting temperature of 333 K.
Usually, the ellipticity at a specific wavelength increases with increasing temperature as the protein unfolds and a melting temperature, T m, may be estimated for the transition between a structured and an unstructured state (Karantzeni et al., 2003 ▶). However, it has recently been observed that the thermal melt for the Nudix hydrolase DR_0079 from the bacterium Deinococcus radiodurans displays the opposite behavior, suggesting an increase in ordered structure with an increase in temperature (Buchko, 2010 ▶). To probe whether this unusual temperature effect observed for DR_0079 is more widespread in Nudix hydrolases, a melting temperature was determined for Bh-MutT. As shown in Fig. 5 ▶(b), the melting profile was typical. A gradual increase in ellipticity at 220 nm was observed for Bh-MutT up to ∼323 K, followed by a more rapid increase in ellipticity that leveled off at ∼335 K. Visual inspection of the sample after heating to 353 K showed evidence of precipitation, indicating that the unfolding was irreversible, and consequently the CD data could not be analyzed thermodynamically (Karantzeni et al., 2003 ▶). A quantitative estimation of the T m for this transition was still possible by assuming a two-state model and taking a first derivative of the curve in Fig. 5 ▶(b) (Greenfield, 2006 ▶). The maximum of this first derivative, shown in Fig. 5 ▶(c), is 333 K.
4. Conclusions
The crystal structure of Bh-MutT is similar to the structures reported for other Nudix hydrolases. The α-helix of the highly conserved Nudix box in Bh-MutT is one of two helices that sandwich a four-stranded mixed β-sheet with the central two β-strands parallel to each other. The structure was determined in the metal-bound form, with the catalytically essential divalent cation Mg2+ observed to be coordinated to the side chains of Glu57 and Glu61. While the crystal structure of Bh-MutT is similar to those of other Nudix hydrolases, these enzymes act on a wide range of substrates. The structural diversity that surrounds the basic Nudix box structure confers the functional diversity of Nudix hydrolases (Ranatunga et al., 2004 ▶; Mildvan et al., 2005 ▶; McLennan, 2006 ▶). The next step in understanding the cellular biochemical role of Bh-MutT in B. henselae is to equate the enzymatic activity of Bh-MutT to specific Nudix substrates (Ranatunga et al., 2004 ▶; Buchko et al., 2008 ▶). Such information, together with the structure provided here for Bh-MutT, will make it possible to better correlate Nudix sequences and structures with specific substrates and facilitate rational structure-based drug design targeting B. henselae.
Supplementary Material
PDB reference: Nudix hydrolase, 3hhj
Acknowledgments
This research was funded by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under Federal Contract No. HHSN272200700057C. The SSGCID internal ID for Bh-MutT is BaheA.00264.a. Part of this research was conducted at the W. R. Wiley Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the US Department of Energy’s Office of Biological and Environmental Research (BER) program located at Pacific Northwest National Laboratory (PNNL). Battelle operates PNNL for the US Department of Energy.
References
- Abeygunawardana, C., Weber, D. J., Gittis, A. G., Frick, D. N., Lin, J., Miller, A. F., Bessman, M. J. & Mildvan, A. S. (1995). Biochemistry, 34, 14997–15005. [DOI] [PubMed]
- Bailey, S., Sedelnikova, S. E., Blackburn, G. M., Abdelghany, H. M., Baker, P. J., McLennan, A. G. & Rafferty, J. B. (2002). Structure, 10, 589–600. [DOI] [PubMed]
- Bessman, M. J., Frick, D. N. & O’Handley, S. F. (1996). J. Biol. Chem. 271, 25059–25062. [DOI] [PubMed]
- Buchko, G. W. (2010). Protein Pept. Lett. 17, 831–835. [DOI] [PubMed]
- Buchko, G. W., Litvinova, O., Robinson, H., Yakunin, A. F. & Kennedy, M. A. (2008). Biochemistry, 47, 6571–6582. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Choi, R., Kelley, A., Leibly, D., Nakazawa Hewitt, S., Napuli, A. & Van Voorhis, W. (2011). Acta Cryst. F67, 998–1005. [DOI] [PMC free article] [PubMed]
- Chomel, B. B. & Kasten, R. W. (2010). J. Appl. Microbiol. 109, 743–750. [DOI] [PubMed]
- Chomel, B. B., Kasten, R. W., Floyd-Hawkins, K. A., Chi, B., Yamamoto, K., Roberts-Wilson, J., Gurfield, A. N., Abbott, R. C., Pedersen, N. C. & Koehler, J. E. (1996). J. Clin. Microbiol. 34, 1952–1956. [DOI] [PMC free article] [PubMed]
- Cowtan, K. (2006). Acta Cryst. D62, 1002–1011. [DOI] [PubMed]
- Deng, J., Davies, D. R., Wisedchaisri, G., Wu, M., Hol, W. G. J. & Mehlin, C. (2004). Acta Cryst. D60, 203–204. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol. 4, 269–275. [DOI] [PubMed]
- Dunn, C. A., O’Handley, S. F., Frick, D. N. & Bessman, M. J. (1999). J. Biol. Chem. 274, 32318–32324. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fisher, D. I., Cartwright, J. L., Harashima, H., Kamiya, H. & McLennan, A. G. (2004). BMC Biochem. 5, 7. [DOI] [PMC free article] [PubMed]
- Florin, T. A., Zaoutis, T. E. & Zaoutis, L. B. (2008). Pediatrics, 121, e1413–e1425. [DOI] [PubMed]
- Gabelli, S. B., Bianchet, M. A., Bessman, M. J. & Amzel, L. M. (2001). Nature Struct. Biol. 8, 467–472. [DOI] [PubMed]
- Galperin, M. Y., Moroz, O. V., Wilson, K. S. & Murzin, A. G. (2006). Mol. Microbiol. 59, 5–19. [DOI] [PubMed]
- Greenfield, N. J. (2006). Nature Protoc. 1, 2527–2535. [DOI] [PMC free article] [PubMed]
- Grollman, A. P. & Moriya, M. (1993). Trends Genet. 9, 246–249. [DOI] [PubMed]
- Holzwarth, G. M. & Doty, P. (1965). J. Am. Chem. Soc. 87, 218–228. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Karantzeni, I., Ruiz, C., Liu, C.-C. & Licata, V. J. (2003). Biochem. J. 374, 785–792. [DOI] [PMC free article] [PubMed]
- Klaus, S. M., Wegkamp, A., Sybesma, W., Hugenholtz, J., Gregory, J. F. & Hanson, A. D. (2005). J. Biol. Chem. 280, 5274–5280. [DOI] [PubMed]
- Koonin, E. V. (1993). Nucleic Acids Res. 21, 4847. [DOI] [PMC free article] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Lawhorn, B. G., Gerdes, S. Y. & Begley, T. P. (2004). J. Biol. Chem. 279, 43555–43559. [DOI] [PubMed]
- Lin, J., Abeygunawardana, C., Frick, D. N., Bessman, M. J. & Mildvan, A. S. (1997). Biochemistry, 36, 1199–1211. [DOI] [PubMed]
- Maki, H. & Sekiguchi, M. (1992). Nature (London), 355, 273–275. [DOI] [PubMed]
- Massiah, M. A., Saraswat, V., Azurmendi, H. F. & Mildvan, A. S. (2003). Biochemistry, 42, 10140–10154. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McLennan, A. G. (2006). Cell. Mol. Life Sci. 63, 123–143. [DOI] [PMC free article] [PubMed]
- Mildvan, A. S., Xia, Z., Azurmendi, H. F., Saraswat, V., Legler, P. M., Massiah, M. A., Gabelli, S. B., Bianchet, M. A., Kang, L.-W. & Amzel, L. M. (2005). Arch. Biochem. Biophys. 433, 129–143. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Ranatunga, W., Hill, E. E., Mooster, J. L., Holbrook, E. L., Schulze-Gahmen, U., Xu, W., Bessman, M. J., Brenner, S. E. & Holbrook, S. R. (2004). J. Mol. Biol. 339, 103–116. [DOI] [PubMed]
- Safrany, S. T., Caffrey, J. J., Yang, X., Bembenek, M. E., Moyer, M. B., Burkhart, W. A. & Shears, S. B. (1998). EMBO J. 17, 6599–6607. [DOI] [PMC free article] [PubMed]
- Stein, N. (2008). J. Appl. Cryst. 41, 641–643.
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Wang, Z., Jiao, X., Carr-Schmid, A. & Kiledjian, M. (2002). Proc. Natl Acad. Sci. USA, 99, 12663–12668. [DOI] [PMC free article] [PubMed]
- Wang, S., Mura, C., Sawaya, M. R., Cascio, D. & Eisenberg, D. (2002). Acta Cryst. D58, 571–578. [DOI] [PubMed]
- Weber, D. J., Bhatnagar, S. K., Bullions, L. C., Bessman, M. J. & Mildvan, A. S. (1992). J. Biol. Chem. 267, 16939–16942. [PubMed]
- Wernimont, A. & Edwards, A. (2009). PLoS One, 4, e5094. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Woody, R. W. (1974). Peptides, Polypeptides and Proteins, edited by E. R. Blout, F. A. Bovey, M. Goodman & N. Loton, pp. 338–350. New York: John Wiley & Sons.
- Zheng, H., Chruszcz, M., Lasota, P., Lukasz, L. & Minor, W. (2008). Inorg. Biochem. 102, 1765–1776. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Nudix hydrolase, 3hhj