

SYNOPSIS
Case studies of patients with familial schizophrenia may help to define the pathophysiology of this illness and indicate potential candidate genes for genetic linkage studies. In this regard, the clinical, radiological and pathological assessments of a 39-year-old affected man from a pedigree with familial schizophrenia are presented. Brain imaging with CT indicated moderate cortical atrophy, particularly of the temporal lobes. Neuropathological examination revealed granular ependymitis, indicating possible past ventricular pathology. Granular ependymitis was reported to occur in genetic developmental disorders with neuronal migration abnormalities. In the present case, heterotopic clusters of neurons were visualized in the entorhinal cortex, suggesting that temporal lobe development was not entirely normal. This case study suggests that genetic factors could be investigated further as one possible aetiology of certain neurodevelopmental abnormalities observed in schizophrenia.
INTRODUCTION
Abnormal processes of brain development that affect the temporal lobe may contribute to the mechanism of illness in schizophrenia. Neuro-imaging studies indicate changes in the lateral temporal lobe; most consistently reported is an increase in the size of the Sylvian fissure (Dewan et al. 1983; Harvey et al. 1993). In the medial temporal lobe, decreased volumes of the hippocampus, amygdala and parahippocampal gyrus are reported (DeLisi et al. 1988; Suddath et al. 1989; Bogerts et al. 1990; Marsh et al. 1994). The presence of certain of these abnormalities at illness onset (Bogerts et al. 1990) and failure to demonstrate a relationship with duration of illness (Marsh et al. 1994) suggest the possibility of a mechanism related to brain development or maturation. Pathological studies indicate evidence for subtle dysgenesis of two medial temporal lobe regions, the entorhinal cortex (Jakob & Beckmann, 1986; Falkai et al. 1988; Arnold et al. 1991) and the hippocampus (Kovelman & Scheibel, 1984; Conrad et al. 1991). Abnormalities of neuronal position or number in the absence of gliosis suggest altered processes of brain development. The lateral temporal lobe is also reported to be abnormal in post-mortem studies, with unusual gyral patterns (Jakob & Beckmann, 1986, 1989), loss of the expected asymmetry in the length of the Sylvian fissure (Falkai et al. 1992), and evidence for abnormally positioned neurons (Akbarian et al. 1993).
Research into the phenotypic characteristics of familial schizophrenia may provide indications of the mechanism of expression of a putative genetic aetiology for the illness. As part of an ongoing study of familial schizophrenia, we have had the opportunity to investigate the brain of an individual with chronic schizoaffective disorder from a multiply-affected pedigree.
CASE REPORT
Clinical history
The patient was the eleventh of 12 children of unrelated parents. The pregnancy was uneventful with spontaneous onset of labour at 40 weeks of gestation. Delivery was aided by forceps; the birth weight was 5100 g (> 97th percentile). The neonatal period and infancy were largely unremarkable, with normal developmental milestones. The patient was described as quiet, and as a loner who could be stubborn and bullied his younger sibling. He left school at age 13 after completing grade six. He did not learn to read or write, but was proficient at mechanical tasks such as auto-repair.
The first psychiatric hospitalization was at the age of 18 for threatening behaviour in the context of alcohol intoxication. He had experienced paranoid and referential ideas for the previous 2 years. On mental status examination the patient was described as being slightly apprehensive with awkward movements, a somewhat constricted affect and concrete thought processes. There were no hallucinations or delusions. Psychological testing with the WAIS indicated: Verbal IQ 86, Performance IQ 72, and Full Scale IQ 79. An EEG was normal. He was discharged after 1 month, without medication.
He married and had a family; however, he remained rather socially isolated. There was no further alcohol or drug abuse. At age 31 he became increasingly suspicious and socially withdrawn over a period of 6 months. He reported hearing and seeing military aircraft, believed there was a war occurring, and that the world was coming to an end. He heard voices talking to one another, giving commands, and commenting on him. There were paranoid and somatic delusions, thought insertion and thought broadcasting. His mood was described as very frightened and depressed, with insomnia, agitation and difficulty concentrating. He was admitted to hospital, where bizarre catatonic-like postures and long pauses in speech were noted. The delusions and hallucinations decreased in response to 10 mg of haloperidol daily during the 3-week admission.
Over the next 2 years his level of functioning deteriorated. He was unable to work, separated from his wife and his self-care worsened. He was hospitalized twice for 2 weeks at a time, with exacerbations of psychosis similar to the first episode. Medications included depot preparations of fluphenazine, pipotiazine, haloperidol and flupenthixol. Trifluoperazine was also used. Imipramine (200 mg daily) did not ameliorate his depressive symptoms. At age 36 he participated in a genetic linkage research study. A SCID-I interview, extensive collateral information from family members and medical records confirmed a diagnosis of schizoaffective disorder, depressed type according to DSM-III-R, and schizoaffective disorder, mainly schizophrenic type by RDC criteria. Negative symptoms were more prominent than positive symptoms according to the Positive and Negative Syndrome Scale (Kay et al. 1987): positive 14 (25th percentile), negative 26 (75th percentile), general psychopathology 48 (85th percentile). Mini-Mental State Examination (Folstein et al. 1975) score was 25 out of 29 (sentence item could not be completed due to illiteracy); immediate recall was impaired although attention was good. The Global Assessment of Functioning score (DSM-III-R Axis V) was 43, indicating serious impairment. He had moderate tardive dyskinesia, parkinsonism and akathisia, as well as some repetitive stereotypic movements. A second-degree relative suffered from schizophrenia. There was also a strong family history of heart disease, but not hypercholesterolaemia.
Over the next few years he developed severe congestive heart failure. Antipsychotic medications were discontinued and the patient required admission to a chronic care facility due to his medical condition. His final admission at age 39 was for 2 days, and he died due to refractory congestive heart failure with oliguria and hypertension. The general autopsy confirmed critical trivessel coronary artery atherosclerosis with ischaemic cardiomyopathy and congestive heart failure.
Radiological findings
A computed tomographic (CT) scan was performed as part of the research study, when the subject was 37 years old. Contiguous 5 mm slices were obtained from the skull base to the vertex at an angle of approximately 12° to the canthomeatal line. Reconstruction used a 512 × 512 matrix, and the field of view was 25 cm. Review by a neuroradiologist (J.S.L.) indicated the presence of moderate cortical atrophy, particularly affecting the temporal lobes (see Fig. 1). The involvement of medial as well as lateral structures of the temporal lobe was suggested by the enlargement of the temporal horns, suprasellar cistern and temporal midbrain cistern. The frontal horn and body (but not the inferior horn) of the left lateral ventricle was larger than the right. Overall, the size of the ventricular system was felt to be within normal limits.
Fig. 1.
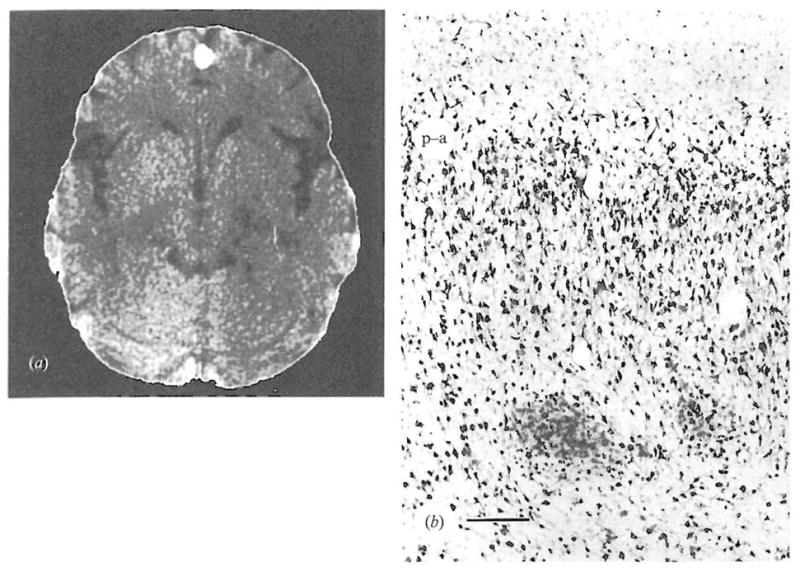
(a) CT scan showed moderate cortical atrophy, particularly affecting the temporal lobes. The temporal horns and suprasellar cistern were somewhat enlarged, as were the Sylvian fissures. The ambient cistern was also prominent. This CT scan illustration was edited with the NIH Image program (written by Wayne Rasband) to remove the bony structures.
(b) Abnormally positioned clusters of pre-alpha neurons in the entorhinal cortex. The pial surface is at the top, and the pre-alpha cell layer is indicated by (p-a). Two clusters of neurons remained deep to their expected position. Staining with antibody TG2, bar represents 250 μ.
Neuropathological findings
The brain weighed 1330 g. The left half was placed in formalin and the right half was frozen. Examination of the left hemibrain by a neuropathologist (T.G.B.) revealed mild frontal sulcal widening. The gross examination was otherwise unremarkable.
Haematoxylin/eosin, Luxol fast blue, cresyl violet, Prussian blue and Von Kossa stains, as well as an immunocytochemical study for ubiquitin were used to examine sections microscopically. The regions studied were: cortex (superior frontal gyrus, frontal pole, cingulate gyrus, temporal pole, superior parietal cortex, visual cortex, anterior and mid-hippocampus), basal ganglia (midbrain, head of caudate and putamen, lenticular nucleus), amygdala, thalamus, pons, cerebellum with dentate and medulla. Granular ependymitis of the ventricular system was noted, possibly representing evidence of temporally remote ventricular pathology. Multiple sections were prepared from a 3 mm block of entorhinal cortex (Duvernoy, 1988) and were stained with cresyl violet. Also, sections were stained using a mouse monoclonal antibody (TG2) generated following immunization with a preparation of neural cytoskeletal proteins (Vincent et al. 1993). This antibody appears to be reactive with the vast majority of neurons in formalin fixed human brain, and is therefore useful for anatomical studies of cell distribution. Entorhinal cortex sections were examined (P.F.) without knowledge of the diagnosis or other specifics of the case, and were graded according to a standardized method (Falkai & Bogerts, 1991). With both staining techniques, moderate numbers (two or more per section) of heterotopic or abnormally positioned pre-alpha cell clusters were noted on multiple sections (see Fig. 1). These heterotopic clusters were positioned deep to their expected location. The presence of Alzheimer type II astroglia in the basal ganglia, thalamus and cerebellum was the only other pathological finding observed, and was consistent with ante-mortem liver failure.
DISCUSSION
Examination of a case of schizoaffective disorder from a multiply affected family permitted identification of several features consistent with imaging and post-mortem studies of larger samples of patients with schizophrenia. The salient findings in the present case were radiological evidence for cortical atrophy, and patho-anatomical evidence for a subtle disturbance of neurodevelopment. Both findings were present in the temporal region.
The clinical features of illness in the present case were typical of schizophrenia and schizoaffective disorder. Previous reports indicated no differences in phenomenological features of affected individuals in families with schizophrenia compared to clinically selected samples of patients (Bassett et al. 1993, 1994). Difficulties with learning to read are reported in schizophrenia (Done et al. 1994); the present individual was severely affected in this regard.
Radiological evidence of enlargement of lateral temporal sulcal spaces and medial temporal ventricular and cisternal spaces was observed in families with schizophrenia (Honer et al. 1994). The individual in the present report was a member of one of these families. In this case the radiological findings were associated with neuropathological abnormalities in the medial temporal lobe.
The neuropathological examination indicated abnormalities in the ependymal cell layer lining the ventricular system, and in the position of pre-alpha cell clusters in the entorhinal cortex. Granular ependymitis indicates a change in cell morphology, often associated with disruption in the continuity of the ependymal cell layer (Duchen, 1992). Granular ependymitis may be the sequela of non-specific inflammatory changes. However, this finding was also observed in several types of neuronal migration disorders (Sarnat et al. 1993) including Miller–Dieker syndrome, which has a defined genetic aetiology (Hattori et al. 1994). The foetal ependyma plays an important role during the developmental phases of neuronal proliferation and migration. Abnormalities in the adult ependymal layer may serve as markers for dysregulation of development. In the present case, abnormally positioned neurons were observed in the entorhinal cortex pre-alpha cell layer. Similar abnormalities in schizophrenia were reported previously (Jakob & Beckmann, 1986, 1989; Arnold et al. 1991). Abnormally positioned cortical neurons (‘ectopic neurons’ found superficial to their expected location) are reported in patients with dyslexia (Galaburda et al. 1985). The finding of abnormally positioned neurons is consistent with impaired neuronal migration. The present case indicates that such findings can occur in familial schizophrenia, and raises the possibility that one mechanism could relate to a genetic aetiology. Non-genetic aetiologies, or gene-environment interactions, are also proposed to explain developmental abnormalities in schizophrenia (Scheibel & Conrad, 1993). More detailed analysis of heterotopic neurons in schizophrenia, dyslexia and temporal lobe epilepsy may define similarities and differences in the pathogenesis of these disorders.
The present case comes from a family in which the genetic phenomena of anticipation, or an increasing severity of illness across generations occurs (Bassett & Honer, 1994). The observation of anticipation in other neuropsychiatric diseases has been associated with an unstable mutation mechanism, involving variable numbers of trinucleotide repeats in the DNA sequence of specific genes. Similar unstable mutations are proposed for schizophrenia (Bassett & Honer, 1994). The present report suggests that as novel triplet repeat containing genes expressed in human brain are described (Li et al. 1993), any of these with homology to genes known to be involved in the process of neuronal migration would be interesting candidates for genetic linkage studies of schizophrenia.
Acknowledgments
The authors are most grateful for the assistance of Drs R. Henderson, D. Toms, P. Forsythe, G. N. Smith and Ms J. McAlduff. Antibody TG2 was kindly provided by Drs P. Davies and I. Vincent. Support was provided by the Medical Research Council of Canada and the Ontario Mental Health Foundation.
REFERENCES
- Akbarian S, Viñuela A, Kim JJ, Potkin SG, Bunney WE, Jones EG. Distorted distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase neurons in temporal lobe of schizophrenics implies anomalous cortical development. Archives of General Psychiatry. 1993;50:178–187. doi: 10.1001/archpsyc.1993.01820150016002. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Hyman BT, Van Hoesen GW, Damasio AR. Some cytoarchitectural abnormalities of the entorhinal cortex in schizophrenia. Archives of General Psychiatry. 1991;48:625–632. doi: 10.1001/archpsyc.1991.01810310043008. [DOI] [PubMed] [Google Scholar]
- Bassett AS, Honer WG. Evidence for anticipation in schizophrenia. American Journal of Human Genetics. 1994;54:864–870. [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Collins EJ, Nuttall SE, Honer WG. Positive and negative symptoms in families with schizophrenia. Schizophrenia Research. 1993;11:9–19. doi: 10.1016/0920-9964(93)90033-f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Bury A, Honer WG. Testing Liddle’s three syndrome model in families with schizophrenia. Schizophrenia Research. 1994;12:213–221. doi: 10.1016/0920-9964(94)90031-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerts B, Ashtari M, Degreef G, Alvir JMJ, Bilder RM, Lieberman JA. Reduced temporal limbic structure volumes on magnetic resonance images in first episode schizophrenia. Psychiatry Research: Neuroimaging. 1990;35:1–13. doi: 10.1016/0925-4927(90)90004-p. [DOI] [PubMed] [Google Scholar]
- Conrad AJ, Abebe T, Austin R, Forsythe S, Scheibel AB. Hippocampal pyramidal cell disarray in schizophrenia as a bilateral phenomenon. Archives of General Psychiatry. 1991;48:413–417. doi: 10.1001/archpsyc.1991.01810290025003. [DOI] [PubMed] [Google Scholar]
- DeLisi LE, Dauphinais ID, Gershon E. Perinatal complications and reduced size of brain limbic structures in familial schizophrenia. Schizophrenia Bulletin. 1988;14:185–191. doi: 10.1093/schbul/14.2.185. [DOI] [PubMed] [Google Scholar]
- Dewan MJ, Pandurangi AK, Lee SH, Ramachandran T, Levy B, Boucher M, Yozawitz A, Major LF. Central brain morphology in chronic schizophrenic patients: a controlled CT study. Biological Psychiatry. 1983;18:1133–1139. [PubMed] [Google Scholar]
- Done DJ, Sacker A, Crow TJ. Childhood antecedents of schizophrenia and affective illness: intellectual performance at ages 7 and 11. Schizophrenia Research. 1994;11:96–97. [Google Scholar]
- Duchen LW. General pathology of neurons and neuroglia. In: Adams JH, Duchen LW, editors. Greenfield’s Neuropathology. Oxford University Press; New York: 1992. pp. 1–68. [Google Scholar]
- Duvernoy HM. The Human Hippocampus. J. F. Bergmann Verlag; Munich: 1988. [Google Scholar]
- Falkai P, Bogerts B. Qualitative and quantitative assessment of pre-alpha-cell clusters in the entorhinal cortex of schizophrenics. A neurodevelopmental model of schizophrenia? Schizophrenia Research. 1991;4:357–358. [Google Scholar]
- Falkai P, Bogerts B, Rozumek M. Limbic pathology in schizophrenia: the entorhinal region – a morphometric study. Biological Psychiatry. 1988;24:515–521. doi: 10.1016/0006-3223(88)90162-x. [DOI] [PubMed] [Google Scholar]
- Falkai P, Bogerts B, Greve B, Pfeiffer U, Machus B, Fölsch-Reetz B, Majtenyi C, Ovary I. Loss of sylvian fissure asymmetry in schizophrenia. Schizophrenia Research. 1992;7:23–32. doi: 10.1016/0920-9964(92)90070-l. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. ‘Mini-Mental State’: a practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Galaburda AM, Sherman GF, Rosen CD, Aboitz F, Geschwind N. Developmental dyslexia: four consecutive patients with cortical anomalies. Annals of Neurology. 1985;18:222–233. doi: 10.1002/ana.410180210. [DOI] [PubMed] [Google Scholar]
- Harvey I, Ron MA, Du Boulay G, Wicks D, Lewis SW, Murray RM. Reduction of cortical volume in schizophrenia on magnetic resonance imaging. Psychological Medicine. 1993;23:591–604. doi: 10.1017/s003329170002537x. [DOI] [PubMed] [Google Scholar]
- Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Miller-Dicker lissencephaly gene encodes a subunit of brain platelet-activating factor. Nature. 1994;370:216–218. doi: 10.1038/370216a0. [DOI] [PubMed] [Google Scholar]
- Honer WG, Bassett AS, Smith GN, Lapointe JS, Falkai P. Temporal lobe abnormalities in multi-generational families with schizophrenia. Biological Psychiatry. 1994;36:737–743. doi: 10.1016/0006-3223(94)90084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob H, Beckmann H. Prenatal developmental disturbances in the limbic allocortex in schizophrenics. Journal of Neural Transmission. 1986;65:303–326. doi: 10.1007/BF01249090. [DOI] [PubMed] [Google Scholar]
- Jakob H, Beckmann H. Gross and histological criteria for developmental disorders in brains of schizophrenics. Journal of the Royal Society of Medicine. 1989;82:466–469. doi: 10.1177/014107688908200808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay S, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophrenia Bulletin. 1987;13:261–276. doi: 10.1093/schbul/13.2.261. [DOI] [PubMed] [Google Scholar]
- Kovelman JA, Scheibel AB. A neurohistological correlate of schizophrenia. Biological Psychiatry. 1984;19:1601–1621. [PubMed] [Google Scholar]
- Li SH, McInnis MG, Margolis RL, Antonarakis SE, Ross CA. Novel triplet repeat containing genes in human brain: cloning, expression, and length polymorphisms. Genomics. 1993;16:572–579. doi: 10.1006/geno.1993.1232. [DOI] [PubMed] [Google Scholar]
- Marsh L, Suddath RL, Higgins N, Weinberger DR. Medial temporal lobe structures in schizophrenia: relationship of size to duration of illness. Schizophrenia Research. 1994;11:225–238. doi: 10.1016/0920-9964(94)90016-7. [DOI] [PubMed] [Google Scholar]
- Sarnat HB, Darwish HZ, Barth PG, Trevenen CL, Pinto A, Kotagal S, Shishikura K, Osawa M, Korobkin R. Ependymal abnormalities in lissencephaly/pachygyria. Journal of Neuropathology and Experimental Neurology. 1993;52:525–541. doi: 10.1097/00005072-199309000-00011. [DOI] [PubMed] [Google Scholar]
- Scheibel AB, Conrad AS. Hippocampal dysgenesis in mutant mouse and schizophrenic man: is there a relationship? Schizophrenia Bulletin. 1993;19:21–33. doi: 10.1093/schbul/19.1.21. [DOI] [PubMed] [Google Scholar]
- Suddath RL, Casanova MF, Goldgerb TE, Daniel DG, Kelsoe JR, Weinberger DR. Temporal lobe pathology in schizophrenia: a quantitative magnetic resonance imaging study. American Journal of Psychiatry. 1989;146:464–472. doi: 10.1176/ajp.146.4.464. [DOI] [PubMed] [Google Scholar]
- Vincent IJ, Katen RN, Isaacs A, Mattiace LA, Davies P. TGI: a marker for nuclear tau with specificity for neuronal nuclei in Alzheimer’s disease. Society for Neuroscience Abstracts. 1993;19:670.4. [Google Scholar]