

Abstract
The extraordinary potency and pathological relevance of Gram-negative bacterial lipopolysaccharides (LPSs) have made them very popular experimental agonists, yet little is known about what happens to these stimulatory molecules within animal tissues. We tracked fluorescent and radiolabeled-LPS from a subcutaneous inoculation site to its draining lymph nodes (DLN), blood and liver. Although we found FITC-labeled LPS in DLN within minutes of injection, drainage of radiolabeled LPS continued for more than six weeks. Within the DLN, most of the LPS was found in the subcapsular sinus or medulla, near or within lymphatic endothelial cells and CD169+ macrophages. Whereas most of the LPS seemed to pass through the DLN without entering B cell follicles, by 24 hrs after injection a small amount of LPS was found in the paracortex. In wildtype mice, ≥70% of the injected radiolabeled-LPS underwent inactivation by deacylation before it left the footpad; in animals that lacked acyloxyacyl hydrolase, the LPS-deacylating enzyme, prolonged drainage of fully acylated (active) LPS boosted polyclonal IgM and IgG3 antibody titers. LPS egress from a subcutaneous injection site thus occurred over many weeks and was mainly via lymphatic channels. Its immunological potency, as measured by its ability to stimulate polyclonal antibody production, was greatly influenced by the kinetics of both lymphatic drainage and enzymatic inactivation.
Keywords: LPS, endotoxin, acyloxyacyl hydrolase, lymph nodes, trafficking, lymph
Introduction
Animals protect themselves from many Gram-negative bacteria by sensing the bacterial cell wall lipopolysaccharide (LPS, also called endotoxin), then mounting inflammatory responses that kill the microbes (1). The host response to LPS also typically includes the production of both polyclonal and anti-LPS antibodies. Whereas much is known about the fates of LPS molecules that have been injected into the bloodstream (2–5), how LPSs traffic from subcutaneous tissues to draining lymph nodes and distant organs has not been studied. It is important to know how, when, and where LPS is inactivated in tissue sites, since infection usually starts in extravascular tissues and LPS that escapes inactivation there may stimulate cells at other sites within the body. It is also of interest to know how the transport and inactivation of LPS molecules influence an animal’s immunological responses to it.
To follow LPS movement from a peripheral tissue site, we tracked the fate of small doses of LPS injected subcutaneously into mice. We used both radiolabeled and fluorescent LPS probes to measure LPS movement from an injection site in a footpad or flank to draining lymph nodes (DLN) and its appearance in the liver, the major organ for clearing blood-borne LPS.
Inactivation of microbial agonists may also influence host immune responses. We reported previously that acyloxyacyl hydrolase (AOAH), a host enzyme that inactivates LPS by deacylation, limits polyclonal antibody responses to LPS in mice (6). In the present studies we defined the magnitude and time course of LPS deacylation in a subcutaneous injection site and assessed its impact on LPS’s ability to stimulate downstream B lymphocytes by measuring polyclonal antibody levels in serum.
Materials and Methods
Reagents
E. coli O14 LPS was prepared by the phenol-chloroform-petroleum ether method (7). N. meningitidis LPS, a generous gift from Michael Apicella at the University of Iowa, was purified from a group B (L3,7,9) strain. Rc S. typhimurium LPS ([3H/14C] LPS; 3H-labeled fatty acyl chains and 14C-labeled glucosamine backbone) was prepared from S. typhimurium PR122 as described previously (8); 1 μg had approximately 150,000 dpm 3H and 10,000 dpm 14C. FITC-LPS or Texas Red-LPS was prepared as described by Tobias et al. (9). In brief, E. coli O14 LPS (Ra chemotype) was resuspended (2 mg/ml) in 0.1 M borate, pH 10.5. Five μg radiolabeled LPS was added so that the concentration of the final product could be calculated. 10 mg of solid FITC was then added to 2.5 ml suspension and incubated for 3 hrs at 37°C. A 10-fold excess of glycine was added to stop the reaction. The suspension was dialyzed (1000 MW cut off) against PBS at 4°C for 2 weeks. The FITC-LPS was then precipitated by adding a two-fold excess of ethanol. The pellet was washed three times with 70% ethanol and resuspended in PBS. The labeling efficiency was 0.76 FITC/LPS (mol/mol) and 0.15 Texas Red/LPS (mol/mol). The LPS lost 15% of its fatty acyl chains during labeling at alkaline pH, resulting in a FITC-LPS that was approximately 10-fold less stimulatory than the LPS used to prepare it. Glycine-FITC was made by mixing glycine with FITC in PBS. The solution was diluted so that its OD494 was the same as that of the FITC-LPS. TNP-FICOLL was purchased from Biosearch Technologies. Trinitrophenol (TNP)-LPS was prepared by the same method used for preparing FITC-LPS. N. meningitidis LPS was incubated with 2,4,6-trinitrobenzenesulfonic acid solution (Sigma) and the TNP-LPS was purified by dialysis and precipitation. The labeling efficiency was 1.13 TNP/LPS (mol/mol).
Antibodies
Murine monoclonal anti-MD-2—TLR4 antibody, UT12 (10), was prepared as described (11). Antibodies used for microscopy were B220 (Clone RA3-6B2, BD), CD169 (Clone: 3D6.112, AbD Serotec Inc.), LYVE1 (polyclonal, Abcam), and CD11c (Clone N418, eBioscience).
Liposomes containing clodronate or PBS (control) were prepared as described (12).
Mice
Aoah−/− C57Bl/6 mice were prepared as described (13). CD11c-DTR (B6.FVB-Tg (Itgax-DTR/EGFP)57Lan/J) and Tlr4−/− (B6.B10ScN-Tlr4lps-del/JthJ) mice were obtained from Jackson Labs, Bar Harbor, ME. Mice were housed in a SPF facility and studied using protocols approved by the Institutional Animal Care and Use Committee, UT-Southwestern Medical Center, or by the Animal Resources Center of the National Institute of Allergy and Infectious Diseases.
Studies using fluorescent LPS
Five μg LPS-FITC, 5 μg Texas Red-LPS, or Gly-FITC with the same OD as LPS-FITC were dissolved in 40 ul PBS and injected into a mouse hind footpad or flank using an insulin syringe. The draining popliteal or brachial node was dissected at various time points after injection. Lymph nodes were immersed in Sakura Tissue-Tek Oct Compound (OCT, IMEB INC) and snap-frozen in isopentane chilled in liquid nitrogen. Eight μm sections were cut using a Leica CM1900 cryostat. The sections were fixed and permeabilized in methanol for 10 min at −20°C and blocked with (a) 1 mg/ml BSA, 0.5 mg/ml normal mouse Ig, 10 μg/ml streptavidin in PBS, and then (b) 1 mg/ml BSA, 2 mM biotin in PBS. Sections were stained with primary antibodies at 4°C overnight and with the secondary reagents at room temperature for 1 hr. After washing, FluorSave™ Reagent Aqueous mounting medium (EMD Chemicals) was applied and then coverslips were affixed. Stained sections were examined by using a Leica SP5 X-WLL confocal microscope and analyzed using LAS AF Lite (Leica) software.
To distinguish between cell-free and cell-mediated transport, LPS-FITC and LPS-Texas Red were injected s.c. at separate locations on the back. Both injection sites drained to the same brachial node. If the two LPS preparations were carried by cells to DLN, they should not co-localize within the node. Four, 24, and 48 hrs later, brachial nodes were dissected and analyzed for co-localization of green and red fluorescence. As a control, 40 μl of a 0.02% suspension of 1.0 μm yellow-green or red fluorescent microspheres (Invitrogen) were injected into the same sites in other mice and their location in the brachial node was determined 2 days later. Microspheres were known to be carried from skin to DLN by monocyte-derived dendritic cells (14).
Quantitation of radiolabeled LPS
To follow the location and deacylation of LPS over time, we inoculated the footpads of mice with 10 μg [3H/14C] LPS (8) (see above). Mice were euthanized at 3, 7, 14 and 41 days. The feet were solubilized by immersion in Solvable (Packard Instruments, Meriden, CT), DLNs and livers were homogenized in PBS. To estimate LPS deacylation, the ratio of 3H to 14C was used (12).
Inoculation site removal experiment
AOAH WT and KO mice were injected with 10 μg N. meningitidis LPS s.c. at a shaved site on the back. Red microspheres were included in the LPS suspension to mark the injection location. On day 1 or day 4 after injection, a skin patch that included the injection site was excised. For control mice, similar sizes of skin patches were excised from the opposite (non-injection) sites. Mice were bled before LPS injection and 7, 14, 21 days thereafter. Serum total IgM and IgG3 levels were measured using ELISA.
Enzyme-linked immunosorbent assays (ELISA)
Standard ELISA methods were used. To assay antibody concentrations, microtiter wells were coated with goat anti-mouse polyvalent immunoglobulins (IgG, IgM and IgA) from Sigma-Aldrich. The detection antibodies were HRP-conjugated goat anti-mouse IgM (Sigma) and anti-mouse IgG3 (Southern Biotechnology Associates). HRP substrate (BD-PharMingen) was used. Plates were read on a MRX Microplate Reader (Dynex Technologies Incorporation, Chantilly, VA). IgM and IgG standards were kindly provided by E. Vitetta (UT-Southwestern Medical Center, Dallas, TX). In all cases, differences between post-and pre-immunization are reported (delta).
Cell depletion experiments
To deplete neutrophils, anti-Gr-1 ascites was injected i.p. to mice on day −1 before LPS injection and again on day 1 after LPS injection. About 65% and 88% of neutrophils were depleted in the footpad and LN respectively, as assessed by myeloperoxidase assay of solubilized tissue (Supplemental Fig. 1A, B). To deplete dendritic cells in CD11c-DTR mice, 100 ng diphtheria toxin was injected i.p. on day −1 before LPS injection on day 0. To deplete resident macrophages, clodronate liposomes (12) (50 ul) were injected into footpads on day −5; control animals received PBS-liposomes. The depletion of DCs or macrophages was documented by a92% decrease in CD11c+ cells in spleens measured by flow cytometry (LSRFortessa, BD) or by the disappearance of subcapsular and medullary CD169+ cells in DLN by immuno-microscopy, respectively (Supplemental Fig. 1C, D–G).
Results
LPS moves rapidly from a subcutaneous injection site to its draining lymph nodes yet drainage continues for many weeks
Previous studies on lymphatic drainage from subcutaneous injection sites to DLN showed that molecules of higher molecular mass, including LPS aggregates, appeared in the subcapsular sinuses within 10–12 min after injection, but were largely excluded from the lymph node cortex (parenchyma) (15). In contrast, Randolph et al. (14) found that fluorescent microspheres, which are carried from skin to DLN by (mainly dendritic) cells, appeared 24 hours or more after injection in the DLN paracortex; co-injection of LPS delayed microsphere transport by preventing the differentiation of monocytes into dendritic cells (16). We found evidence for both cell-free and cell-mediated transport of LPS from a subcutaneous site to DLN. Confirming Gretz et al’s results, we found FITC-LPS in the ipsilateral popliteal node within 3 to 10 minutes after injecting it into a hind footpad, strongly suggesting that the first LPS molecules that arrived in the DLN were not transported by cells (Fig. 1A–D). Some of the FITC-LPS co-localized with LYVE1+ lymphatic endothelium (Fig. 1A, B, D). FITC-LPS was also located in, or on the surfaces of, CD169+ macrophages in the subcapsular sinus yet it did not extend into the B cell zone (Fig. 1, A–D, E–G). In sections obtained as long as 5 days after injection, most of the FITC appeared within the subcapsular sinus and medulla (as in Fig. 1A), and the amount of detectable FITC gradually diminished over time.
FIGURE 1. Cell-free and cell-mediated LPS transport to DLN.
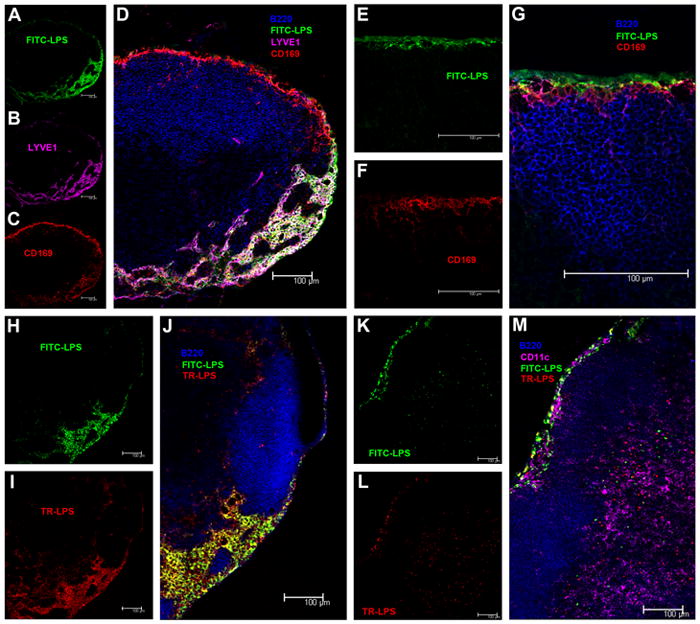
A–D,10 minutes after injection into a footpad, FITC-LPS (green, A), lymphatic endothelium, LYVE+ (purple, B) and CD 169+ macrophages (red, C) co-localize in the subcapsular sinus and medulla (overlap, D) of the popliteal node. No LPS is seen in B cell follicles (B220, blue) (D). E–G 10 minutes after injection into a footpad, FITC-LPS is found in the subcapsular sinus (green, E) where it contacts CD169+ macrophages (F) but does not enter the B cell follicle (B220, blue) (overlap, G). H–J Four hours after injection into adjacent sites on the back, FITC-LPS (green, H) and TR-LPS (red, I) had moved to the brachial node and been taken up by cells in the subcapsular sinus and medulla. Note co-localization (yellow) of the two LPS preparations in or on some cells in J (overlap). K–M Twenty-four hrs post-injection of FITC-LPS and TR-LPS into adjacent subcutaneous sites on the back, most of the FITC-LPS (K) and TR-LPS (L) are still found in the subcapsular sinus of the brachial node (upper left in images), with some co-localization (M, yellow), yet small aggregates of FITC-LPS and TR-LPS are found in the paracortex (lower right in these images) but do not co-localize (M). In M, CD11c+ cells are shown in purple.
To distinguish cell-free from cell-mediated transport from the injection site to DLN, we injected FITC-LPS and Texas Red (TR)-LPS into adjacent intradermal sites on a flank. Because LPS moves more slowly to DLN when injected into sites on the back than when injected into a footpad, we excised the draining brachial node four hours after LPS injection. The distribution of the two fluorescent labels overlapped in the subcapsular sinus and medulla, where many cells had taken up both labels (Fig. 1H–J). Although it seems likely that the two LPS preparations traveled cell-free to the DLN, where they could then be taken up by the same cells in the subcapsular sinus and medulla, it is also possible that the LPS was carried by cells and passed from cell to cell after arriving in the DLN; the slow rate with which LPS is released from macrophages in vitro argues against the latter interpretation (17) (18). One or two days after injection, in contrast, there were small, discrete collections of FITC-LPS and TR-LPS in the paracortex of the brachial node (Fig. 1K–M). Fluorescent microspheres had a similar paracortical location two days after they were injected into adjacent subcutaneous sites (Supplemental Fig. 2). This LPS was probably carried to the paracortex by cells (19); in keeping with Randolph et al. (14), we found some of it within, or in close proximity to, CD11c+ cells (Fig. 1M). When we injected glycine-FITC into a footpad as a control, we did not find FITC in the DLN.
We conclude that LPS can traffic from a subcutaneous site to DLN both cell-free and carried by cells. Most of the injected LPS passed through the DLN via the subcapsular sinus and medulla, beginning within minutes of injection and continuing for weeks; a smaller fraction of the injected LPS made its way to the paracortex, where contact with B and T cells would be expected (20).
We used radiolabeled LPS to quantitate LPS movement from the footpad to the DLN over time. The amount of 14C is a reliable marker for the number of LPS molecules because 14C is in the glucosamine backbone of lipid A and this part of the molecule does not undergo catabolism in vivo. The 14C dpm remaining in the footpad declined for at least 6 weeks following injection, as did the amount of 14C in the popliteal and inguinal nodes (Fig. 2A). At each time point from 3 to 41 days, the nodes contained 3% to 8% of the 14C dpm recovered from the footpad. At no point after day 3 was accumulation of 14C radioactivity evident in the node. LPS deacylation did not alter this pattern (Aoah+/+ vs. Aoah−/− mice) (Fig. 2A).
FIGURE 2.

A. LPS traffics from footpad injection site to DLN. Recovery of 14C dpm from LPS injected into footpads (Foot) and from their draining popliteal and inguinal lymph nodes (DLN). Symbols show the mean +/− SE of 3 measurements per time point. Decay curves were generated using PRISM (two phase exponential decay). The results from a second experiment (larger symbols) are plotted alongside the best-fit curves from the first experiment (smaller symbols), which has been previously reported (6). Closed symbols and solid lines = Aoah+/+, open symbols and interrupted lines = Aoah−/−. Blue = feet, red = DLN. B. LPS traffics to the liver. 14C radioactivity (dpm, as % of the injected dose) recovered from the plasma and liver following footpad injection of 10 μg [3H/14C] LPS. Fewer than 1% of the injected 14C counts were recovered from the plasma at any time point (round symbols, right axis label), whereas 14C gradually accumulated in the livers of both Aoah+/+ and Aoah−/− mice (triangles, left axis label). Open symbols = Aoah−/−, closed symbols = Aoah+/+. n = 3 to 6 mice/time point. Data combined from two independent experiments with similar results. Error bar =1 SEM.
LPS slowly traffics to the liver
We also measured the amount of 14C-LPS in plasma and liver at different time points after footpad injection (Fig. 2B). At no point was more than 1% of the injected dose found in the plasma. Whereas we found in previous studies that over 80% of an intravenous dose of the same radiolabeled LPS appeared in the liver within 5 minutes (12), accumulation of subcutaneously-injected LPS by the liver was much more gradual, so that by 14 days after injection the liver contained approximately 20% of the injected dose (at this time point, ~40% of the inoculum was still in the footpad (Fig. 2A) and ~10% had been excreted in the urine or feces [data not shown]).
Most LPS molecules are deacylated at the injection site
In wildtype mice, almost all of the LPS that remained in the footpad had been deacylated by one week following injection (Fig. 3A). The amount of fully acylated LPS in the footpad thus became quite low at a time when over half of the injected LPS was still in the footpad (Fig. 2A). LPS recovered from DLNs and the liver had been even further deacylated (Fig. 3B and C). Although LPS may be deacylated by Kupffer cells, which express AOAH (12), it seems likely that most of the LPS recovered from the liver had undergone deacylation within the footpad or in the DLN. Very little deacylation occurred in Aoah−/− mice (6) (Fig. 3A–C), so fully acylated LPS continued to drain to the DLN for many weeks (Fig. 3D).
FIGURE 3. LPS undergoes deacylation at the injection site.

A–C. LPS deacylation time course in the footpad, DLN and liver. Circles, Aoah−/−; boxes, Aoah+/+. Maximum deacylation is 33% (AOAH removes 2 of the 6 fatty acyl chains from the LPS backbone). In Aoah+/+ mice, almost all of the LPS in the footpad had been deacylated by day 7 after injection. D. The amount of fully acylated LPS remaining in the feet of Aoah−/− mice (open circles) and Aoah+/+ mice (closed boxes), expressed as a percentage of the LPS inoculum. The figure was plotted using data from Figs. 2A and 3A. Error bars=1 SEM. In mice that lack AOAH, fully acylated LPS continued draining from the footpad long after almost all of the LPS had been deacylated in wildtype animals.
Prolonged movement of fully acylated LPS from a peripheral site to draining lymph nodes promotes exaggerated Ab responses
If LPS deacylation by AOAH takes over a week to reach completion, how could such as low reaction limit LPS-induced polyclonal antibody production by B cells? We hypothesized that fully acylated LPS must reach the DLN over a prolonged period of time to be able to produce maximal antibody responses. If this hypothesis is correct, the exaggerated antibody responses seen in Aoah−/− animals should be prevented by excising the injection site, thus removing the supply of fully acylated LPS. To test this idea, we injected LPS into a subcutaneous site on the back on day 0. We then excised the injection site skin from Aoah−/− mice on day 1 or day 4 after injection and measured serum antibody levels 7, 14, and 21 days after injection. Aoah−/− mice that underwent skin excision on day 1 had IgM and IgG3 responses that were similar to those of Aoah+/+ mice and significantly lower than those observed in control Aoah−/− mice (subcutaneous LPS with mock skin excision) (Fig. 4, A and B). Aoah−/− mice that underwent excision of the injection site on day 4 had slightly lower Ab responses than did control Aoah−/− mice. The robust antibody responses observed in Aoah−/− mice thus require a continuous source of LPS at the inoculation site from days 1 to ~4. Although LPS deacylation at the inoculation site occurs over several days in wildtype mice, it is sufficient to limit their B cell responses. (In contrast, AOAH does not act quickly enough to be able to diminish LPS-induced cytokine production, which occurs within minutes to hours after LPS injection (11).)
FIGURE 4. Prolonged LPS drainage increases polyclonal antibody responses in Aoah−/− mice.

Mice received 10 μg LPS s.c. at a site on the right flank on day 0. The overlying skin and subcutaneous fat in the Aoah−/− mice was excised on day 1 (open triangles) or day 4 (open circles). Titers of IgM (A) and IgG3 (B) were measured before and 7, 14 and 21 days after LPS inoculation. Controls included Aoah−/− (open boxes) and Aoah+/+ (closed boxes) mice that received sham excision (from the left flank) on day 1 after LPS inoculation. In Aoah−/− mice, removing the inoculation site one day after injection reduced antibody titers almost to levels seen in Aoah+/+ mice. Data combined from two experiments, each with n = 4 mice/group. Significantly different from Aoah−/− response: *, p < 0.05, **, p< 0.01, ***, p < 0.001. Delta, change from pre-immune sera. Error bar =1 SEM.
AOAH does not modulate antibody responses to a non-LPS TLR4 agonist or to a TI-II antigen
It is important to be sure that phenomena attributed to AOAH are indeed due to its ability to deacylate LPS. Accordingly, we have previously injected mice with UT12, an agonistic monoclonal Ab to MD-2—TLR4 that, like LPS, induces murine B cell proliferation in vitro and in vivo and is a potent activator of TLR4-dependent intracellular signaling (10). We found virtually identical IgM and IgG3 responses in Aoah+/+ and Aoah−/− mice (21), suggesting strongly that the enzyme’s action on LPS accounts for its ability to modulate antibody responses in vivo. Here we asked whether Aoah−/− mice have exaggerated responses to a TI-II antigen, Ficoll-TNP. Unlike LPS and UT12, Ficoll-TNP did not induce polyclonal antibody responses, so we measured anti-TNP responses. Ficoll-TNP elicited similar anti-TNP IgM and IgG3 responses in Aoah+/+ and Aoah−/− mice, while anti-TNP responses were much higher in LPS-TNP immunized Aoah−/− mice than in Aoah+/+ mice (Supplemental Fig. 3). These results confirm that AOAH modulates antibody responses to its substrate, LPS, but not to a different TI antigen.
Multiple cell types contribute to LPS deacylation in vivo
AOAH is produced by monocytes-macrophages, neutrophils and dendritic cells. To identify the cells that contribute to deacylation in the footpad, we depleted macrophages by giving clodronate-liposomes, neutrophils by pre-treatment with anti-Gr-1 mAb, and dendritic cells by injecting diphtheria toxin into mice engineered to produce the diphtheria toxin receptor downstream of the CD11c promoter (Supplemental Fig. 1). We found that each of these cell types partially contributes to LPS deacylation in the footpad (Fig. 5A); depleting macrophages had the greatest effect. In contrast, depleting each cell type did not increase the amount of LPS that remained in feet or alter the amount of LPS that was recovered from DLNs 3 days after injection (Fig. 5B and C). These phagocytes thus may not play a critical role in transporting LPS, or perhaps other cells can compensate for the absence of a single cell type.
FIGURE 5.

A. Multiple cell types contribute to LPS deacylation in the footpad. Wildtype PBS-treated (control), macrophage-depleted, neutrophil-depleted, ordendritic cell-depleted mice were injected with [3H][14C] LPS on day 0 and their feet were harvested for study on day 3. Results are expressed relative to the amount of deacylation measured in the wildtype mice that were pre-treated with PBS. n = 6–10 mice/group. *** = p<0.001. B and C. Radiolabeled LPS was recovered in the ipsilateral foot (B) and DLN (C) 3 days after LPS injection into footpads of mice that had undergone depletion of macrophages, neutrophils, or DCs. Recovery relative to PBS controls (100%) is shown. No significant differences from the PBS-injected controls were seen. n= 6–10. Error bar =1 SEM. D and E. Macrophage depletion increases LPS-induced IgM (C) and IgG3 (D) in wild type mice. Clodronate-liposomes were injected into the footpads of Aoah+/+ and Aoah−/− mice 5 days prior to injecting LPS. Both IgM and IgG3 titers were higher in macrophage-depleted Aoah+/+ mice (closed triangles) than in Aoah+/+ controls (PBS liposomes before LPS, closed squares). Clodronate pre-treatment did not alter responses in Aoah−/− mice (open triangles, clodronate; open squares, PBS). Data combined from 2 experiments, each with n = 5/group. **, significantly different from PBS liposome control, p < 0.01; ***, p <0.001. Delta is the change from the pre-immune value. Error bar =1 SEM.
To characterize further the role played by macrophages in LPS inactivation, we studied the antibody responses of wildtype and Aoah−/− mice after depleting macrophages using clodronate-liposomes. If footpad macrophages deacylate LPS, depleting them should allow a larger fraction of the LPS injected to remain acylated, drain to regional nodes and elicit antibody production. Alternatively, LPS-responsive macrophages might produce cytokines or other mediators that would stimulate B cells and indirectly augment antibody production. When we injected footpads of Aoah+/+ mice with clodronate-liposomes 5 days before injecting LPS, we found elevated total IgM and IgG3 levels 7 and 14 days later (Fig. 5D, E), in keeping with a prominent role for macrophages in LPS inactivation. Clodronate-liposome pre-treatment did not alter LPS-induced antibody production in Aoah−/− mice. Since subcapsular CD169+ macrophages in the DLN were depleted by clodronate (Supplementary Fig. 1, D–G), this finding is also evidence that these cells do not play an essential role in LPS-induced polyclonal antibody-production, despite their close association with LPS in the sinus (Fig. 1E–G) and their ability to transfer lymph-born virus or immune complexes to B cells and promote B cell activation (22,23).
TLR4 influences LPS trafficking and deacylation in vivo
In previous studies we found that TLR4-deficient macrophages, which are unable to mount an inflammatory response to LPS, take up extracellular LPS and deacylate it at the same rates observed for wildtype macrophages (17). When we injected LPS into the footpads of Tlr4−/− mice, the amount of LPS remaining in the footpad 2 to 3 days after injection was significantly less than that observed in wildtype mice, whereas the amount recovered from the DLN was significantly higher (Fig. 6A). The local inflammatory response thus may slow the rate at which the LPS leaves a subcutaneous site. In other experiments we found that TLR4-deficient mice had deacylated about 50% less of the LPS in their feet on day 3 after inoculation than had wildtype mice (Fig. 6B), raising the possibility that TLR4 signaling may enhance AOAH expression in resident phagocytes and/or promote recruitment of AOAH-expressing cells such as neutrophils and monocytes. When we tested this hypothesis, we found that injecting LPS into one footpad of wildtype mice increased AOAH enzyme activity (Fig. 6C) and enhanced the deacylation of [3H/14C] LPS that was given into the same footpad 2 days later (Fig. 6D).
FIGURE 6. TLR4 influences LPS trafficking and deacylation in vivo.

A. Recovery of 14C-LPS from footpad and DLN 3 days after footpad injection into Tlr4−/− and wildtype mice. Data from day 2 after injection were similar. B. LPS deacylation in footpad 3 days after injection. n = 10–12 mice/group. ***= p < 0.001. Error bar =1 SEM. C and D. Pre-treatment with LPS enhances LPS deacylation in the footpad. Aoah+/+ mice were injected with PBS (n = 3) or 4 μg nonradioactive LPS (n = 3) in both footpads on day −3. On day 0, [3H/14C] LPS was injected to the right footpads of all mice. Two days later, the left feet were harvested to measure AOAH activity in tissue lysates (C) and the right feet were used to measure deacylation of the injected 3H/14C LPS (D). Maximal deacylation = ~33%. **, p < 0.01; ***, p < 0.001. Error bar =1 SEM.
Discussion
This study of LPS trafficking in vivo yielded 3 note worthy findings. First, we found that LPS moves from a local injection site principally via lymphatics. Endotoxemia, or the presence of LPS in the circulating blood, is a much-feared complication of many Gram-negative bacterial diseases. LPS in an extravascular tissue might enter the blood directly, via venous capillaries, or indirectly, by draining first through lymphatic channels to the thoracic duct. Our results indicate that lymphatics provide an important conduit from an inflamed subcutaneous site to the circulating blood, just as lymphatic drainage is a major route of LPS clearance from infected peritoneal fluid (24). The slow course of lymphatic drainage not only provides greater time for LPS inactivation by deacylation prior to entering the blood, but the LPS may also be exposed to inhibitory factors in lymph (25,26). It is possible that some LPS moved directly from the footpad into the blood, or that it was internalized by footpad phagocytes that then left the injection site and entered the bloodstream, but these seemed to be minor modes of LPS egress from the injection site. Although almost five per cent of the LPS injectate was found in the liver on day 1 after injection, the LPS recovered from the livers of wildtype mice had lost over half of its secondary acyl chains, consistent with AOAH-mediated deacylation/inactivation either prior to, or after (12), hepatic uptake of LPS from the circulation.
Second, most of the injected LPS passed through the DLN without entering the paracortex or B follicles. Radiolabeled LPS did not accumulate in the DLN. FITC-LPS that entered the DLN was found principally within the subcapsular sinus, where it was associated with macrophages (CD169+) and lymphatic endothelial cells (LYVE1+), and also in the medulla. With time, discrete foci of FITC-LPS and TR-LPS appeared in the paracortex, closely resembling the pattern observed for subcutaneously-injected fluorescent beads, suggesting that some of the LPS is carried to the DLN by cells. This was more prominent at later time points (1 to 2 days after injection). Although a minor fraction of the injected LPS was found in the paracortex, this may be sufficient to bring LPS into direct contact with B cells (20), a prerequisite for B cell stimulation by LPS in mice (MF Lu, unpublished results).
Third, our results document the important role that AOAH plays in modulating the bioactivity of LPS in vivo. Acyloxyacyl hydrolase is a lipase, found principally in myeloid cells, that removes two of the six fatty acids that are required for LPS to be sensed by its receptor on animal cells, the MD-2—TLR4 complex. In three different experimental settings – involving B cells, Kupffer cells, and peritoneal macrophages – mice that lacked AOAH were unable to restore homeostasis after they were exposed to small amounts of LPS in vivo (6,11,12). In each instance, the response to LPS exposure was exaggerated and prolonged in the absence of AOAH. Here we found that over 70% of the injected LPS was deacylated by AOAH before it left the injection site in Aoah+/+ mice. There was strong evidence that phagocytes took part in deacylating LPS, but we were unable to distinguish extracellular from intracellular deacylation. AOAH has an acid pH optimum and resides within intracellular granules, yet it may be secreted and taken up by other cells in a mannose-6-phosphate-dependent fashion (27). In addition, Gioannini et al. found that soluble CD14 and LPS binding protein (LBP) can bind LPS in a manner that makes it accessible to deacylation by AOAH (28). Although there is evidence that LPS can be released after being processed by cultured macrophages (18), the absence of a defined mechanism for this phenomenon makes it seem less likely than extracellular deacylation within the footpad by secreted AOAH.
To be able to follow LPS molecules quantitatively in vivo, we injected LPS subcutaneously into a footpad or a site on the back. Although this approach was meant to mimic the LPS released by bacteria into an infected tissue site, it required relatively high doses of LPS and utilized purified LPS instead of intact bacteria. Using rough (Ra or Rc) LPS or lipooligosaccharide (N. meningitidis) offered several advantages over smooth (long polysaccharide-containing) LPS preparations: each of the LPS preparations used here has a relatively uniform structure, with 6 fatty acyl chains attached to the lipid A moiety of most molecules, and each potently activates MD-2—TLR4 (29). A disadvantage is the tendency of rough LPS preparations to form aggregates or micelles that, while possibly resembling the size of bacterial outer membrane fragments, are clearly artificial. LPS is not soluble in methanol, yet it is possible that cell-free FITC-LPS was washed away during tissue fixation for microscopy. Although our FITC-LPS was partially deacylated during the labeling process, there is strong evidence that partially deacylated LPS structures bind to LPS binding protein (LBP), CD14 and MD-2—TLR4 (30–32). It thus seems unlikely that this degree of deacylation would alter the FITC-LPS’s ability to interact normally with cells.
To our knowledge, these are the first studies to track the fates of LPS molecules in tissues other than the bloodstream. We did not expect to find the very slow disappearance kinetics from the injection site, the prominent role played by lymphatics in removing LPS from the subcutaneous tissue, the passage of a large fraction of the LPS through DLN without entering the parenchyma of the node, or the inactivation of such a large fraction of the LPS before it entered the lymph. The immunological potency of LPS in vivo, measured here as B cell activation to produce polyclonal antibodies, was greatly influenced by the kinetics of drainage and enzymatic inactivation as well as by lymph node anatomy.
Supplementary Material
Footnotes
Funded by extramural NIH grant AI18188 to the University of Texas Southwestern Medical School, Dallas, TX, and by the Division of Intramural Research, NIAID, NIH.
Reference List
- 1.Munford RS. Sensing Gram-negative bacterial lipopolysaccharides: a human disease determinant? Infect Immun. 2008;76:454–465. doi: 10.1128/IAI.00939-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ge Y, Ezzell RM, Tompkins RG, Warren HS. Cellular distribution of endotoxin after injection of chemically purified lipopolysaccharide differs from that after injection of live bacteria. J Infect Dis. 1994;169:95–104. doi: 10.1093/infdis/169.1.95. [DOI] [PubMed] [Google Scholar]
- 3.Mathison JC, Ulevitch RJ. The clearance, tissue distribution, and cellular localization of intravenously injected lipopolysaccharide in rabbits. J Immunol. 1979;123:2133–2143. [PubMed] [Google Scholar]
- 4.Munford RS, Hall CL, Lipton JM, Dietschy JM. Biological activity, lipoprotein-binding behavior, and in vivo disposition of extracted and native forms of Salmonella typhimurium lipopolysaccharides. J Clin Invest. 1982;70:877–888. doi: 10.1172/JCI110684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skarnes RC. In vivo distribution and detoxification of endotoxins. In: Berry LJ, editor. Handbook of Endotoxin, vol. 3: Cellular Biology of Endotoxin. Elsevier Science Publishers B.V.; Amsterdam: 1985. pp. 56–81. [Google Scholar]
- 6.Lu M, Zhang M, Takashima A, Weiss J, Apicella MA, Li XH, Yuan D, Munford RS. Lipopolysaccharide deacylation by an endogenous lipase controls innate antibody responses to Gram-negative bacteria. Nat Immunol. 2005;6:989–994. doi: 10.1038/ni1246. [DOI] [PubMed] [Google Scholar]
- 7.Galanos C, Luderitz O, Westphal O. A new method for the extraction of R lipopolysaccharides. Eur J Biochem. 1969;9:245–249. doi: 10.1111/j.1432-1033.1969.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 8.Munford RS, Erwin AL. Eucaryotic lipopolysaccharide deacylating enzyme. Meth Enzymol. 1992;209:485–492. doi: 10.1016/0076-6879(92)09059-c. [DOI] [PubMed] [Google Scholar]
- 9.Tobias PS, Soldau K, Gegner JA, Mintz D, Ulevitch RJ. Lipopolysaccharide binding protein-mediated complexation of lipopolysaccharide with soluble CD14. J Biol Chem. 1995;270:10482–10488. doi: 10.1074/jbc.270.18.10482. [DOI] [PubMed] [Google Scholar]
- 10.Ohta S, Bahrun U, Shimazu R, Matsushita H, Fukudome K, Kimoto M. Induction of Long-Term Lipopolysaccharide Tolerance by an Agonistic Monoclonal Antibody to the Toll-Like Receptor 4/MD-2 Complex. Clin Vaccine Immunol. 2006;13:1131–1136. doi: 10.1128/CVI.00173-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu M, Varley AW, Ohta S, Hardwick J, Munford RS. Host inactivation of bacterial lipopolysaccharide prevents prolonged tolerance following gram-negative bacterial infection. Cell Host Microbe. 2008;4:293–302. doi: 10.1016/j.chom.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shao B, Lu M, Katz SC, Varley AW, Hardwick J, Rogers TE, Ojogun N, Rockey DC, DeMatteo RP, Munford RS. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007;282:13726–13735. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 13.Lu M, Zhang M, Kitchens RL, Fosmire S, Takashima A, Munford RS. Stimulus-dependent deacylation of bacterial lipopolysaccharide by dendritic cells. J Exp Med. 2003;197:1745–1754. doi: 10.1084/jem.20030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–761. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- 15.Gretz JE, Norbury CC, Anderson AO, Proudfoot AE, Shaw S. Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J Exp Med. 2000;192:1425–1440. doi: 10.1084/jem.192.10.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rotta G, Edwards EW, Sangaletti S, Bennett C, Ronzoni S, Colombo MP, Steinman RM, Randolph GJ, Rescigno M. Lipopolysaccharide or whole bacteria block the conversion of inflammatory monocytes into dendritic cells in vivo. J Exp Med. 2003;198:1253–1263. doi: 10.1084/jem.20030335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munford RS, Hall CL. Uptake and deacylation of bacterial lipopolysaccharides by macrophages from normal and endotoxin-hyporesponsive mice. Infect Immun. 1985;48:464–473. doi: 10.1128/iai.48.2.464-473.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duncan RL, Jr, Morrison DC. The fate of E. coli lipopolysaccharide after the uptake of E. coli by murine macrophages in vitro. J Immunol. 1984;132:1416–1424. [PubMed] [Google Scholar]
- 19.Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5:617–628. doi: 10.1038/nri1670. [DOI] [PubMed] [Google Scholar]
- 20.Qi H, Egen JG, Huang AYC, Germain RN. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science. 2006;312:1672–1677. doi: 10.1126/science.1125703. [DOI] [PubMed] [Google Scholar]
- 21.Munford R, Lu M, Varley A. Chapter 2: Kill the bacteria…and also their messengers? Adv Immunol. 2009;103:29–48. doi: 10.1016/S0065-2776(09)03002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, Van RN, Mempel TR, Whelan SP, von Andrian UH. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature. 2007;450:110–114. doi: 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- 23.Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, Whelan SP, Guidotti LG, von Andrian UH. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature. 2010;465:1079–1083. doi: 10.1038/nature09118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olofsson P, Nylander G, Olsson P. Endotoxin: routes of transport in experimental peritonitis. Am J Surg. 1986;151:443–446. doi: 10.1016/0002-9610(86)90098-x. [DOI] [PubMed] [Google Scholar]
- 25.Lemaire LCJM, Van Lanschot JB, van der Poll T, Buurman WA, van Deventer SJH, Gouma DJ. Lymph of patients with a systemic inflammatory response syndrome inhibits lipopolysaccharide-induced cytokine production. J Infect Dis. 1998;178:883–886. doi: 10.1086/515348. [DOI] [PubMed] [Google Scholar]
- 26.Munford RS. Detoxifying endotoxin: time, place, person. J Endotoxin Res. 2005;11:69–84. doi: 10.1179/096805105X35161. [DOI] [PubMed] [Google Scholar]
- 27.Feulner JA, Lu M, Shelton JM, Zhang M, Richardson JA, Munford RS. Identification of acyloxyacyl hydrolase, a lipopolysaccharide-detoxifying enzyme, in the murine urinary tract. Infect Immun. 2004;72:3171–3178. doi: 10.1128/IAI.72.6.3171-3178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gioannini TL, Teghanemt A, Zhang D, Prohinar P, Levis EN, Munford RS, Weiss JP. Endotoxin-binding Proteins Modulate the Susceptibility of Bacterial Endotoxin to Deacylation by Acyloxyacyl Hydrolase. J Biol Chem. 2007;282:7877–7884. doi: 10.1074/jbc.M605031200. [DOI] [PubMed] [Google Scholar]
- 29.Huber M, Kalis C, Keck S, Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Beutler B, Galanos C, Freudenberg MA. R-form LPS, the master key to the activation of TLR4/MD-2-positive cells. Eur J Immunol. 2006;36:701–711. doi: 10.1002/eji.200535593. [DOI] [PubMed] [Google Scholar]
- 30.Kitchens RL, Ulevitch RJ, Munford RS. Lipopolysaccharide (LPS) partial structures inhibit responses to LPS in a human macrophage cell line without inhibiting LPS uptake by a CD14-mediated pathway. J Exp Med. 1992;176:485–494. doi: 10.1084/jem.176.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kitchens RL, Munford RS. Enzymatically deacylated lipopolysaccharide (LPS) can antagonize LPS at multiple sites in the LPS recognition pathway. J Biol Chem. 1995;270:9904–9910. doi: 10.1074/jbc.270.17.9904. [DOI] [PubMed] [Google Scholar]
- 32.Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.