

Abstract
Paraneoplastic cerebellar degeneration (PCD) is a disorder in which breast or ovarian tumors express an onconeural antigen termed cdr2, which normally is expressed in cerebellar Purkinje neurons. This leads to an immune response to cdr2 that is associated with tumor immunity and autoimmune cerebellar degeneration. We have found that cdr2, a cytoplasmic protein harboring a helix–leucine zipper (HLZ) motif, interacts specifically with the HLZ motif of c-Myc. Both proteins colocalize in the cytoplasm of adult cerebellar Purkinje neurons, and coimmunoprecipitate from tumor cell lines and cerebellar extracts. cdr2 down–regulates c-Myc-dependent transcription in cotransfection assays, and redistributes Myc protein in the cytoplasm. Disease antisera from six of six PCD patients specifically blocked the interaction between cdr2 and c-Myc in vitro. These data indicate that cdr2 normally sequesters c-Myc in the neuronal cytoplasm, thereby down-regulating c-Myc activity, and suggest a mechanism whereby inhibition of cdr2 function by autoantibodies in PCD may contribute to Purkinje neuronal death.
Keywords: Purkinje neurons, cerebellar degeneration, c-Myc function, paraneoplastic neurological disease, leucine zipper protein
Paraneoplastic neurologic disorders are immune-mediated neuronal degenerations that develop in the setting of malignancy. They provide perhaps the clearest examples of naturally occurring tumor immunity in humans. It is believed that the disorders are initiated when neuron-specific proteins, normally immunologically privileged, are expressed ectopically in tumor cells and thereby, are recognized as foreign (tumor) antigens. Such patients are asymptomatic until this immune response becomes competent to recognize antigens expressed in the nervous system, leading to an immune-mediated neuronal death. Therefore, these rare disorders touch on important questions of tumor immunity, autoimmune neurologic disease, and neuron-specific protein function.
Paraneoplastic cerebellar degeneration (PCD) is one of the best-studied PNDs. Characterization of 55 PCD patients (Peterson et al. 1992) revealed that they almost invariably have breast or ovarian cancer, and that their cerebellar degeneration is characterized pathologically by Purkinje cell death. The immune response in PCD is characterized by high titers of serum and cerebrospinal fluid antibodies that recognize a ∼55–kD antigen in the patient’s tumors and in cerebellar Purkinje neurons. PCD antisera has been used to clone cDNAs encoding several related target antigens (Dropcho et al. 1987; Sakai et al. 1990; Fathallah-Shaykh et al. 1991), only one of which, cdr2, is expressed in PCD tumor specimens (Corradi et al. 1997). The cdr2 gene is widely transcribed, but the protein has only been found to be expressed in cerebellar Purkinje neurons, some brainstem neurons, and spermatogonia (Corradi et al. 1997), all immune-privileged sites.
The major insight to the biologic function of cdr2 has been the identification of structural motifs in the predicted amino acid sequence. The cdr2 amino terminus harbors an acidic domain followed by an extended amphipathic helix that ends in a typical leucine zipper. Recognition of the leucine zipper domain, which is present in a number of proteins, including some transcription factors, initially led to the suggestion that cdr2 might be involved in the regulation of gene expression (Fathallah-Shaykh et al. 1991). However, the antigen was found to be localized to the cytoplasm, where it can be found both free and associated with membrane-bound ribosomes (Hida et al. 1994).
We have used the cdr2 helix-leucine zipper (HLZ) dimerization domain in a yeast two-hybrid screen to identify an interaction between cdr2 and c-Myc. cdr2 and c-Myc interact specifically, colocalize in Purkinje neuronal cytoplasm, and coimmunoprecipitate from cerebellar extracts. Cotransfection experiments indicate that cdr2 inhibits c-Myc-dependent transcription, most likely by sequestering the protein in the cytoplasm. Finally, we find that the interaction between cdr2 and c-Myc is abrogated by PCD antisera. These data suggest a model whereby the PCD immune response blocks the ability of cdr2 to down–regulate c-Myc, leading to excessive signaling along a pathway known to lead to Purkinje neuronal apoptosis.
Results
cdr2 binds selectively to c-Myc
The amino-terminal 150 amino acids of cdr2 contain an acidic region of 30 amino acids, followed by an extended amphipathic helix of 100 amino acids and a classic leucine zipper dimerization motif. We tested several amino-terminal constructs for activation in a yeast two-hybrid system, and found that constructs containing the cdr2 HLZ domain without the acidic domain were suitable for screening. Because cdr2 is expressed in HeLa cells (Fathallah-Shaykh et al. 1991), we performed a yeast two-hybrid screen of a HeLa cell cDNA library using the cdr2 HLZ domain as bait, and identified c-Myc as a specifically interacting clone (Table 1). This clone encoded the carboxylterminus of c-Myc, a region that includes the HLZ c-Myc interaction domain. To test the specificity of this interaction, we assayed different HLZ constructs for cdr2 amino-terminal binding. cdr2 bound strongly to c-Myc but did not bind constructs expressing Max or bicoid. Thus, cdr2 binds specifically to c-Myc in the yeast two-hybrid system.
Table 1.
Specificity of cdr2 yeast two-hybrid interactions
| Bait
|
GAL4 cDNA
|
LacZ
|
Leu−
|
|---|---|---|---|
| cdr2 | c-Myc | +++ | +++ |
| Bicoid | c-Myc | − | − |
| cdr2 | Max | − | N.D. |
| cdr2 | Mxi1 | − | N.D. |
The EGY48 host strain was transformed with the indicated bait plasmids and B42 activation domain fusion plasmids, and interactions scored by induction of LacZ expression and growth on Leu− media (see Materials and Methods). (N.D.) Not determined.
To confirm these findings, and to demonstrate that the interaction between cdr2 and c-Myc is not dependent on additional yeast proteins, we assayed directly the protein interactions in vitro using GST fusion proteins and 35S-labeled in vitro translation products. 35S-Labeled c-Myc showed equally robust interactions with Max and cdr2, but failed to react with control proteins (Fig. 1A). Conversely, 35S-labeled cdr2 reacted with c-Myc and cdr2 itself, suggesting that the protein may be able to homodimerize through its leucine zipper domain (Fig. 1B). cdr2 failed to interact with Max or with c-Myc deletion constructs lacking the carboxy-terminal HLZ domain (Fig. 1B). These results demonstrate a direct and specific interaction between cdr2 and the c-Myc HLZ domain.
Figure 1.
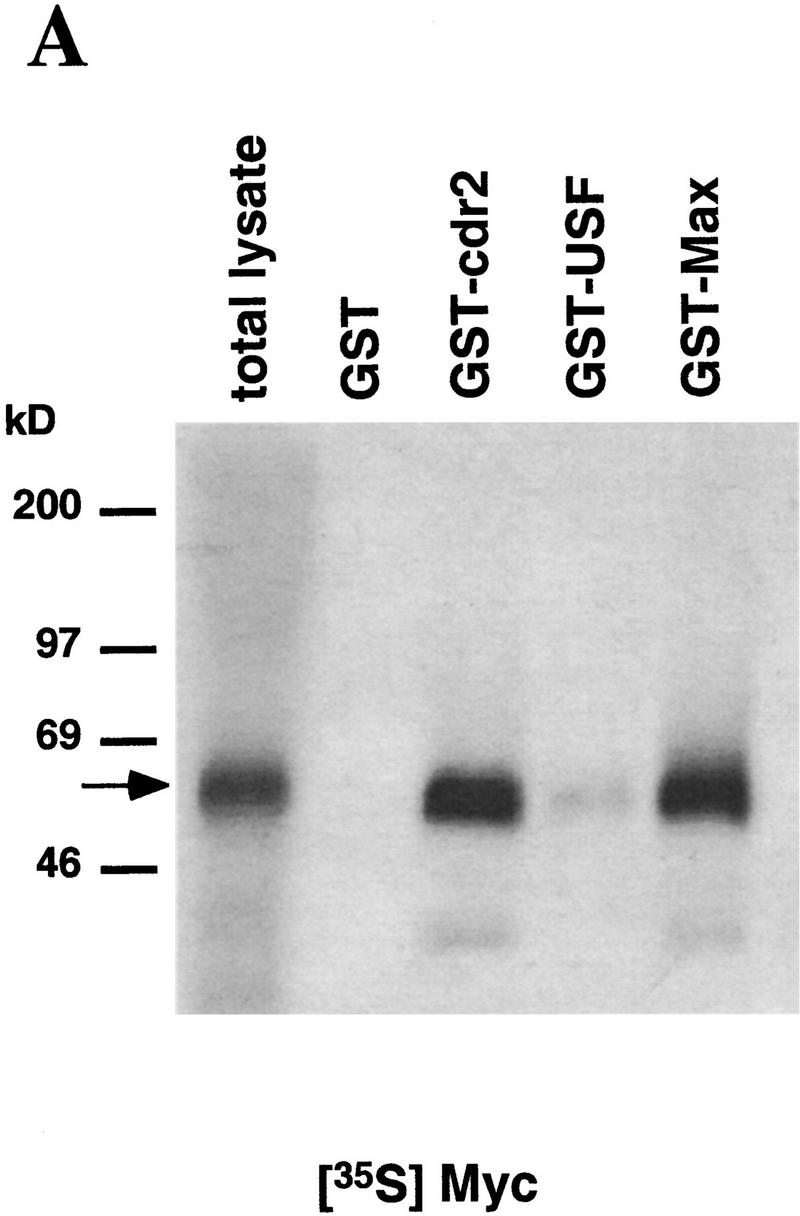

cdr2 and c-Myc interact in vitro. A GST pull-down assay was used to examine the in vitro binding of cdr2 and c-Myc. Immobilized GST fusion proteins were incubated with full–length in vitro translated c-Myc and cdr2. (A) Full–length 35S-labeled c-Myc binds to GST–cdr2 in vitro. c-Myc did not bind to GST alone, and weakly bound to GST–USF, another HLZ protein. GST–Max was used as a positive control. (B) Full–length 35S-labeled cdr2 interacted with a truncated c-Myc fusion protein that contained the HLZ domain (Myc439) and also bound to itself (GST–cdr2). Cdr2 did not bind to GST alone, GST–Max, or Myc353, which lacks the HLZ region. The arrows indicate the size of the full-length 35S-labeled proteins.
Colocalization of cdr2 and c-Myc in cerebellar Purkinje neurons
cdr2 is a cytoplasmic protein found specifically in Purkinje neurons in the cerebellum. Although c-Myc is a nuclear transcription factor, its distribution can vary between the cytoplasm and the nucleus in relation to cellular proliferation and differentiation in cell lines (Vriz et al. 1992; Craig et al. 1993), in vivo (Bai et al. 1994; Wang et al. 1997), and in pathologic specimens of human tumors (Royds et al. 1992; Sasano et al. 1992; Bai et al. 1994). In addition, several studies have reported c-Myc mRNA and protein in developing Purkinje neurons (Ruppert et al. 1986; McCormack et al. 1992; Miyazawa et al. 1993; Takahashi et al. 1993), and perhaps very low levels of c-Myc mRNA in some adult Purkinje neurons (Ruppert et al. 1986).
To examine whether adult cerebellar Purkinje neurons express c-Myc protein and to assess where it is localized, we examined rat brain sections by immunohistochemistry using a panel of c-Myc antibodies, and compared this with the staining obtained with cdr2 antibody. Figure 2 demonstrates that c-Myc and cdr2 show a striking colocalization in the cytoplasm of Purkinje neurons. c-Myc expression was high in sharply demarcated groups of Purkinje neurons, typically groups of 12–16 neurons, and absent or weakly expressed in most (∼80%) of Purkinje neurons (Fig. 2F; data not shown), whereas cdr2 expression was high in all Purkinje neurons (Fig. 2D,E). Double labeling with cdr2 and c-Myc antibodies confirmed that individual Purkinje neurons expressing c-Myc coexpress cdr2 (Fig. 2G–I). The same pattern of c-Myc expression was seen with four different anti-c-Myc antibodies, and one available blocking peptide abrogated binding (Fig. 2A-C; data not shown). As a positive control for nuclear reactivity in these fixation conditions, we stained serial sections for the Nova protein, which is expressed abundantly in the Purkinje cell nucleus (data not shown). The overlap in cdr2 and c-Myc localization is consistent with a direct association of the proteins in vivo.
Figure 2.
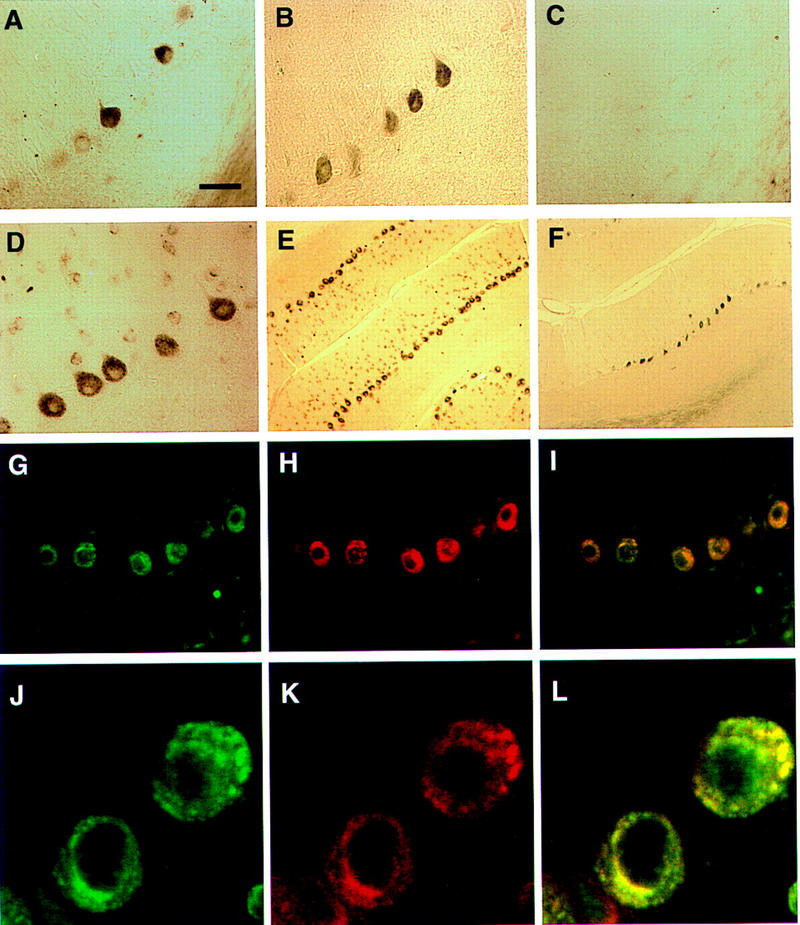
Immunohistochemical colocalization of cdr2 and c-Myc in the cytoplasm of rat cerebellar Purkinje neurons. (A) A section of adult rat cerebellar cortex stained with anti-c-Myc mouse polyclonal antibody N262 shows strong reactivity with some but not all cerebellar Purkinje neurons. (B) Purkinje neuronal reactivity with a different anti-c-Myc antibody, C-19. (C) Reactivity with C-19 after preincubation with immunizing peptide (2 μg/ml). (D) Reactivity with PCD CSF (anti-cdr2) reveals cdr2 immunoreactivity in the cytoplasm of Purkinje cells. (E), Low power view of cerebellum reacted with PCD CSF, reveals that Purkinje neurons are uniformly labeled. (F) A section obtained near (∼24 μm) the section in (E) stained with anti-c-Myc antibody C-19, illustrating isolated groups of c-Myc expressing Purkinje neurons. (G) Immunofluorescent photograph of rat Purkinje neurons reactive with PCD CSF visualized with Cy-2-conjugated anti-human antibody. (H) Same neurons as (G) visualized with anti-c-Myc antibody and with Cy-3-conjugated anti-rabbit antibody. (I) Fusion of images in G and H showing overlap in the expression of both proteins within individual neurons; some neurons in the molecular layer (presumably stellate neurons) stain lightly with cdr2 antibody but not with c-Myc antibody. (J), Confocal laser image of cdr2 expression in a single 2–μm optical section, imaged with Cy-2-conjugated anti-human antibody. (K) Same 2–μm section as J visualized with anti-c-Myc polyclonal C-19 antibody and with Cy-5-conjugated anti-rabbit antibody. (L) Fusion of images in Jand K showing colocalization of both cdr2 and c-Myc within the Purkinje neuronal cytoplasm. (A–D, G–IBar, 20 μm; (E,F) bar, 80 μm; (J–L) bar, 3.4 μm.
We next examined whether cdr2 and c-Myc colocalize by confocal microscopy. Rat cerebellar sections were stained with cdr2 and c-Myc antibodies and protein localization examined by immunofluorescence confocal microscopy. Figures 2J–L demonstrate that in a single confocal optical section, cdr2 and c-Myc immunoreactivities colocalize largely, although not completely, within the Purkinje neuronal cytoplasm. This colocalization is consistent with a direct association of cdr2 and c-Myc in vivo.
cdr2 and c-Myc interact directly
To evaluate further whether cdr2 and c-Myc interact directly, we performed coimmunoprecipitation experiments in transfected cell lines, using either c-Myc antibody or a T7 antibody specific to an epitope tag encoded in the cdr2 expression construct. c-Myc antibody was able to precipitate c-Myc protein and coprecipitate cdr2 protein (Fig. 3A). Conversely, T7 antibody was able to precipitate the T7–tagged cdr2 protein and coprecipitate c-Myc protein (Fig. 3A). We examined the cytoplasmic and nuclear localization of both T7-tagged cdr2 and c-Myc in these cells by Western blot analysis (Fig. 3B). We detected c-Myc protein in both cytoplasm and nuclear fractions, but were able to detect T7–tagged cdr2 protein only in the cytoplasmic fraction. Taken together, these data indicate that expressed cdr2 protein interacts directly with c-Myc in the cytoplasm of heterologous transfected cells.
Figure 3.



Cytoplasmic cdr2 coimmunoprecipitates with c-Myc in vitro and in mouse cerebellum. (A) c-Myc coprecipitates with T7-tagged cdr2 in HTC-75 cells. Expression of T7-tagged cdr2 protein was induced by removing doxycyclin from the media. Cell extracts were run on Western blot before (data not shown) or after immunoprecipitation with the indicated antibodies. Blots were then probed with monoclonal antibodies to c-Myc or the T7-tag, as indicated. Similar coimmunoprecipitations were obtained after cotransfection of Rat-1A cells (data not shown). (B) Western blot analysis of cell extracts after induction of cdr2 expression in HTC-75 cells. Total cell lysates (T), cytoplasmic fractions (C), or nuclear fractions (N) were run on Western blots and probed with either cdr2 antibody or c-Myc antibody as indicated. cdr2 expression is restricted to the cytoplasm; c-Myc is expressed in both compartments. (C) In vivo coimmunoprecipitation of cdr2 and c-Myc. (Lanes 1–4) Mouse cerebellar homogenate precipitated with normal rabbit sera (lane 2), anti-c-Myc rabbit polyclonal antibody (lane 3) or anti-cdr2 PCD patient’s CSF (lane 4). Immune complexes were analyzed by Western blot using PCD (anti-cdr2) patient’s serum. (Lane 1) Western blot of the cerebellar lysate. Two cdr2 immunoreactive proteins are indicated, the lower of which may preferentially be coimmunoprecipitated by c-Myc (lane 3). (Lanes 5–8) Homogenates immunoprecipitated as for lanes 1–4; Western blot probed with anti-c-Myc antibody.
To examine whether cdr2 and c-Myc interact in vivo, we performed coimmunoprecipitation experiments from mouse cerebellum. c-Myc antibodies were able to precipitate c-Myc itself, and to coimmunoprecipitate cdr2 from solubilized whole cerebellar extracts, demonstrating the existence of a cdr2:c-Myc complex in vivo (Fig. 3C). In these SDS–polyacrylamide gels, which were run under highly resolving conditions to separate cdr2 from IgG, cdr2 was detected as a doublet of ∼55 kD (Fig. 3C). Interestingly, c-Myc antibody appeared to immunoprecipitate preferentially the smaller of the two cdr2 bands, suggesting that c-Myc may interact specifically with a unique cdr2 species. Neither of these cdr2 reactive proteins were immunoprecipitated by nonspecific rabbit immunoglobulin (Fig. 3C). Although cdr2 antibody was able to precipitate cdr2 protein from cerebellar extracts, it failed to coimmunoprecipitate c-Myc protein under a variety of conditions (Fig. 3C; data not shown), although we were able to coimmunoprecipitate T7-tagged cdr2 with c-Myc in transfected cells using a T7 monoclonal antibody (Fig. 3A; see Fig. 6 and discussion below).
Figure 6.

Inhibition of cdr2:c-Myc interaction by PCD patients’ sera. The effect of PCD antisera on the cdr2:c-Myc interaction was assessed using GST pull-down assays. GST–cdr2 or GST–Max fusion proteins in solution were immobilized on glutathione–Sepharose beads, preincubated with sera obtained from six different PCD patients or non-PCD control sera, washed, and then mixed with in vitro-translated [35S]-methionine–labeled c-Myc protein. Specifically bound c-Myc protein was assessed by SDS-PAGE and fluorography. Non-PCD sera (from patients with irrelevant paraneoplastic neurologic disorders) do not affect the interactions of c-Myc with either cdr2 or Max. However, PCD patients’ sera inhibited the interaction of c-Myc with cdr2, but not with Max. Quantitation of the ratio of cdr2 to Max protein precipitated in the presence of each serum sample indicated that PCD sera inhibited 35S-labeled c-Myc pull-downs by an average of 5.5-fold (range, 4.9–7.1) relative to the average effect of non-PCD sera.
cdr2 induces redistribution of Myc in the cytoplasm
To begin to evaluate the functional consequences of the cdr2:c-Myc interaction, we transiently expressed cdr2 under the control of a cytomegalovirus (CMV) promoter into N2A cells (a mouse neuroblastoma cell line). In cells that were not transfected with cdr2, no significant amount of cdr2 immunoreactive protein could be detected, and c-Myc showed predominantly nuclear staining (Fig. 4). In cells transfected with cdr2, cdr2 protein was readily detected in the cytoplasm, predominantly in a perinuclear location, although some protein could be detected diffusely in the cytoplasm; no protein was detected within the nucleus. Under these conditions, the Myc protein showed a striking redistribution into the cytoplasm, where it colocalized with cdr2 protein (Fig. 4). We found similar results in experiments in which cdr2 expression was induced under the control of a tetracycline promoter in stably transfected HTC-75 cells (data not shown), the same cell line used for coimmunoprecipitation experiments in Figure 3A. These data indicate that in heterologous transfected cells the cdr2 protein is capable of interacting with and inducing a change in the subcellular localization of c-Myc to the cytoplasm.
Figure 4.
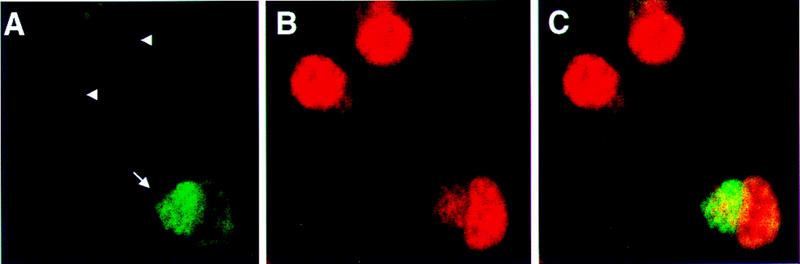
Transfected cdr2 redistributes c-Myc to the cytoplasm of N2A cells where it colocalizes with cdr2 protein. N2A cells were transfected transiently with pcDNA3-cdr2 plasmid, fixed and stained with anti-cdr2 patient CSF (A, green, Cy2) and anti-Myc polyclonal C-19 antibody (B, red, Cy5), and imaged by confocal microscopy (Zeiss). Fusion of images in A and B shows overlap in the cytoplasmic expression of both proteins (C, yellow). Nontransfected cells (arrowheads) show little or no cdr2 staining and nuclear c-Myc expression; transfected cells (arrow) show strong cytoplasmic cdr2 staining and a redistribution of c-Myc reactivity to the cytoplasm, where it colocalizes with cdr2. The same result was seen when a different anti-c-Myc antibody (N262) was used (data not shown).
cdr2 down–regulates the ability of c-Myc to induce transcription in heterologous cells
To determine whether the cdr2:c-Myc interaction is able to alter the ability of c-Myc to induce gene expression, we examined c-Myc–induced transcription of a reporter construct in the presence or absence of cdr2. We transiently transfected Rat 1A fibroblast cell lines with a reporter gene that either had (M4 minCAT) or did not have (minCAT) c-Myc–binding sites (E-box elements) upstream of a CAT reporter gene. In the absence of transfected c-Myc, baseline transcription from endogenous cellular c-Myc proteins leads to specific CAT induction from the M4 minCAT construct (Fig. 5A, lane−; Kretzner et al. 1992). Transfection of a c-Myc–expressing plasmid led to a 2.6–fold increase in E-box–dependent CAT activity, which was suppressed by cotransfection with cdr2 (Fig. 5A).
Figure 5.


cdr2 represses c-Myc transcriptional activity. (A) Rat 1A fibroblasts were transfected transiently with the minCAT or M4minCAT reporter plasmids. Cells were cotransfected with either no additional plasmid (−), a c-Myc expressing plasmid (myc; 0.5 μg SpMyc plasmid), or C-Myc together with a cdr2-expressing plasmid (myc + cdr2; 0.5 μg of SpMyc plasmid plus 0.75 μg of pcDNA3-cdr2). Transfection with c-Myc alone resulted in an average 2.6-fold induction of CAT activity. Cotransfection with cdr2 inhibited the c-Myc-induced CAT activity to near baseline levels. No effect of cdr2 alone on M4CAT expression was seen with up to 1 μg of pcDNA 3-cdr2 (data not shown). The results shown represent the average of two independent experiments, each performed in triplicate (n = 6). Error bars indicate 2 s.d. (B) NIH-3T3 cells were transfected transiently with 1.5 μg of reporter plasmid that did not harbor Myc E-box-binding elements (−E-box; pGL3, Promega) or reporter plasmid that did harbor E-box binding elements (+E-box; pM4luc, a pGL3 derivative), or pM4luc in the presence of 1.0 μg of pCMV-Myc and increasing amounts of pcDNA3-cdr2 plasmid as indicated. Cotransfected cdr2 inhibited c-Myc-dependent luciferase activity in a titratable manner. The results shown represent the average of transfections performed in triplicate, and error bars indicate the standard deviation. Relative transfection efficiency was determined in all experiments by measuring activity from a cotransfected reporter plasmid (see Materials and Methods). No effect of cdr2 alone on pM41uc expression was seen with up to 2 μg of pcDNA3-cdr2 (data not shown).
To confirm and extend these observations, we examined whether titrating increasing amounts of cdr2 yielded a dose-dependent effect on c-Myc–dependent transcription. For these experiments, we assayed transcription using a luciferase reporter construct in NIH-3T3 cells transfected with a Myc–expressing plasmid. We found that a luciferase reporter harboring E-box elements (pM4luc) showed approximately four to five fold more activity than a basal promoter construct (Fig. 5B, lanes 1,2). Cotransfection of increasing concentrations of cdr2 together with the pM4luc reporter led to a linear decrease in c-Myc-dependent transcriptional activity over a fourfold range (Fig. 5B). These results indicate that cdr2 was able to block the action of cotransfected c-Myc to induce E-box–dependent transcription.
PCD disease antisera inhibit the cdr2:c-Myc interaction
We next examined whether PCD antisera could affect the interaction between cdr2 and c-Myc. PCD antisera have been reported to recognize an epitope in the region of the cdr2 HLZ domain (Sakai et al. 1993). Moreover, we were unable to coprecipitate cdr2 and c-Myc from mouse brain or transfected tissue culture cells using PCD antisera (see Fig. 3C), although were able to coprecipitate both proteins using T7–tagged cdr2 (see Fig. 3A). These observations suggested that PCD antisera might inhibit the interaction between cdr2 and c-Myc.
To test this hypothesis, we incubated identical amounts of 35S-labeled c-Myc with GST–Max or GST–cdr2 fusion proteins in the presence of six different PCD disease antisera or six different non-PCD disease control antisera. All six PCD disease antisera significantly inhibited the interaction of cdr2 with c-Myc (Fig. 6) relative to the non-PCD control sera. Moreover, the PCD antisera failed to affect the interaction of Max with c-Myc. Quantitation of these data revealed a 5.5–fold inhibition of the cdr2:c-Myc interaction by PCD antisera, relative to control sera. Comparable results were obtained with PCD cerebrospinal fluid (data not shown). These results indicate that a hallmark of PCD disease antisera is an ability to disrupt the interaction of cdr2 with c-Myc in vitro.
Discussion
cdr2 regulation of the transcription factor c-Myc
We have found that the cytoplasmic paraneoplastic cerebellar degeneration antigen cdr2 binds to c-Myc in vivo and down–regulates c-Myc function in cotransfection assays. Previous studies have described a variety of proteins that bind to and regulate c-Myc transcriptional activity in the nucleus. The canonical c-Myc–binding protein is Max, which binds nuclear c-Myc through its HLZ domain to mediate transcriptional activation (Blackwood et al. 1992). In addition, a series of HLZ proteins, including Mad1, Mxi1, Mad3, Mad4, and Mnt (Ayer et al. 1993; Hurlin et al. 1996,1997), down–regulate c-Myc activity indirectly, by competing with nuclear c-Myc for binding to Max. Other nuclear c-Myc–binding partners have been described, most of which bind outside of the HLZ, including Bin1 (Sakamuro et al. 1996), a protein related to the paraneoplastic breast tumor antigen amphiphysin, and TRRAP, which interact with the c-Myc amino-terminal domain (McMahon et al. 1998), as well as YY1 (Shrivastava et al. 1993) and Miz-1 (Peukert et al. 1997). Other than cdr2 and Max, the only known protein that might interact with the c-Myc HLZ is Nmi, a protein identified in a yeast two-hybrid screen that is expressed in all tissues except brain (Bao and Zervos 1996) and that localizes to the cytoplasm of tumor cell lines (Lebrun et al. 1998).
Our findings suggest that cdr2 acts to down–regulate c-Myc function as a nuclear transcription factor, most likely by a mechanism that involves sequestering c-Myc in the cytoplasm. There is abundant precedent for cytoplasmic proteins sequestering and down–regulating transcription factor activity. Transcription factors belonging to the NFκB/Rel/NFAT family, termed IκB proteins, interact with cytoplasmic regulatory proteins to inhibit nuclear translocation and thereby down–regulate transcription factor function (Schmitz and Baeuerle 1995; Rao et al. 1997). Similarly, cytoplasmic glucocorticoid receptors are believed to bind heat shock proteins, which thereby inhibit their nuclear translocation and transcriptional activation (Groyer et al. 1987).
There have been previous reports that c-Myc protein can be detected in the cytoplasm of certain cell types. c-Myc has been detected previously in the cytoplasm of cell lines (Craig et al. 1993) and tumor samples (Lipponen 1995; Pietilainen et al. 1995; Boni et al. 1998). In neurons, the detection of c-Myc in Purkinje cell cytoplasm parallels the observation that N-Myc, which is expressed in the nuclei of cerebellar Purkinje neurons during development, but localizes to Purkinje cell cytoplasm in adults (Wakamatsu et al. 1993). The colocalization of the cytoplasmic protein cdr2 and c-Myc and the coimmunoprecipitation of these proteins from cerebellar extracts indicates that cdr2 binds to cytoplasmic c-Myc in Purkinje neurons.
Nonetheless, c-Myc presumably functions as a nuclear transcription factor in neurons. Detection of steady–state c-Myc expression in Purkinje neuronal cytoplasm suggests that regulation of its subcellular localization may control c-Myc activity in neurons. One means by which c-Myc localization might be regulated is through control of its interaction with cdr2. Serine phosphorylation of the inhibitor IκB regulates its interaction with cytoplasmic NF-κB, thereby regulating NF-κB nuclear entry and transcriptional activity (DiDonato et al. 1997). Similarly, post-translational modifications of cdr2 could provide a mechanism for the regulated entry of c-Myc into Purkinje neuronal nuclei. However, despite the suggestion of two closely migrating cdr2 species, which may interact differentially with c-Myc (Fig. 3), and one report of in vitro phosphorylation of cdr2 by PKN (Takanaga et al. 1998), we have not yet been able to demonstrate evidence of cdr2 phosphorylation in vivo (W.-Y. Park and R.B. Darnell, unpubl.).
The rapid regulation of basic-leucine zipper transcription factors, such as fos and jun that mediate activity-dependent changes in neuronal gene expression (Morgan et al. 1987; Morgan and Curran 1995), suggests that a similar transient action, albeit by a different mechanism, may underlie c-Myc function in neurons. c-Myc expression in adult neurons previously has not been clearly established, perhaps in part as a result of the transient nature of its expression. We found strong c-Myc expression only in small sets of Purkinje neurons in the cerebellum, and similar small pockets of c-Myc expression in some other areas of the brain [ventral spinal cord, scattered cortical neurons(H. Okano and R. Darnell, unpubl.)]. These groups of neurons may correspond to activated sets of cells, such as the functional microzones of cerebellar Purkinje neurons described physiologically (Ito 1990).
The role of the cdr2:c-Myc interaction in Purkinje neuronal death
The observations that cdr2 inhibits c-Myc–dependent transcriptional activity and that PCD disease antisera disrupt the cdr2:c-Myc interaction lead to a model for the role of cdr2 antibodies in PCD disease pathogenesis. PCD patients harbor high titer cdr2 antibodies in their cerebrospinal fluid (CSF) early in the course of the disease (Peterson et al. 1992; M.L. Albert and J.C. Darnell, unpubl. data), and these antibodies are actively synthesized within the CSF compartment by B cells (Furneaux et al. 1990). In addition, the pathologic hallmark of PCD brains is the destruction of Purkinje neurons (Posner 1995; Verschuuren et al. 1996; Corradi et al. 1997), although the mechanism of cell death is not known. Thus, a correlation can readily be made between the local synthesis of cdr2 antibodies in the brain and Purkinje neuronal degeneration.
There is one particularly relevant animal model of cerebellar degeneration that implicates dysregulation of cell cycle pathways in Purkinje neuronal death. Orr and colleagues found that transgenic mice expressing the SV40 T antigen from the Purkinje neuron-specific promoter pcp-2 developed cerebellar degeneration as a result of Purkinje neuronal apoptosis (Feddersen et al. 1992). Cell death was preceded by an abortive attempt of neurons to enter the cell cycle (Feddersen et al. 1995). These observations have led to the hypothesis that inappropriate cell cycle signals in neurons may mediate neuronal apoptosis (Heintz 1993; Feddersen et al. 1995). The ability of nerve growth factor (NGF) to prevent c-Myc–induced apoptosis in fibroblast cell lines expressing trkA receptor (Ulrich et al. 1998) lends support to the suggestion that c-Myc itself is capable of mediating neuronal apoptosis.
Taken together these observations suggest that antibody-mediated abrogation of the cdr2:c-Myc interaction could contribute to Purkinje neuronal degeneration in paraneoplastic cerebellar degeneration (Fig. 7). All PCD disease antisera target the cdr2 leucine zipper epitope (Sakai et al. 1993; J.P. Corradi, W.-Y. Park, and R.B. Darnell, unpubl.) and disrupt its ability to bind c-Myc in vitro (Fig. 6). This is predicted to antagonize the actions of cdr2 to bind c-Myc in the cytoplasm (Figs. 2, 3, and 6) and down–regulate c-Myc-dependent transcription (Fig. 5), culminating in increased c-Myc activity. The phenotype of Purkinje promoter SV40 T antigen transgenic mice, which activate a signaling pathway closely tied to the c-Myc signaling pathway (Fig. 7), demonstrates that Purkinje neurons respond to aberrant activation of such pathways by inducing apoptotic cell death.
Figure 7.
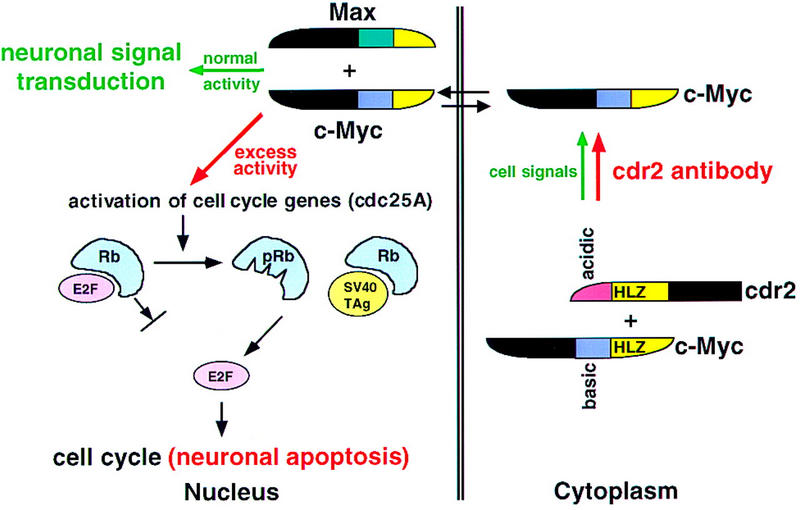
A model for the role of cdr2 antibodies in PCD pathogenesis. Cdr2 and c-Myc form a complex in Purkinje cell cytoplasm. It is presumed that this interaction is normally regulated by signals that allow c-Myc entry into the nucleus, where it acts to promote transcription and transduce neuronal signaling. In PCD, cdr2 antibodies are proposed to disrupt normal regulation of the cdr2:c-Myc interaction, leading to unregulated entry of c-Myc into the nucleus and aberrant c-Myc-induced gene transcription. c-Myc can drive cell cycle pathways or induce apoptosis in dividing cells (Evan and Littlewood 1998). For example, c-Myc activates cdc25A (Galaktionov et al. 1996), and cdc25 family members act on a number of downstream proteins leading to the phosphorylation of the retinoblastoma gene product (pRb) and the release of the transcription factor E2F, a common final step in S–phase entry. c-Myc also can induce ARF (Zindy et al. 1998), a protein that induces p53-mediated apoptosis (Prives 1998). Excess nuclear c-Myc in neurons is proposed to induce inappropriate cell cycle signaling and Purkinje cell apoptosis in a manner analogous to that observed in Purkinje promoter SV40 TAg transgenic mice (Feddersen et al. 1992, 1995). In these mice, T antigen binds Rb, leading to the release of E2F, S–phase entry, and Purkinje apoptosis. We suggest that cdr2 antibody-mediated disruption of the cdr2:c-Myc interaction leads to aberrant nuclear c-Myc activity, activation of these cell cycle and apoptotic pathways, and contributes to the pathogenesis of PCD.
Several unresolved issues regarding PCD bear on this model. First, the role that antibodies play in disease pathogenesis is uncertain. Although there is a nearly absolute correlation between the presence of high titer cdr2 antibodies and the presence of both gynecologic malignancies and Purkinje neuronal degeneration, attempts to reproduce the disease in animals by passive or active immunization with antibodies have failed, and treatments aimed at reducing serum antibody titers are ineffectual. Nonetheless, the invariant presence of CSF cdr2 antibodies in PCD, the active synthesis of cdr2 antibodies within the spinal fluid, and reports that neurons, especially Purkinje neurons, can efficiently take up IgG (Borges et al. 1985; Fabian and Petroff 1987; Graus et al. 1991), emphasize the possibility that antibodies may be involved in disease pathogenesis.
One suggestion from these observations is that cdr2 antibodies are necessary but not sufficient to cause PCD. Recent studies have demonstrated the presence of cdr2-specific cytotoxic T lymphocytes (CTLs) in PCD patients (Albert et al. 1998). CTL activation and killing requires both antigen presentation by antigen–presenting cells (APCs), and recognition of targets by way of major histocompatibility complex 1 (MHC I) molecules (Lenschow et al. 1996). Antigen presentation to T cells within the CSF compartment may be mediated by microglia (probably corresponding to dendritic cells; Flaris et al. 1993). Although neurons have not generally been thought to express MHC I molecules, recently Shatz and colleagues have reported significant MHC I expression in neurons, including Purkinje neurons (Corriveau et al. 1998; C. Shatz, pers. comm.).
However, missing in a scenario of T–cell–mediated neuronal death in PCD are the triggers–events that initially allow APCs to take up and present intracellular Purkinje antigens. Our findings, together with the demonstration that apoptotic bodies from dying cells are an extremely potent antigen stimulus for APCs, suggest a new model of PCD pathogenesis. cdr2 antibody-mediated Purkinje apoptosis, even as a rare event, could trigger APC-mediated CTL activation of cdr2-specific CTLs within the CSF compartment and lead to CTL-mediated elimination of Purkinje neurons.
cdr2:c-Myc interactions in gynecologic tumors
It is unclear why cdr2, whose function appears antagonistic to c-Myc, is coexpressed in gynecologic tumors together with c-Myc, whose activity promotes tumor progression. Although deregulated c-Myc activity in tumor cells clearly drives cell proliferation, c-Myc may also induce tumor cell apoptosis under some circumstances (for review, see Evan and Littlewood 1998). These observations have suggested that c-Myc overexpression is a two-edged sword in tumor cells, and have implied the necessity of tumor cells to express proteins that can antagonize c-Myc–induced apoptosis (Green 1997). The inhibition of c-Myc–dependent transcription by cdr2 suggests that cdr2 may attenuate c-Myc function in vivo. It will be of interest to examine whether a common function for cdr2 in tumor cells and neurons is to inhibit the ability of c-Myc to induce apoptosis. The importance of understanding this interaction is underscored by the observation that cdr2 expression is induced in large numbers of gynecologic tumors (>60% ovarian tumors, and >25% of breast tumors; R.B. Darnell et al., unpubl.).
Materials and methods
Yeast two-hybrid screen
Dr. Roger Brent (Fox Chase Cancer Center, Philadelphia, PA) kindly provided a pJG4-5-based HeLa cDNA library encoding B42 activation domain fusion proteins under control of the Gal1 promoter. The host strain for all assays was EGY48 (MATa trp1 ura3 his3 LEU2::pLexAop6–LEU2), in which the endogenous LEU2 gene has been replaced by a LEU2 reporter harboring six LexA–binding sites. The cdr265-140 bait was tested for its ability to activate the LEU2 reporter gene independently and to enter the nucleus before library screening. Specificity of the yeast two-hybrid interaction was tested in yeast by the amount of growth on Leu− media and β-galactosidase (β-gal) expression. Significant growth and β-gal expression were evident when c-Myc was present with the LexA/cdr265-140 bait construct in the presence of galactose but not glucose. Similarly, there was no interaction of c-Myc with a Drosophila bicoid bait construct. Conversely, cdr265-140 interacted with c-Myc but not Max or Mxi1 constructs (data not shown). pJG4-5 plasmids encoding Max and Mxi1 activation domain fusion proteins were kindly provided by Dr. Erica Golemis (Fox Chase Cancer Center, Philadelphia, PA).
Recombinant proteins, in vitro translation, and in vitro–binding assay
Plasmids encoding GST fusion with Max, upstream stimulating factor (USF), Myc353 (c-Myc amino acids 250–353) and Myc439 (c-Myc amino acids 250–439), and a plasmid containing the full–length cDNA of mouse c-Myc were kindly provided by Dr. K. Calame (Shrivastava et al. 1993).
Full–length mouse c-Myc and mouse cdr2 RNAs were transcribed in vitro using T7 RNA polymerase (Promega), and those RNAs were translated in vitro using rabbit reticulocyte lysate system (Promega) with [35S-L]-methionine (Amersham). All fusion proteins were expressed in bacteria and affinity purified with glutathione–Sepharose (Pharmacia). The GST–cdr2 fusion protein containing amino acids 16–192 of human cdr2 was also purified using glutathione–Sepharose affinity chromatography. In vitro binding assays were performed essentially as described (Harper et al. 1993). Briefly, GST fusion proteins were immobilized to glutathione–Sepharose and washed with binding buffer [50 mm Tris HCl (pH 7.5), 120 mm NaCl, 2 mm EDTA, 0.1% NP-40, 1 mm NaF, 2 μg/ml aprotinin, 100 μg/ml PMSF]. Labeled proteins were incubated with immobilized fusion proteins and after washing unbound proteins, the samples were separated by SDS-PAGE and analyzed by fluorography.
Immunohistochemical staining
Adult rats (male Sprague-Dawley, ∼3 months, ∼280 grams) were used. Rat brains were perfused with formalin/PBS, paraffin embedded, and sectioned at 14 μm. After deparaffinization, sections were boiled in 0.01 m citric acid (pH 6.0) in a microwave for 10 min. All immune reactions were preceded by a blocking step in PBS/0.05% Triton X-100/2% normal horse serum, and were carried out at 4°C overnight. The following polyclonal antisera were used at 1.0 μg/ml: anti-c-Myc antibody N262 (Santa Cruz Biotechnology), C-19 (Santa Cruz Biotechnology), and anti-human c-Myc polyclonal antibody (Upstate Biotechnology; data not shown).
In addition c-Myc reactivity was seen in sections made from rat brains perfused with 4% paraformaldehyde/PBS, postfixed in the same fixative at 4°C for 4 hr and stored in 10% sucrose/PBS overnight (data not shown). Floating sections (30 μm) were blocked in PBS/0.05% Triton X-100/2% normal horse serum (blocking buffer). c-Myc monoclonal antibody C-33 (Santa Cruz Biotechnology) diluted in blocking buffer was used at 1 μg/ml, and PND antisera was used at 1:200 dilution.
All sections were washed with PBS/0.05% Triton X-100, incubated with biotinylated secondary antibodies (Vector Laboratories) and washed again. Signals were enhanced by addition of horseradish peroxidase (HRP)-conjugated avidin (Vector Laboratories), developed with diaminobenzidene (DAB) in the presence of H2O2, and visualized by light microscopy using a Zeiss Axioplan microscope.
Immunofluorescent staining
Tissue culture cells were grown in chamber slides and fixed with 2% paraformaldehyde in PBS. After permeabilization using 0.5% NP-40, cells were blocked in 0.2% gelatin and 0.5% bovine serum albumin in PBS. Primary anti-cdr2 CSF from patients (1:10 dilution) and anti-Myc (C-19, N262, Santa Cruz Laboratory) were incubated with the cells. Each protein was visualized using Cy2 anti-human IgG and Cy5 anti-rabbit IgG (Jackson ImmunoResearch) by confocal microscopy (Zeiss).
For immunofluorescent staining of rat brain (Fig. 2G–I), tissue sections were fixed in formalin/PBS and paraffin embedded as described above, and visualization was performed using Cy2 anti-human IgG or Cy3 anti-rabbit IgG (Jackson ImmunoResearch) followed by fluorescence microscopy (Zeiss Axioplan using a Hamamatsu ORCA CCD camera). Confocal immunofluorescent microscopy (Figs. 2J-K and 4) was performed using a Zeiss confocal microscope and Cy2 anti-human IgG or Cy5 anti-rabbit IgG (Jackson ImmunoResearch). We confirmed that sections stained with cdr2/Cy2 gave no signal when detected at the wavelength of Cy5 in the absence c-Myc/Cy5 antibody; similarly, c-Myc/Cy5 staining gave no signal when detected at the wavelength of Cy2 in the absence of cdr2/Cy2 antibody (data not shown).
Immunoprecipitation and Western blot
Mouse cerebellum was homogenized in LS lysis buffer [20 mm HEPES (pH 7.5), 100 mm KCl, 10 mm MgCl2, 5 mm dithiothreitol (DTT), 0.2% NP-40, 2 μg/ml aprotinin, 0.2 mm phenylmethylsulfonyl fluoride (PMSF)] using a dounce homogenizer. Homogenates were sonicated briefly and soluble fractions were collected after spinning. The lysates were precleared with protein A–Sepharose and normal rabbit sera, and then precipitated with PCD CSF or anti-human c-Myc rabbit polyclonal antibody (Upstate Biotechnology). Tissue culture nuclear and cytoplasmic fractions were prepared by hypotonic lysis and centrifugation. Immunoprecipitated proteins were separated by SDS-PAGE and transferred to nitrocellulose filters. Blotted proteins were analyzed using anti-cdr2 sera from patients or anti-c-Myc mouse monoclonal antibody (C-33, Santa Cruz Biotechnology). The identification of c-Myc protein on Western blots was confirmed with two additional c-Myc antibodies (C-19 polyclonal and C-8 monoclonal antibodies; Santa Cruz Biotechnology). Each protein was visualized in the blot using HRP-conjugated secondary IgG and enhanced chemiluminescence kit (Amersham). In vivo coimmunoprecipitation experiments were repeated in three independent experiments.
Transcription assay (CAT assay and luciferase assay)
Dr. R. Eisenman (FHCRC, Seattle, WA) kindly provided the pSpMyc and (+/−) M4minCAT plasmids used in transfection assays. For CAT assays, transfected cells were lysed in 0.25 m Tris HCl (pH 7.5) by repeated freezing and thawing. Cytoplasmic extracts were mixed with CAT assay buffer [2 μCi/ml 14C-chloramphenicol, 0.25 mg/ml n-butyryl CoA, 16.6 mm Tris HCl (pH 8.0)], and incubated at 37°C. To isolate the acetylated 14C-chloramphenicol samples were extracted by 2:1 mixture of tetramethyl pentadecane (TMPD)/xylene. Radioactivity in the extracted organic phase was measured using a liquid scintillation counter.
Luciferase assays were done using a luciferase assay kit (Promega) as described by the manufacturer. Transfection efficiency was normalized by measuring the β-gal activity derived from cotransfection with a CMV–lacZ reporter construct, and, in some instances normalizing the number of cells transfected using a pEGFP reporter (Clontech). To measure the β-gal activity, cytoplasmic extracts were mixed with buffer A [100 mm NaH2PO4 (pH 7.5), 10 mm KCl, 1 mm MgSO4, 50 mm β-mercaptoethanol] and 4 mg/ml O-nitrophenyl d-β-galactopyranoside (ONPG). After incubation, reactions were stopped by adding 1 m Na2CO3 and the absorbance at 420 nm was measured.
Competition assay
GST–cdr2 or GST–Max fusion proteins in solution were immobilized on glutathione–Sepharose beads and incubated with patients’ sera. After washing, in vitro–translated 35S-labeled c-Myc proteins were added to each tube. After washing again, proteins present on GST–cdr2 or GST–Max Sepharose beads were analyzed by SDS-PAGE and fluorography.
cdr2 expression in cell lines
N2A cells were obtained from the ATCC (no. CCL-131). N2A cells were grown in DMEM supplemented with glutamine, 10% fetal bovine serum, and antibiotics, and were transfected by using Fugene 6 (Roche). HTC-75 cells and the HT1080-derived tet-suppressive cell line were provided by Dr. T. de Lange (van Steensel and de Lange, 1997[0015]). These cells were grown in DME media supplemented with 10% fetal bovine serum and antibiotics. Full–length mouse cdr2 cDNA linked to the T7 tag sequence was cloned into pUHD10-3 plasmid. DNA was transferred to HTC-75 cells using lipofectamine (GIBCO-BRL) as described by the manufacturer. Stable transformants were selected and cloned by adding Geneticin (GIBCO-BRL) to 400 μg/ml and examining cdr2 expression in individual clones by Western blot analysis.
Acknowledgments
We authors are grateful to Drs. R. Eisenman and K. Calame for antibodies and expression vectors, to T. deLange for HTC-75 cells and to M. Young for helpful discussions. We thank members of the laboratory and Nathaniel Heintz for critical reading of the manuscript. This work was supported by an American Cancer Society Research Award RPG-98-033-CIM, Irma T. Hirschl Career Scientist Award (R.B.D.), National Research Service Award Training Grant GM07982-12 (J.P.C.), the Korea Research Foundation (W.-Y. P.), and the AT Children’s Project (H.J.O.). Clinical specimens were obtained with support from the Rockefeller University Hospital General Clinical Research Center grant M01 RR00102 following approval by institutional International Review Board RDA-148.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL darnelr@rockvax.rockefeller.edu; FAX (212) 327-7147
References
- Albert ML, Darnell JC, Bender A, Francisco LM, Bhardwaj N, Darnell RB. Tumor-specific killer cells in paraneoplastic cerebellar degeneration. Nature Med. 1998;4:1321–1324. doi: 10.1038/3315. [DOI] [PubMed] [Google Scholar]
- Ayer DE, Kretzner L, Eisenman RN. Mad: A heterodimeric partner for Max that antagonizes Myc transcriptional activity. Cell. 1993;72:211–222. doi: 10.1016/0092-8674(93)90661-9. [DOI] [PubMed] [Google Scholar]
- Bai MK, Costopoulos JS, Christoforidou BP, Papadimitriou CS. Immunohistochemical detection of the c-myc oncogene product in normal, hyperplastic and carcinomatous endometrium. Oncology. 1994;51:314–319. doi: 10.1159/000227356. [DOI] [PubMed] [Google Scholar]
- Bao J, Zervos AS. Isolation and characterization of Nmi, a novel partner of Myc proteins. Oncogene. 1996;12:2171–2176. [PubMed] [Google Scholar]
- Blackwood EM, Luscher B, Eisenman RN. Myc and Max associate in vivo. Genes & Dev. 1992;6:71–80. doi: 10.1101/gad.6.1.71. [DOI] [PubMed] [Google Scholar]
- Boni R, Bantschapp O, Muller B, Burg G. c-myc is not useful as prognostic immunohistochemical marker in cutaneous melanoma. Dermatology. 1998;196:288–291. doi: 10.1159/000017922. [DOI] [PubMed] [Google Scholar]
- Borges LF, Elliott PJ, Gill R, Iversen SD, Iversen LL. Selective extraction of small and large molecules from the cerebrospinal fluid by Purkinje neurons. Science. 1985;228:346–348. doi: 10.1126/science.2580350. [DOI] [PubMed] [Google Scholar]
- Corradi JP, Yang CW, Darnell JC, Dalmau J, Darnell RB. A post-transcriptional regulatory mechanism restricts expression of the paraneoplastic cerebellar degeneration antigen cdr2 to immune privileged tissues. J Neurosci. 1997;17:1406–1415. doi: 10.1523/JNEUROSCI.17-04-01406.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21:505–520. doi: 10.1016/s0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- Craig RW, Buchan HL, Civin CI, Kastan MB. Altered cytoplasmic/nuclear distribution of the c-myc protein in differentiating ML-1 human myeloid leukemia cells. Cell Growth Diff. 1993;4:349–357. [PubMed] [Google Scholar]
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Dropcho E, Chen Y, Posner J, Old L. Cloning of a brain protein identified by autoantibodies from a patient with paraneoplastic cerebellar degeneration. Proc Natl Acad Sci. 1987;84:1–5. doi: 10.1073/pnas.84.13.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1321. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- Fabian RH, Petroff G. Intraneuronal IgG in the central nervous system: Uptake by retrograde axonal transport. Neurology. 1987;37:1780–1784. doi: 10.1212/wnl.37.11.1780. [DOI] [PubMed] [Google Scholar]
- Fathallah-Shaykh H, Wolf S, Wong E, Posner J, Furneaux H. Cloning of a leucine-zipper protein recognized by the sera of patients with antibody-associated paraneoplastic cerebellar degeneration. Proc Natl Acad Sci. 1991;88:3451–3454. doi: 10.1073/pnas.88.8.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT. Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron. 1992;9:955–966. doi: 10.1016/0896-6273(92)90247-b. [DOI] [PubMed] [Google Scholar]
- Feddersen RM, Clark HB, Yunis WS, Orr HT. In vivo viability of postmitotic Purkinje neurons requires pRb family member function. Mol Cell Neurosci. 1995;6:153–167. doi: 10.1006/mcne.1995.1014. [DOI] [PubMed] [Google Scholar]
- Flaris NA, Densmore TL, Molleston MC, Hickey WF. Characterization of microglia and macrophages in the central nervous system of rats: Definition of the differential expression of molecules using standard and novel monoclonal antibodies in normal CNS and in four models of parenchymal reaction. Glia. 1993;7:34–40. doi: 10.1002/glia.440070108. [DOI] [PubMed] [Google Scholar]
- Furneaux HF, Reich L, Posner JB. Autoantibody synthesis in the central nervous system of patients with paraneoplastic syndromes. Neurology. 1990;40:1085–1091. doi: 10.1212/wnl.40.7.1085. [DOI] [PubMed] [Google Scholar]
- Galaktionov K, Chen X, Beach D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature. 1996;382:511–517. doi: 10.1038/382511a0. [DOI] [PubMed] [Google Scholar]
- Graus F, Illa I, Agusti M, Ribalta T, Cruz-Sanchez F, Juarez C. Effect of intraventricular injection of an anti-Purkinje cell antibody (anti-Yo) in a guinea pig model. J Neurol Sci. 1991;106:82–87. doi: 10.1016/0022-510x(91)90198-g. [DOI] [PubMed] [Google Scholar]
- Green DR. A Myc-induced apoptosis pathway surfaces. Science. 1997;278:1246–1247. doi: 10.1126/science.278.5341.1246. [DOI] [PubMed] [Google Scholar]
- Groyer A, Schweizer-Groyer G, Cadepond F, Mariller M, Baulieu EE. Antiglucocorticosteroid effects suggest why steroid hormone is required for receptors to bind DNA in vivo but not in vitro. Nature. 1987;328:624–626. doi: 10.1038/328624a0. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Heintz N. Cell death and the cell cycle: A relationship between transformation and neurodegeneration? Trends Biochem Sci. 1993;18:157–159. doi: 10.1016/0968-0004(93)90103-t. [DOI] [PubMed] [Google Scholar]
- Hida C, Tsukamoto T, Awano H, Yamamoto T. Ultrastructural localization of anti-Purkinje cell antibody-binding sites in paraneoplastic cerebellar degeneration. Arch Neurol. 1994;51:555–558. doi: 10.1001/archneur.1994.00540180033010. [DOI] [PubMed] [Google Scholar]
- Hurlin PJ, Queva C, Koskinen PJ, Steingrimsson E, Ayer DE, Copeland NG, Jenkins NA, Eisenman RN. Mad3 and Mad4: Novel Max-interacting transcriptional repressors that suppress c-myc dependent transformation and are expressed during neural and epidermal differentiation. EMBO J. 1996;15:2030. [PMC free article] [PubMed] [Google Scholar]
- Hurlin PJ, Queva C, Eisenman RN. Mnt, a novel Max-interacting protein is coexpressed with Myc in proliferating cells and mediates repression at Myc binding sites. Genes & Dev. 1997;11:44–58. doi: 10.1101/gad.11.1.44. [DOI] [PubMed] [Google Scholar]
- Ito M. A new physiological concept on cerebellum. Rev Neurol. 1990;146:564–569. [PubMed] [Google Scholar]
- Kretzner L, Blackwood EM, Eisenman RN. Myc and max proteins possess distinct transcriptional activities. Nature. 1992;359:426–429. doi: 10.1038/359426a0. [DOI] [PubMed] [Google Scholar]
- Lebrun SJ, Shpall RL, Naumovski L. Interferon-induced upregulation and cytoplasmic localization of Myc- interacting protein Nmi. J Interferon Cytokine Res. 1998;18:767–771. doi: 10.1089/jir.1998.18.767. [DOI] [PubMed] [Google Scholar]
- Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- Lipponen PK. Expression of c-myc protein is related to cell proliferation and expression of growth factor receptors in transitional cell bladder cancer. J Pathol. 1995;175:203–210. doi: 10.1002/path.1711750208. [DOI] [PubMed] [Google Scholar]
- McCormack MA, Rosen KM, Villa-Komaroff L, Mower GD. Changes in immediate early gene expression during postnatal development of cat cortex and cerebellum. Brain Res Mol Brain Res. 1992;12:215–223. doi: 10.1016/0169-328x(92)90087-r. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998;94:363–374. doi: 10.1016/s0092-8674(00)81479-8. [DOI] [PubMed] [Google Scholar]
- Miyazawa A, Takahashi M, Horikoshi T, Yoshioka T. Immunohistochemical analysis of signal transduction system in developing rat Purkinje cell by using antibodies for signaling molecules. Ann N Y Acad Sci. 1993;707:533–535. doi: 10.1111/j.1749-6632.1993.tb38119.x. [DOI] [PubMed] [Google Scholar]
- Morgan JI, Curran T. Immediate-early genes: Ten years on. Trends Neurosci. 1995;18:66–67. [PubMed] [Google Scholar]
- Morgan JI, Cohen DR, Hempstead JL, Curran T. Mapping patterns of c-fos expression in the central nervous system after seizure. Science. 1987;237:192–197. doi: 10.1126/science.3037702. [DOI] [PubMed] [Google Scholar]
- Peterson K, Rosenblum MK, Kotanides H, Posner JB. Paraneoplastic cerebellar degeneration. I. A clinical analysis of 55 anti-Yo antibody-positive patients. Neurology. 1992;42:1931–1937. doi: 10.1212/wnl.42.10.1931. [DOI] [PubMed] [Google Scholar]
- Peukert K, Staller P, Schneider A, Carmichael G, Hanel F, Eilers M. An alternative pathway for gene regulation by Myc. EMBO J. 1997;16:5672–5686. doi: 10.1093/emboj/16.18.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietilainen T, Lipponen P, Aaltomaa S, Eskelinen M, Kosma VM, Syrjanen K. Expression of c-myc proteins in breast cancer as related to established prognostic factors and survival. Anticancer Res. 1995;15:959–964. [PubMed] [Google Scholar]
- Posner JB. Neurologic complications of cancer. Philadelphia, PA: F.A. Davis; 1995. [Google Scholar]
- Prives C. Signaling to p53: Breaking the MDM2-p53 circuit. Cell. 1998;95:5–8. doi: 10.1016/s0092-8674(00)81774-2. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: Regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Royds JA, Sharrard RM, Wagner B, Polacarz SV. Cellular localisation of c-myc product in human colorectal epithelial neoplasia. J Pathol. 1992;166:225–233. doi: 10.1002/path.1711660304. [DOI] [PubMed] [Google Scholar]
- Ruppert C, Goldowitz D, Wille W. Proto-oncogene c-myc is expressed in cerebellar neurons at different developmental stages. EMBO J. 1986;5:1897–1901. doi: 10.1002/j.1460-2075.1986.tb04442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Mitchell DJ, Tsukamoto T, Steinman L. Isolation of a complementary DNA clone encoding an autoantigen recognized by an anti-neuronal antibody from a patient with paraneoplastic cerebellar degeneration. Ann Neurol. 1990;28:692–698. doi: 10.1002/ana.410280515. [DOI] [PubMed] [Google Scholar]
- Sakai K, Ogasawara T, Hirose G, Jaeckle KA, Greenlee JE. Analysis of autoantibody binding to 52-kd paraneoplastic cerebellar degeneration-associated antigen expressed in recombinant proteins. Ann Neurol. 1993;33:373–380. doi: 10.1002/ana.410330407. [DOI] [PubMed] [Google Scholar]
- Sakamuro D, Elliott KJ, Wechsler-Reya R, Prendergast GC. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nature Genet. 1996;14:69–77. doi: 10.1038/ng0996-69. [DOI] [PubMed] [Google Scholar]
- Sasano H, Nagura H, Silverberg SG. Immunolocalization of c-myc oncoprotein in mucinous and serous adenocarcinomas of the ovary. Hum Pathol. 1992;23:491–495. doi: 10.1016/0046-8177(92)90125-m. [DOI] [PubMed] [Google Scholar]
- Schmitz ML, Baeuerle PA. Multi-step activation of NF-kappa B/Rel transcription factors. Immunobiology. 1995;193:116–127. doi: 10.1016/s0171-2985(11)80534-6. [DOI] [PubMed] [Google Scholar]
- Shrivastava A, Saleque S, Kalpana GV, Artandi S, Goff SP, Calame K. Inhibition of transcriptional regulator Yin-Yang-1 by association with c-Myc. Science. 1993;262:1889–1892. doi: 10.1126/science.8266081. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Toyoshima S, Miyazawa A, Horikoshi T, Yoshioka T. Regulation of c-MYC protein expression in the developing rat cerebellum by phosphoinositide turnover. Biochem Biophys Res Commun. 1993;197:278–286. doi: 10.1006/bbrc.1993.2472. [DOI] [PubMed] [Google Scholar]
- Takanaga H, Mukai H, Shibata H, Toshimori M, Ono Y. PKN interacts with a paraneoplastic cerebellar degeneration-associated antigen, which is a potential transcription factor. Exp Cell Res. 1998;241:363–372. doi: 10.1006/excr.1998.4060. [DOI] [PubMed] [Google Scholar]
- Ulrich E, Duwel A, Kauffmann-Zeh A, Gilbert C, Lyon D, Rudkin B, Evan G, Martin-Zanca D. Specific TrkA survival signals interfere with different apoptotic pathways. Oncogene. 1998;16:825–832. doi: 10.1038/sj.onc.1201842. [DOI] [PubMed] [Google Scholar]
- van Steensel B, de Lange T. Control of telomere length by the human telomeric protein TRF1. Nature. 1997;385:740–743. doi: 10.1038/385740a0. [DOI] [PubMed] [Google Scholar]
- Verschuuren J, Chuang L, Rosenblum MK, Lieberman F, Pryor A, Posner JB, Dalmau J. Inflammatory infiltrates and complete absence of Purkinje cells in anti-Yo-associated paraneoplastic cerebellar degeneration. Acta Neuropathol. 1996;91:519–525. doi: 10.1007/s004010050460. [DOI] [PubMed] [Google Scholar]
- Vriz S, Lemaitre JM, Leibovici M, Thierry N, Mechali M. Comparative analysis of the intracellular localization of c-Myc, c-Fos, and replicative proteins during cell cycle progression. Mol Cell Biol. 1992;12:3548–3555. doi: 10.1128/mcb.12.8.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamatsu Y, Watanabe Y, Shimono A, Kondoh H. Transition of localization of the N-Myc protein from nucleus to cytoplasm in differentiating neurons. Neuron. 1993;10:1–9. doi: 10.1016/0896-6273(93)90236-k. [DOI] [PubMed] [Google Scholar]
- Wang Y, Toury R, Hauchecorne M, Balmain N. Expression and subcellular localization of the Myc superfamily proteins: c-Myc, Max, Mad1 and Mxi1 in the epiphyseal plate cartilage chondrocytes of growing rats. Cell Mol Biol. 1997;43:175–188. [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]