

Amyotrophic lateral sclerosis (ALS) is a paralytic and usually fatal disorder caused by motor neuron degeneration in the brain and spinal cord. Most cases of ALS are sporadic (SALS), but about 5–10% are familial (FALS). Mutations in superoxide dismutase 1 (SOD1) 1,2, TAR DNA-binding protein (TDP43) 3,4 and fused in sarcoma/translated in liposarcoma (FUS/TLS) 5,6 account for approximately 30% of classic FALS. Mutations in several other genes have also been reported as rare causes of ALS or ALS-like syndromes 7–15. The causes for the rest of familial ALS and the vast majority of sporadic ALS are unknown. Despite extensive studies of previously identified ALS-causing genes, the pathogenic mechanism underlying motor neuron degeneration in ALS remains largely obscure. Dementia, usually of the frontotemporal lobar type (FTD), may occur in some ALS cases. It is unclear if ALS and dementia share common etiology and pathogenesis in ALS/dementia. Here, we show that mutations in UBQLN2, which encodes a ubiquitin-like protein, ubiquilin2, cause dominantly inherited chromosome X-linked ALS and ALS/dementia. We describe novel ubiquilin2 pathology in the spinal cords of ALS cases and in the brains of ALS/dementia cases with or without UBQLN2 mutations. Ubiquilin2 is a member of the ubiquilin family (ubiquilins), which regulate degradation of ubiquitinated proteins. Functional analysis showed that mutations in UBQLN2 lead to an impairment of protein degradation. Our findings, therefore, link abnormalities in ubiquilin2 to defects in the protein degradation pathway, abnormal protein aggregation and neurodegeneration, implying a common pathogenic mechanism that can be exploited for therapeutic intervention.
We identified a five-generation family (Family #186) with ALS, including 19 affected individuals (Supplementary information). The disease is transmitted in a dominant fashion with reduced penetrance in females. Mutations in the known ALS-linked genes were excluded. No evidence of genetic linkage was found with genome-wide set of autosomal microsatellite markers. There was no evidence of male-to-male transmission of the disease, so we screened the family with markers from the X chromosome. Linkage was established with several X chromosome microsatellite markers, with the highest two-point LOD score of 5.0 with marker DXS9736 at θ=0 (Supplementary Table 1). Detailed mapping with dense microsatellite markers and Illumina’s Sentrix HumanHap300 Genotyping BeadChip defined the disease-causing gene in a 21.3Mb minimum candidate region (MCR) between markers rs6417786 and DXS1275, which is located in the pericentric region from Xp11.23 to Xq13.1.
No additional large ALS families without male to male transmission were available to us to narrow down the MCR. We therefore focused on finding the causative gene in Family #186. Of the 206 genes in this MCR, 191 genes were protein coding. Genes in this MCR were analyzed based on their expression profile, function, structure and potential relevance of their encoded proteins to disease. Forty-one genes were sequenced and a unique mutation in UBQLN2 was identified. This mutation, c.1490C>A, is predicted to result in an amino acid substitution of proline by histidine at codon 497 (P497H) (Fig. 1a). The c.1490C>A mutation co-segregated with the disease in this large X-ALS pedigree (Fig. 1a). This mutation was not present in the SNP database nor was it present in 928 ethnically matched control samples (representing 1332 X chromosomes).
Fig. 1.

Mutations of UBQLN2 in patients with ALS and ALS/dementia. (a) A mutation, c.1490C>A, resulting in p.P497H, was identified in a large family with ALS (F#186). This family was used to map X-chromosome-linked ALS. The pedigree is shown on the left and sequences are shown on the right. The wild-type sequence is shown in the upper panel. A representative hemizygous mutation in a male patient (V3) is shown in the lower panel. All of the affected members whose DNA samples were available for sequencing analysis had the mutation. Two obligate carriers (III4 and IV2) were identified to have the same mutation. For simplicity and clarity, more than one unaffected individual of both genders is represented by a single diamond and more than one unaffected male individual is represented by a single square. Individuals with mutation in the UBQLN2 are labeled by (m) and those without mutation are labeled by (n). (b) A mutation c.1516C>A (p.P506T) was identified in F#6316. Shown in the right lower panel is a heterozygous mutation from a female obligate carrier (II1). (a–b) Probands are indicated with arrows and patients with dementia are indicated with asterisks. (c) Evolutionary conservation of amino acids in the mutated region of ubiquilin2 in different species. Comparison of human (H. sapiens) ubiquilin and its orthologues in chimpanzee (P. troglodytes), dog (C. lupus familiaris), cattle (B. taurus), mouse (M. musculus) and rat (R. norvegicus). Amino acids identical to human UBQLN2 are in black letters and non-identical ones are denoted in red letters. The positions of the C-terminal amino acids are shown on the right. The mutated amino acids are indicated by arrows on the top. (d) Predicted structural and functional domains of ubiquilin2. Ubiquilin2 is a protein of 624 amino acids. Predicted structural and functional domains include a UBL (ubiquitin-like domain, 33–103), four STI1 (heat shock chaperonin-binding motif), a 12 PXX repeats (491–526) and a UBA (ubiquitin-associated domain). ALS- and ALS/dementia-linked mutations are clustered in the 12 PXX repeats.
UBQLN2 is an intronless gene. To test if mutations of UBQLN2 are causative for other ALS patients, we analyzed 188 probands from families with ALS or ALS/dementia, but without male-to-male transmission. Mutations in SOD1, TDP43 and FUS were excluded in this cohort. The sequenced region covered the entire coding sequence (Materials and Methods). We found four other UBQLN2 mutations in four unrelated families, including c.1489C>T (p.P497S), c.1516C>A (p.P506T), c.1525C>T (p.P509S) and c.1573C>T (p.P525S) (Fig. 1 and Supplementary Fig.1). All the amino acids residues at the mutated sites are conserved (Fig. 1c). None of these mutations were present in the SNP database and 928 control samples. Remarkably, all the five ALS-linked UBQLN2 mutations identified in this study involved proline residues within a unique PXX repeat region (Fig. 1c and 1d).
Clinical data were obtained from 40 individuals in the five families with UBQLN2 mutations, including 35 patients and five obligate carriers. We estimated a penetrance of approximately 90% by the age of 70 years. The age of onset of the disease ranged from 16 to 71 years. A significant difference in age at onset was noted between male and female patients, with male patients having earlier age of onset (33.9 ± 14.0 vs. 47.3 ± 10.8 years, p=0.003, two-tailed Student t-test) (Supplementary Table 2). However, differences in the duration of the disease were not statistically significant (43.1 ± 42.1 vs. 48.5 ± 19.9 months, p=0.61). Eight patients with both ALS and dementia were identified. Dementia in these patients was similar to FTD type, including abnormalities in both behavior and executive function. The dementia was progressive, and eventually global in most ALS/dementia patients. In some cases the dementia preceded motor symptoms, but all patients eventually developed motor disability. Pathological analysis of spinal cord autopsy samples from two patients with either P497H or P506T mutation revealed axonal loss in the corticospinal tract, loss of anterior horn cells, and astrocytosis in the anterior horn of the spinal cord (Supplementary Fig. 2).
Protein aggregates/inclusions have been recognized as a pathological hallmark in several neurodegenerative disorders, such as extracellular amyloid-beta plaques and intracellular tau neurofibrillary tangles in Alzheimer disease (AD), and α-synuclein-containing Lewy bodies in Parkinson disease (PD) 16. In ALS protein aggregates/inclusions are most common in spinal motor neurons, and are typically skein-like in morphology. These ubiquitin-positive inclusions among others are considered to be a hallmark of ALS pathology. Notably, several proteins that are mutated in a small subset of ALS, such as SOD1, TDP43, FUS and optineurin (OPTN) are prominent components of these inclusions 6,12,17–20. To test if ubiquilin2 is present in the characteristic skein-like inclusions, we performed immunohistochemical analysis of the postmortem spinal cord sections from two patients with a P497H or P506T mutation. Two different ubiquilin2 antibodies were used. One was a commercially available mouse monoclonal antibody raised with a polypeptide of 71 amino acids (aa) at the C-terminus (554aa-624aa, ubiquilin2-C). The other was a rabbit polyclonal antibody that we generated using a polypeptide of 17aa at the N-terminus (8aa-24aa, ubiquilin2-N). The polypeptide of 17aa is unique to ubiquilin2 and not present in other members of the ubiquilin family or any known protein. The ubiquilin2-N antibody recognized human and mouse ubiquilin2 (Supplementary Fig. 3). We also detected a single band of the expected size by Western blots using ubiquilin2-N and ubiquilin2-C antibodies with human spinal cord autopsy tissues (Supplementary Fig. 3). Using immunohistochemistry, we observed skein-like inclusions that were immunoreactive with both ubiquilin2-C and ubiquilin2-N antibodies (Supplementary Fig. 4), suggesting that ubiquilin2 is involved in inclusion formation in X-ALS. We then examined if the inclusions in X-ALS cases were also immunoreactive with antibodies against other proteins that are known to be involved in the formation of the inclusions in other types of ALS. We found that the skein-like inclusions in the X-ALS patients were also immunoreactive with antibodies to ubiquitin, p62, TDP43, FUS and OPTN (Fig. 2a-c and Supplementary Figures 4 and 5), but not SOD1.
Fig. 2.
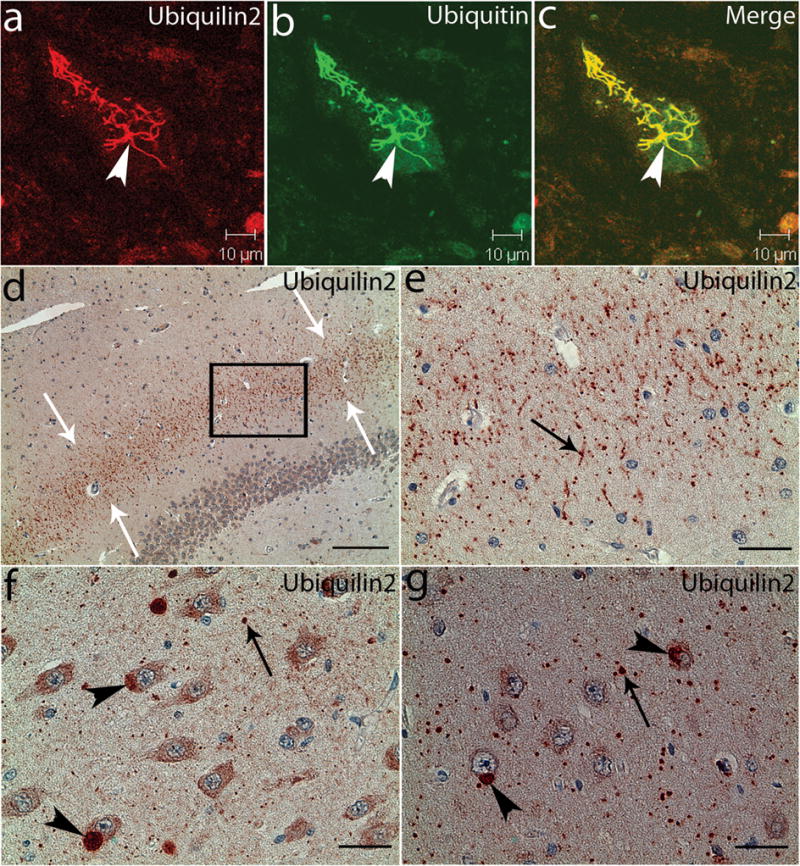
Ubiquilin2-immunoreactive inclusions in the spinal cord and hippocampus. Spinal cord (a–c) and hippocampal (d–g) sections from a patient with a UBQLN2P506T mutation were analyzed with confocal microscopy (a–c) and immunohistochemistry (d–g) using a monoclonal antibody against ubiquilin2 (ubiquilin2-C). The ubiquilin2-positive and skein-like inclusions (arrowhead) are shown in a spinal motor neuron (a). These inclusions are also ubiquitin-positive (b and c). In the hippocampus, the ubiquilin2-positive inclusions are shown in the molecular layer of the fascia dentate (d and e), CA3 (f) and CA1 (g). White arrows in the panel (d) indicate the middle region of the molecular layer with ubiquilin2-positive inclusions. The higher magnification image of the boxed area in panel (d) is shown in panel (e). Black arrows indicate the representative inclusions in neurites (e–g), and arrowheads indicate cytoplasmic inclusions in the cell bodies (f and g). Scale bar, 200μm in panel (d), 50μm in panel (e) and 25 μm in panels (f and g).
Mutations in TDP43, FUS or optineurin occur in a small fraction of FALS, but these proteins have been found in the inclusions of a wide spectrum of ALS 6,12,17,18,20. To test if ubiquilin2 is involved in inclusion formation of other types of ALS, we examined 47 post-mortem spinal cord samples, including SALS (n=23), FALS without mutations in SOD1, TDP43 and FUS (n=5), ALS with dementia (n=5), FALS with SOD1 mutations [n=7, (A4V, n=4; G85R, n=2; E100G, n=1)], FALS with a G298S mutation in TDP43 (n=1), and controls without ALS (n=6). We observed ubiquilin2-positive and skein-like inclusions in all ALS cases (Supplementary Figures 6 and 7), suggesting that ubiquilin2 is a common component in the skein-like inclusions of a wide variety of ALS as well.
Dementia was a prominent feature in eight UBQLN2-linked cases. To examine if ubiquilin2-immunoreactive inclusions are present in brain, and to explore the potential link between ubiquilin2 inclusions and dementia, we analyzed brain autopsy samples from two patients with the P506T mutation. We observed ubiquilin2 pathology, which was most prominent in the hippocampus (Fig. 2d–g and Supplementary Fig. 8). Small ubiqulin2 inclusions were predominantly situated in the neuropil (1–5 μm in diameter). The fascia dentata presented with a band of radially oriented dendritic and neuropil inclusions in the intermediate region of the molecular layer (Supplementary Fig. 8). In addition to the small neuropil inclusions, large inclusions (up to 20 μm in diameter) were observed in some pyramidal neurons, especially those in the CA3 and CA1 (Fig. 2f and 2g, and Supplementary Fig. 8). Co-localization of ubiquilin2 and ubiquitin in these inclusions were confirmed with confocal microscopy (Supplementary Fig. 8). This type of hippocampal pathology has not been previously observed in any other neurodegenerative disorder. The ubiquilin2/ubiquitin positive inclusions did not appear to be co-localized with major glial markers (Supplementary Fig. 9). In addition, we also observed a novel membrane-bound perikaryal structure, which contained eosinophilic granules of varying sizes in some hippocampal pyramidal neurons. These structures were strongly immunoreactive for ubiquilin2 (Supplementary Fig. 10).
To test if ubiquilin2 pathology is present in the hippocampus of ALS/dementia cases without UBQLN2 mutations, and to explore the correlation of ubiquilin2 pathology with dementia in ALS, we further examined hippocampal sections of 15 pathologically characterized ALS cases without UBQLN2 mutation, including five cases of ALS/dementia with pathological signatures corresponding to frontotemporal lobar degeneration of motor neuron disease type (FTLD-MND/FTLD-U). We found prominent ubiquilin2 pathology in the hippocampus of all five cases with ALS/dementia (Supplementary Fig. 11). Similar to the ubiquilin2 inclusions in UBQLN2-linked ALS/dementia cases, the ubiquilin2 inclusions in these non-UBQLN2-linked ALS/dementia cases were also positive with ubiquitin and p62 (Supplementary Fig. 11), but negative with FUS. Although there was no apparent TDP43 neuritic pathology in the dentate molecular layer, we observed variable numbers of cytoplasmic TDP43 inclusions in dentate granule cells, which have been previously shown in ALS/dementia 18 (Supplementary Fig. 11). However, we noted that a significant number of the ubiquilin2/ubiquitin/p62 inclusions were negative for TDP43 (Supplementary Figures 11 and 12). The presence of TDP43-negative inclusions was further confirmed with an antibody specific to phosphorylated TDP43 that is only present in the cytoplasmic inclusions 18 (Supplementary Fig. 13). We also observed that the ubiquilin2/ubiquitin/p62 inclusions were largely negative for TDP43 in the CA regions in the non-UBQLN2-linked ALS/dementia cases (Supplementary Fig. 12). We did not observe the ubiquilin2 pathology in the hippocampus of the 10 ALS cases without dementia. The correlation of the hippocampal ubiquilin2 pathology and dementia in ALS cases with or without UBQLN2 mutations suggest that ubiquilin2 is widely involved in ALS-related dementia, even without UBQLN2 mutations.
TDP43 inclusions have been observed in dentate granule cells of the hippocampus in most of the cases with FTLD 18. FUS inclusions have been shown in most of the TDP43-negative FTLD cases 21,22. To test if ubiquilin2 co-aggregates with these two known ALS- and dementia-linked proteins in vitro, we generated 10 expression constructs (Supplementary information) and co-transfected Neuro2a cells with different combinations. Both wild-type and mutant ubiquilin2 were largely distributed in the cytosol. We did not observe obvious differences in the distribution between the wild type and mutant ubiquilin2. The wtFUS and wtTDP43 were located almost exclusively in the nuclei (Fig. 3 and Supplementary Fig. 14); whereas, mutant FUS showed prominent cytoplasmic distribution (Supplementary Fig. 13) and the C-terminal fragment (218–414, C-TDP43) of TDP43 that has been linked to ALS and FTLD 18,23 was almost exclusively located in the cytosol (Fig. 3). We did not observe cytoplasmic inclusions in cells transfected with wtFUS and mutant FUS (Supplementary Fig. 14) or wtTDP43 (Fig. 3). However, cytoplasmic inclusions were observed in cells expressing either wild-type or mutant ubiquilin2. Notably, the C-TDP43 was co-localized with either wild-type or mutant ubiquilin2 in the cytoplasmic inclusions (Fig. 3). We obtained consistent data using two expression systems, either tagged ubiquilin2 or tag-free ubiquilin2 (Fig. 3 and Supplementary Figures 14 and 15). These data suggest that both ALS- and dementia-linked ubiquilin2 and TDP43 are prone to co-aggregation. We also noted that inclusion formation was apparently dose-dependent, because the cells with low expression of wild-type and mutant ubiquilin2, or C-TDP43 did not show cytoplasmic inclusions. Moreover, ubiquilin2-positive, but C-TDP43-negative inclusions were frequently observed in cells with relatively lower levels of expression (Fig. 3). This phenomenon suggests that ubiquilin2 may be more prone to aggregation than TDP43. This notion is consistent with the pathology observed in ALS/dementia cases, where the ubiquilin2-containing inclusions in the molecular layer and in some dentate granule cells were TDP43-negative.
Fig. 3.

Co-localization of ubiquilin2 and ALS- and dementia-linked TDP43. Neuro2a cells were transfected with various combinations of wild-type ubiquilin2, wild-type TDP43, mutant ubiquilin2 (P497H), and C-terminal fragment of TDP43 that is linked to ALS and FTLD. Ubiquilin2 is GFP-tagged and TDP43 is mCherry-tagged. Wild-type and mutant ubiquilin2 are largely cytoplasmic. WtTDP43 are almost exclusively distributed in the nuclei. The C-TDP43, an ALS- and dementia-linked TDP43 fragment (aa218-414) is almost exclusively cytoplasmic. TDP43 inclusions are co-localized with wild-type (g–i) and mutant (P497H) ubiquilin2 (j–l) (arrows). Some ubiquilin2-positive inclusions are TDP43-negative (arrowhead).
Ubiquilin2 is a member of the ubiquitin-like protein family (ubiquilins). Humans have four ubiquilin genes, each encoding a separate protein. Ubiquilins are characterized by the presence of a N-terminal ubiquitin-like (UBL) domain and a C-terminal ubiquitin-associated (UBA) domain (Fig. 1d). The middle part of ubiquilins is highly variable. This structural organization is characteristic of proteins that function to deliver ubiquitinated proteins to the proteasome for degradation. In accordance with this function, the UBL domain of the ubiquilins binds subunits of the proteasome, and its UBA domain binds to polyubiquitin chains that are typically conjugated onto proteins marked for degradation by the proteasome 24. In addition to the UBL and UBA domains that are shared by all ubiquilins, ubiquilin2 has a unique repeat region containing 12 PXX tandem repeats (Fig.1d). Remarkably, all the five ALS-linked mutations identified in this study involve proline residues within this short PXX repeat region (Fig. 1c and 1d), suggesting that these mutations may confer on ubiquilin2 a common property that may be related to the pathogenic mechanism of the disease.
Based on the involvement of ubiquilin2 in the protein degradation pathway, we then investigated the functional consequences of mutant ubiquilin2 in protein degradation through the UPS. We used the UPS reporter substrate UbiquitinG76V-Green Fluorescent Protein (UbG76V-GFP) 25 to test the effects of mutant ubiquilin2 on ubiquitin-mediated protein degradation. Two mutations at two different sites were tested (P497H and P506T) using the UbG76V-GFP reporter system. The G76V substitution prevents removal of the N-terminal-fused ubiquitin by cellular de-ubiquitinating enzymes, leading to efficient proteasomal degradation of the UbG76V-GFP reporter 25. We first tested the transfection efficiency of wild-type and mutant ubiquilin2 constructs, and observed similar levels of exogenous ubiquilin2 expression (Supplementary Fig. 16). We also tested the functionality of the UbG76V-GFP reporter system using proteasomal inhibitor MG-132 in transiently transfected cells. As expected, incubation with MG-132 resulted in remarkable accumulation of the UbG76V-GFP signal (Supplementary Figure 17). We then examined the accumulation of UbG76V-GFP reporter in Neuro2a cells transiently transfected with either wild-type (WT) or mutant ubiquilin2 constructs. Expression of mutant ubiquilin2 resulted in significantly higher accumulation of UbG76V-GFP than WT-ubiquilin2 (Fig. 4a). Similar data were obtained using SH-SY5Y cells (Supplementary Fig. 18).
Fig. 4.
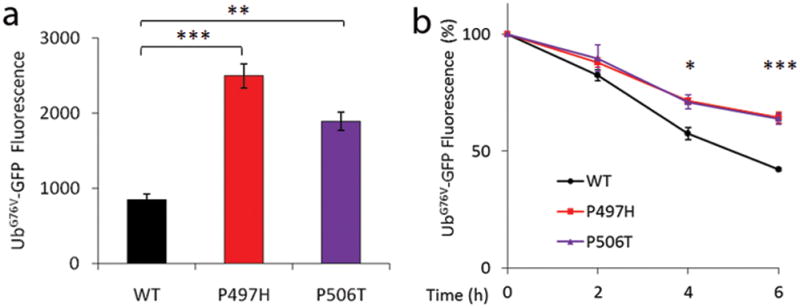
Mutations in ubiquilin2 lead to ubiquitin-mediated impairment of proteasomal degradation. UbG76V-GFP fluorescence intensity was quantified by FACS 48 hours post-transfection in Neuro2a cells (a) transiently transfected with either wild-type (WT) or mutant ubiquilin2. The dynamics of UbG76V-GFP reporter degradation after blockage of protein synthesis with cycloheximide for 0, 2, 4, and 6 hours in Neuro2a cells are shown in panel (b). The rates of UPS reporter degradation were significantly slower in both ubiquilin2-P497H and ubiquilin2-P506T mutants when compared to the wild-type ubiquilin2 at 4 and 6 hours (b). The mean fluorescence intensity was determined at the indicated time points by flow cytometry. Mean fluorescence before cycloheximide administration was standardized as 100%. Data are averaged from at least three independent experiments. *p <0.05, **p <0.01, and ***p <0.001 indicating significant differences when compared to WT-UBQLN2 (two-tailed Student t test). Error bars, means ± s.e.m.
We further analyzed the dynamics of UbG76V-GFP reporter degradation after new protein synthesis was blocked with cycloheximide for 0, 2, 4, and 6 hours in Neuro2a cells. We found that the rates of reporter degradation were significantly slower in both ubiquilin2-P497H and ubiquilin2-P506T mutants when compared to wild-type ubiquilin2 at 4 hours (p <0.05) and 6 hours (p <0.001) (Fig. 4b), further supporting the notion that the ubiquilin2 mutants impair the protein degradation pathway.
It is interesting to note that all the five ALS-linked UBQLN2 mutations identified in the present study involve four proline residues in the PXX region. Proline is a unique amino acid in that it has a side-chain cyclized onto the backbone nitrogen atom, leading to sterical restriction of its rotation, and thus, hindering the formation of major known secondary structures. Moreover, among the primary structures of many ligands for protein-protein interactions, a proline residue is often critical 26. Some protein-protein interaction domains, such as SH3, prefer ligand sequences containing tandem PXXP motifs, as noted in the PXX domain of ubiquilin2, for high affinity and selectivity of such interactions 27. Further studies of the consequences of proline mutations in ubiquilin2 may shed light on the understanding of the pathogenic mechanism.
The exact function of ubiquilin2 is not well understood. However, increasing lines of evidence have shown that ubiquilins, together with their interactions with other proteins, are involved in a broad spectrum of neurodegenerative disorders. Ubiquilin1, another member of the ubiquilins, is associated with AD and it interacts with presenilins 1 and 2 28, and TDP43 29. We observed that ubiquilin2 formed cytoplasmic inclusions with ALS- and FTLD-linked TDP43, implicating that an interaction of TDP43 and ubiquilin2 underlies the pathogenesis of ALS and ALS/dementia, and possibly other neurodegenerative disorders as well.
The removal of misfolded or damaged proteins is critical for optimal cell functioning. In the cytosol and the nucleus, a major proteolytic pathway to recycle misfolded or damaged proteins is the UPS. Though impaired UPS is thought to be associated with the formation of proteinaceous inclusions in many neurodegenerative disorders, direct evidence of mutations in the UPS pathway has been limited 30. In this study, we show mutations of ubiquilin2, a ubiquitin-like protein in five families with ALS and ALS/dementia. We also show that ubiquilin2-containing inclusions are a common pathological feature in a wide spectrum of ALS and ALS/dementia. Functional studies indicate an impairment of ubiquitin-mediated proteasomal degradation in cells expressing mutant ubiquilin2. These data provide robust lines of evidence for an impairment of protein turnover in the pathogenesis of ALS and ALS/dementia, and possibly for other neurodegenerative disorders as well. Further elucidation of these processes may be central to the understanding of pathogenic pathways. These pathways should provide novel molecular targets for designing rational therapies for these disorders.
Methods Summary
Genomic DNA was PCR-amplified and Sanger-sequenced using a CEQ 8000 Genetic Analysis System (Beckman Coulter, Fullerton, CA). Western blot, immunohistochemistry and confocal microscopy were performed using previously established methods17. Construction of expression vectors, cell culture and flow cytometry were performed according to standard protocols. For statistical analysis, all graphs show mean ± s.e.m. Significance was calculated using Student t-test (*, p <0.05; **, p <0.01; ***, p <0.001).
Full methods and associated references are available in the online version of the paper at www.nature.com/nature.
Materials and Methods
Patients and samples
This study was approved by the local institutional review boards. ALS patients met the diagnosis of probable or definite ALS as defined in the revised EL-Escorial 31. Patients with dementia met the criteria for FTD or FTLD proposed by Neary et al 32 or Cairns et al 33. The dementia was similar to FTD on inception and was progressive, and eventually global in most patients. One patient had mild mental retardation before onset of dementia. There were eight patients with both ALS and dementia. Dementia preceded motor symptoms in some patients, but no patient remained free of motor involvement. The FTLD symptoms included abnormalities in both behavior and executive function, although the degree of severity varied depending on the individuals in different stages of the disease. Pedigrees and clinical data were collected by specialists in neuromuscular medicine and were verified by medical records or decent examination to establish diagnosis (Supplementary Table 2). DNA and other samples were taken after obtaining written informed consent. Overall, DNA from over 200 cases with ALS and 928 controls were used for genetic analysis. Spinal cord autopsy samples from two X-linked ALS cases (P497H or P506T each), 41 cases with ALS or ALS dementia and six non-ALS controls were studied. In addition, available autopsy samples from the motor cortex region of a patient with the P497H mutation (F#186, IV-7), brain regions (including hippocampus, cerebellum, optic nerve, visual cortex, pons and midbrain) of two patients with the P506T mutation (F#6316, II-3 and III-4), and the hippocampal region of 10 ALS and five ALS/dementia cases were also used for pathological and immunohistological studies. These five ALS/dementia cases were classified as having frontotemporal lobar degeneration of motor neuron disease type (FTLD-MND/FTLD-U), including four cases with pathological type 3 and one case with pathological type 2 according to the classification system proposed by Mackenzie et al.34. These cases were evaluated by a neuropathologist (E.H.B).
Genetic analysis
Genomic DNA was extracted from whole peripheral blood, transformed lymphoblastoid cell lines or available tissues by standard methods (QIAGEN, Valencia, CA). Intronic primers flanking exons were designed at least 50 nucleotides away from the intron/exon boundary. When a PCR product was over 500 bp, multiple overlapping primers were designed with an average of 50 bp overlap. Forty nanograms of genomic DNA were used for PCR amplification. The amplification protocol consisted of the following steps: incubation at 95°C for 1 min, 32 cycles of 95°C (30s), 55°C (30s) and 72°C (1 min) and a final 5 min extension at 72°C, with modifications when necessary. Unconsumed dNTPs and primers were digested with Exonuclease I and Shrimp Alkaline Phosphatase (ExoSAP-IT, USB, Santa Clara, CA). When nonspecific PCR amplification occurred, the PCR products were separated by 1.5% agarose gel and the specific PCR product was cut out from the gel and purified using QIAquick Gel Extraction Kit (QIAGEN, Valencia, CA). For sequencing of a PCR product, fluorescent dye labeled single-strand DNA was amplified using Beckman Coulter sequencing reagents (GenomeLab DTCS Quick Start Kit) followed by single-pass bidirectional sequencing with a CEQ 8000 Genetic Analysis System (Beckman Coulter, Fullerton, CA). We sequenced the entire protein-coding exons and 30–50 bp intronic sequences flanking the exons. UBQLN2 is an intronless gene. We divided the UBQLN2 gene into five overlapping PCR fragments to carry out sequencing analysis. These five overlapping PCR fragments cover the entire coding sequence (1,872bp), 125bp of the 5′ UTR and 293bp of the 3′UTR.
Ubqln2-1F: 5′-cttcatcacagaggtaccgtg-3′; Ubqln2-1R: 5′-gtgtggagttactcctgggag-3′
Ubqln2-2F: 5′-catgatgggctgactgttcac-3′; Ubqln2-2R: 5′-ctcttgtgcggcattcagcatc-3′
Ubqln2-3F: 5′-gacctggctcttagcaatctag-3′; Ubqln2-3R: 5′-gtgtctggattctgcatctgc-3′
Ubqln2-4F: 5′-cacagatgatgctgaatagcc-3′; Ubqln2-4R: 5′-gctgaatgaactgctggttgg-3′
Ubqln2-5F: 5′-ctgcacctagtgaaaccacgag-3′; Ubqln2-5R: 5′-aacagcattgattcccaccac-3′
For fragments 4 and 5, the PCR protocol consisted of the following steps: incubation at 96°C for 2 min, 32 cycles of 96°C (30s), 56°C (30s) and 72°C (1min) and a final 5 min extension at 72°C. The PCR products were separated on a 1.5% agarose gel and purified with QIAquick Gel Extraction Kit (QIAGEN, Valencia, CA) before sequencing.
Antibodies
Two anti-ubiquilin2 antibodies were used. One was a mouse monoclonal antibody (5F5, Cat# H00029978-M03, Novus Biologicals Inc. Littleton, CO). We made the other, which was raised in rabbit using a polypeptide of human ubiquilin2 (8aa-24 aa, NH2-SGPPRPSRGPAAAQGSA-COOH). The antiserum was affinity purified. Other polyclonal and monoclonal antibodies that were used in this study included those against: ubiquitin (PRB-268C, Covance, Emeryville, CA; 10R-U101b, Fitzgerald Industries International, Concord, MA; Ub (N-19): sc-6085, Santa Cruz Biotechnology, Santa Cruz, CA), p62 (H00008878-M01, Abnova, Taipei, Taiwan; NB110-74805, Novus Biologicals, Inc., Littleton, CO), TDP43 (TIP-PTD-P01, Cosmo Bio Co, Tokyo, Japan; 10782-2-AP, ProteinTech Group, Chicago, IL; 60019-2-Ig; ProteinTech Group, Chicago, IL; WH0023435M1-100UG, Sigma-Aldrich, St. Louis, MO), FUS (11570-1-AP, ProteinTech Group, Chicago, IL), OPTN (100000, Cayman, Ann Arbor, MI) and SOD1 35, c-myc (MMS-150P, Covance, Emeryville), GFAP (Z0334, Dako North America, Carpinteria, CA; G3893, Sigma-Aldrich, St. Louis, MO), Iba1 (019-19741, Wako Pure Chemical Industries, Osaka, Japan), and CNPase (MAB326R, Millipore, Temecula CA).
Immunohistochemistry and Confocal Microscopy
The basic protocols for immunohistochemistry and confocal microscopy have been described in detail in a previous study 17. In brief, 6 μm sections were cut from formalin-fixed, paraffin-embedded spinal cord and brain regions containing the frontal lobe or the hippocampus. The sections were deparaffinized with xylene and rehydrated with a descending series of diluted ethanol and water. Antigens in the sections were retrieved using a high pressure decloaking chamber. For immunohistochemistry, endogenous peroxidase activity was blocked with 2% hydrogen peroxide. Non-specific background was blocked with 1% bovine serum albumin. The titers of the antibodies were determined based on preliminary studies using serial dilution of the antibodies. Various antibodies against ubiquilin2 or other proteins were used as primary antibodies. These antibodies included rabbit polyclonal anti-ubiquilin2 (ubiquilin2-N; 0.5 μg/ml; generated by us), mouse monoclonal anti-ubiquilin2 antibody (ubiquilin2-C; 0.2 μg/ml; H00029978-M03; Novus Biologicals, Littleton, CO), rabbit polyclonal anti-FUS antibody (3 μg/ml; 11570-1-AP; ProteinTech Group, Chicago, IL), mouse monoclonal anti-TDP43 antibody (1 μg/ml; 60019-2-Ig; ProteinTech Group, Chicago, IL), rabbit polyclonal anti-TDP43 antibody (0.1 μg/ml; 10782-2-AP; ProteinTech Group), mouse monoclonal anti-ubiquitin antibody (0.5 μg/ml; 10R-U101B; Fitzgerald Industries International, Concord, MA), rabbit polyclonal anti-ubiquitin antibody (0.5 μg/ml; PRB-268C, Covance, Emeryville, CA), goat polyclonal anti-ubiquitin (0.5 μg/ml; Ub (N-19):sc-6085; Santa Cruz Biotechnology, Santa Cruz, CA), mouse monoclonal anti-p62 antibody (1 μg/ml; H00008878-M01; Abnova Coporation, Taipei, Taiwan), rabbit polyclonal anti-optineurin antibodies (C-term, 0.2 μg/ml, 100000, Cayman Chemical, Ann Arbor, Michigan), Biotinylated goat anti-rabbit and anti-mouse IgG, biotinylated mouse anti-goat IgG or biotinylated rabbit anti-mouse IgG were used as the secondary antibodies. Immunoreactive signals were detected with peroxidase-conjugated streptavidin (BioGenex, San Ramon, CA) using 3-amino-9-ethylcarbazole as a chromogen. The slides were counterstained with hematoxylin and sealed with Aqua Poly/Mount (Polyscience, Warrington, PA).
For confocal microscopy, antibodies generated in different species were used in various combinations. These antibodies included those against ubiquilin2, FUS, TDP43, p62, optineurin, ubiquitin, c-myc, GFAP, Iba1 and CNPase. Fluorescence signals were detected with appropriate secondary anti-rabbit, anti-mouse or anti-goat IgG conjugated with rhodamine or fluorescein isothiocyanate (Invitrogen, Carlsbad, CA; Thermo Scientific, Rockford, IL) using an LSM 510 META Laser Scanning Confocal Microscope with the multi-tracking setting 17. The same pinhole diameter was used to acquire each channel.
Western Blot
Western blotting was performed using the protocol previously described 17. Briefly, spinal cord tissues from lumbar segments were processed and homogenized. Cell lysates or the supernatants of tissue homogenates were subjected to total protein quantification, gel electrophoresis and blotted on PVDF membranes. Ubiquilin2 was detected using ubiquilin2-N or ubiquilin2-C antibody. The membranes were stripped and blotted with an antibody against β-actin, (A5441, Sigma-Aldrich, St. Louis, MO).
Expression constructs
A full length human cDNA clone (Homo sapiens ubiquilin 2, IMAGE:4543266) was used as a template for construction of the expression constructs. Two primers anchored with an XhoI (ubiquilin2-TP1, 5′-ttctcgagggccgccatggctgagaat-3′) and BamHI (ubiquilin2-TP2, 5′-catggatcctgtatgtctgtattacc-3′) were used to amplify the full length coding sequence. The amplified fragment was cloned into plasmid vector pBluescript M13. The ubiquilin2 sequence was verified by direct sequencing. Each of the P497H and P506T mutations was introduced into the plasmid vector by site-directed mutagenesis using a primer containing each respective mutation. The XhoI/BamHI fragment containing wild-type UBQLN2, UBQLN2P497H or UBQLN2P506T was released from the pBluescript M13 vector and cloned into the XhoI and BamHI sites of dual expression vectors pIRES2-DsRed2 or pIRES2-ZsGreen1, to create such constructs as wtUBQLN2, ZsGreen1; mutant ubiquilin2 (P497H, or P506T) (mUBQLN2, ZsGreen1) (Clontech, Mountain View, CA).
In addition, we generated seven other expression constructs, including wild-type ubiquilin2 tagged with GFP (wtUBQLN2-GFP), mutant ubiquilin2 (P497H or P506T) tagged with GFP (mUBQLN2-GFP), wild-type TDP43 tagged with mCherry (wtTDP43-mCherry), an ALS- and dementia-linked C-terminal fragment of TDP43 (amino acids 218–414, C-TDP43) 18,23 tagged with mCherry (C-TDP43-mCherry), wild-type FUS tagged with myc (myc-wtFUS) and mutant FUS (R495X) tagged with myc (myc-mFUS).
Expression of wild-type and mutant ubiquilin2
Cells of SH-SY5Y, Neuro2a and HEK293 lines were grown on collagen-coated plates in Dulbecco’s modified Eagle’s medium containing 10% (v/v) human serum, 2 mM L-glutamine, 2 U/ml penicillin, and 2 mg/ml streptomycin at 37°C in a humidity-controlled incubator with 5% CO2. The cells were transiently transfected with different combinations of expression vectors using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions.
UPS Reporter Assay
SH-SY5Y and Neuro2a cells were grown in 24-well plates and double-transfected with a UPS reporter vector containing UbG76V-GFP 25 (Addgene plasmid #11941), and a dual expression vector containing DsRed2 with either wild-type or mutant ubiquilin2. Forty-eight hours post-transfection, cells were harvested and resuspended in PBS. UbG76V-GFP transfected cells were used for control experiments to test the functionality of the UPS reporter. In these control experiments, media was changed 24 hours post-transfection to that containing 5 μM of the proteasomal inhibitor MG-132 (A.G. Scientific, Inc, San Diego, CA). Cells were incubated in this media for 24 hours and then harvested and resuspended in PBS. For cycloheximide chase of UBG76V-GFP, transiently transfected Neuro2a cells were used. Twenty-four hours post-transfection, the cells were transferred to medium containing 5 μM MG-132. After incubation with MG-132 for 16 hours to accumulate the UbG76V-GFP reporter, cells were washed in sterile PBS and incubated with medium containing 100 μg/ml cycloheximide (Sigma, St. Louis, MO) for 0, 2, 4, and 6 hours. At each time point, cells were washed, harvested and resuspended in ice-cold PBS supplemented with 100 μg/ml cycloheximide. The fluorescence intensities at each time point were measured by FACS. The fluorescence intensity at time=0 hours was taken to be maximal fluorescence (100%). All flow cytometric data were collected and analyzed using a MoFlo cell sorter and Summit software (DakoCytomation, Fort Collins, CO). Argon-ion (488 nm) and yellow (565 nm) lasers were used for excitation. The GFP and DsRed2 signals were collected using 530/40 nm and 600/30 nm bandpass filters, respectively. In all experiments data were gated on GFP/DsRed2 dual-labeled cells. At least 500–1,000 such events were recorded in each experiment. The DsRed2 expression levels and profiles were similar across experiments. Data were collected from three independent experiments. Two-tailed unpaired Student t-test (p <0.05) was used for statistical analysis.
Supplementary Material
Acknowledgments
This study was supported by the National Institute of Neurological Disorders and Stroke (NS050641), the Les Turner ALS Foundation, the Vena E. Schaff ALS Research Fund, the Harold Post Research Professorship, theHerbert and Florence C. Wenske Foundation, the David C. Asselin MD Memorial Fund, the Help America Foundation, and the Les Turner ALS Foundation/Herbert C. Wenske Foundation Professorship. F.F. has support from NIH (T32 AG20506). K.A. is a post-doctoral fellow of the Blazeman Foundation for ALS. We thank Dr. Nico Dantuma for the UPS reporter plasmid (through Addgene) and the staff of the Northwestern University Robert H. Lurie Comprehensive Cancer Center Flow Cytometry Core Facility for technical assistance.
Footnotes
Author contribution
T.S. conceived and supervised this project. W.C., S.-T.H., Y.Y., H.J. M.H., H.-X.D. and T.S. did the sequencing analysis. S.T.H., E.R., J.L.H., M.A.P-V and T.S performed linkage analysis. K.M.B., G.H.G., F.F., G.H.J., H.Z., E.H.B., K.A., E.M. H.-X.D., and T.S. performed immunohistochemical, confocal and pathological analysis. F.F., Y.S. and H.-X.D. performed functional analysis. N.S., S.D. and T.S. collected family information and coordinated this study. K.M.B., G.H.J., B.R.B., R.L.S., and T.S. did clinical studies. H.-X.D. and T.S. analyzed the data and wrote the paper.
Competing interest declaration: None
References
- 1.Deng HX, et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 2.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 3.Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. ng.132 [pii] [DOI] [PubMed] [Google Scholar]
- 4.Sreedharan J, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. 1154584 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwiatkowski TJ, Jr, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. 323/5918/1205 [pii] [DOI] [PubMed] [Google Scholar]
- 6.Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. 323/5918/1208 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen YZ, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74:1128–1135. doi: 10.1086/421054. S0002-9297(07)62840-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenway MJ, et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–413. doi: 10.1038/ng1742. ng1742 [pii] [DOI] [PubMed] [Google Scholar]
- 9.Nishimura AL, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–831. doi: 10.1086/425287. S0002-9297(07)63787-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29:160–165. doi: 10.1038/ng1001-160. [DOI] [PubMed] [Google Scholar]
- 11.Chow CY, et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009;84:85–88. doi: 10.1016/j.ajhg.2008.12.010. S0002-9297(08)00631-9 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maruyama H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226. doi: 10.1038/nature08971. nature08971 [pii] [DOI] [PubMed] [Google Scholar]
- 13.Ticozzi N, et al. Paraoxonase gene mutations in amyotrophic lateral sclerosis. Ann Neurol. 2010;68:102–107. doi: 10.1002/ana.21993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell J, et al. Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc Natl Acad Sci U S A. 2010;107:7556–7561. doi: 10.1073/pnas.0914128107. 0914128107 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson JO, et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. S0896-6273(10)00978-5 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. nature05290 [pii] [DOI] [PubMed] [Google Scholar]
- 17.Deng HX, et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol. 2010;67:739–748. doi: 10.1002/ana.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. 314/5796/130 [pii] [DOI] [PubMed] [Google Scholar]
- 19.Shibata N, et al. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol. 1996;55:481–490. doi: 10.1097/00005072-199604000-00011. [DOI] [PubMed] [Google Scholar]
- 20.Mackenzie IR, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 21.Neumann M, et al. Frontotemporal lobar degeneration with FUS pathology. Brain. 2009 doi: 10.1093/brain/awp214. awp214 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urwin H, et al. FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 2010;120:33–41. doi: 10.1007/s00401-010-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet. 2009;18:3353–3364. doi: 10.1093/hmg/ddp275. ddp275 [pii] [DOI] [PubMed] [Google Scholar]
- 24.Ko HS, Uehara T, Tsuruma K, Nomura Y. Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett. 2004;566:110–114. doi: 10.1016/j.febslet.2004.04.031. S0014579304004612 [pii] [DOI] [PubMed] [Google Scholar]
- 25.Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18:538–543. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- 26.Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- 27.Aitio O, et al. Recognition of tandem PxxP motifs as a unique Src homology 3-binding mode triggers pathogen-driven actin assembly. Proc Natl Acad Sci U S A. 2010;107:21743–21748. doi: 10.1073/pnas.1010243107. 1010243107 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haapasalo A, et al. Emerging role of Alzheimer’s disease-associated ubiquilin-1 in protein aggregation. Biochem Soc Trans. 2010;38:150–155. doi: 10.1042/BST0380150. BST0380150 [pii] [DOI] [PubMed] [Google Scholar]
- 29.Kim SH, et al. Potentiation of amyotrophic lateral sclerosis (ALS)-associated TDP-43 aggregation by the proteasome-targeting factor, ubiquilin 1. J Biol Chem. 2009;284:8083–8092. doi: 10.1074/jbc.M808064200. M808064200 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237–248. doi: 10.1038/nrd3050. nrd3050 [pii] [DOI] [PubMed] [Google Scholar]
- 31.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 32.Neary D, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 33.Cairns NJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mackenzie IR, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–549. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng HX, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. 0602046103 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.