

Abstract
Fas ligand (FasL) belongs to the tumor necrosis factor (TNF) family of death ligands, and its binding to the Fas receptor leads to activation of several downstream signaling pathways and proteins, including nuclear factor-kappa B (NF-κB) and phosphatidylinositol 3-kinase (PI3K)/Akt. However, it is not known whether cross talk exists between NF-κB and PI3K/Akt in the context of FasL signaling. We demonstrate using both human renal epithelial 293T cells and Jurkat T-lymphocyte cells that although FasL activates both Akt and NF-κB, Akt inhibits FasL-dependent NF-κB activity in a ROS-dependent manner. FLICE-inhibitory protein (c-FLIP), an antioxidant and an important component of the DISC, also represses NF-κB upstream of the regulatory I kappa B kinase-gamma (IKKγ) protein sub-unit in the NF-κB signaling pathway, and positive cross talk exists between Akt and c-FLIP in the context of inhibition of FasL-induced NF-κB activity. The presence of two death effector domains (DED) of c-FLIP, and S-nitrosylation of its caspase-like domain were found to be important for mediating c-FLIP-dependent down-regulation NF-κB activity. Taken together, our study reveals a novel link between NF-κB and PI3K/Akt, and establishes c-FLIP as an important regulator of FasL-mediated cell death.
INTRODUCTION
Apoptotic cell death plays an important role in a number of physiological and pathophysiological conditions (1). Apoptosis is mediated primarily by the tumor necrosis factor (TNF) family of proteins, and Fas (APO-1 or CD95) is an important member of this death receptor family, which triggers cell death primarily by binding Fas ligand (FasL) (2). FasL binds Fas as a trimer at the cell surface, initiating the apoptosis cascade by the formation of a death-inducing signaling complex (DISC) (3–5). Caspase-8 is recruited to the DISC, where its gets self-activated by proteolytic cleavage, subsequently activating downstream effector caspases (6). In addition to the formation of the DISC and subsequent activation of pro-apoptotic proteins, FasL binding also causes the production of reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) and superoxide (·O2−−) leading to cell death via oxidative stress signaling (7, 8).
An important mediator of caspase-8 activation at the DISC is a structurally related protein known as cellular FLICE inhibitory protein (c-FLIP) (9). Initially identified as an inhibitor of caspase-8, it is now known that c-FLIP can also exert pro-apoptotic effects (10, 11). In addition to the full-length form of c-FLIP (55 kDa) or p55-FLIP (c-FLIPL) that contains two death effect domains (DED) and a caspase-like domain, two main proteolytically processed forms of c-FLIPL have been characterized – p43-FLIP (43kDa), and the shorter p22-FLIP (22 kDa), which contains only the two DED domains (12, 13). In addition to proteolytic fragments mentioned above, splice variants of cFLIPL, known as cFLIPR and cFLIPS also exist, which have been shown to play an important role in FasL-induced apoptosis (14, 15).
A growing number of reports demonstrate a direct correlation between carcinogenesis and the ectopic expression levels of c-FLIP. However, studies quantifying total c-FLIP in various cells lines show that the physiological level of c-FLIP expression is extremely low, and approximates to only 1% of endogenous pro-caspase 8 levels (12, 16). The functional role of c-FLIP depends upon its expression levels; extremely low levels of c-FLIP typically induce apoptosis whereas higher levels are cytoprotective (17–23).
Apart from proteins at the DISC, activation of NF-κB by Fas receptor (FasR) stimulation has been reported to occur in several cellular systems (16, 24–26). Upon FasL-induced DISC formation, the I kappa B kinase (IKK) complex is activated by receptor interacting protein (RIP), leading to the phosphorylation of I kappa B (IκB) and its subsequent degradation via the ubiquitin-proteosome pathway (27). Degradation of IκB releases the NF-κB complex (composed of p50 and p65 subunits) into the nucleus, leading to subsequent activation of a variety of target genes (28, 29). Mice knockout studies that involve silencing of NF-κB and IKKβ genes showed massive apoptosis of hepatocytes and embryonic lethality (30–32), indicating that NF-κB plays a critical anti-apoptotic role in death-receptor induced signaling.
An important mediator of NF-κB activity that also regulates cell death is the serine/threonine protein kinase Akt/PKB, which is activated via phospho-inositides produced by phosphotidylinositol 3-kinase (PI3K) (33, 34). Depending upon a variety of factors, Akt may have either pro- or anti-apoptotic effects in response to FasL stimulation (35). Akt can stimulate expression of anti-apoptotic proteins such as c-FLIP and inhibitors of apoptosis (IAPs), and suppress pro-apoptotic proteins such as caspase-9 and cytochrome c, thus exhibiting a dual role (33, 36–38).
Although both Akt and NF-κB have been extensively studied for their role in death-receptor signaling, and both are sensitive to FasL, it is not known whether cross talk exists between the two pathways in the context of FasL-induced cell death. Here, we report that FasL can activate both Akt and NF-κB; however, Akt exerts an inhibitory effect on NF-κB in a ROS- and c-FLIP-dependent manner. c-FLIP is also capable of independently inhibiting FasL-mediated NF-κB activity through downregulation of ROS, which occurs upstream of the IKK complex. S-nitrosylation and processing of c-FLIP are also important factors in determining c-FLIP-dependent NF-κB down-regulation.
This study may broaden the scope of the signaling potential of c-FLIP in the regulation of apoptosis at extremely low levels, and establish a hitherto unknown link between the PI3K/Akt and NF-κB pathway in FasL-mediated cell death. Further, it expands the role of c-FLIP to include a novel role in mediating Akt–NF-κB cross talk, which has significant implications in inflammation, cytotoxicity and cellular homeostasis.
MATERIALS AND METHODS
Chemicals and Reagents
Recombinant Fas ligand (SuperFasL) and monoclonal antibody to c-FLIP (Dave-2) were purchased from Alexis Biochemicals (San Diego, CA). Wortmannin, N-acetyl cysteine (NAC) and 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) were obtained from Calbiochem (La Jolla, CA). The oxidative probes, dichlorofluorescein diacetate (DCF-DA) and dihydroethidium (DHE) were from Molecular Probes (Eugene, OR). Antibodies for IκB, phospho-IκB, gylceraldehyde 3-phosphate dehydrogenase (GAPDH), myc and peroxidase-labeled secondary antibodies were obtained from Cell Signaling Technology (Danvers, MA). The transfecting reagent LipofectAMINE 2000 was from Invitrogen (Carlsbad, CA). For transfection of Jurkats, Metafectene Pro was procured from Biontex (San Diego, CA) and transfections carried out using manufacturer-recommended protocol. The dual-luciferase assay kit was purchased from Promega Corporation (Madison, WI).
Plasmids
The pcDNA3-FLIP (c-FLIP) plasmid was generously provided by Dr. Christian Stehlik (Northwestern University, Chicago, IL). The open reading frame of c-FLIP was amplified by high fidelity PCR (Stratagene, Cedar Creek, TX) from the corresponding expressed sequence tags (ESTs) and cloned into pcDNA3 expression vectors containing the N-terminal myc epitope tag. FLIP domain mutants containing either the first death effector domain (Δ3), both death domains (Δ2/p22-FLIP) and part of the caspase-like domain (Δ1/p43-FLIP) were generous gifts from Dr. Robert C. Thome (Burnham Institute, La Jolla, CA). Myc-tagged non-nitrosylable mutant of FLIP (FLIP-C2A) was generated using the Quik-Change mutagenesis kit (Stratagene, Cedar Creek, TX). Authenticity of all constructs was verified by DNA sequencing. The IKKγ mutant plasmids for both the constitutively active form (IKKγ-WT) and the dominant negative form (IKKγ-DN) were a generous gift from Dr. Fei Chen (National Institute for Occupational Safety and Health, Morgantown, WV). The constitutively active Akt plasmid (Akt-WT) was generated by inserting the open reading frame of Akt into the pcDNA3 expression vector. The NF-κB reporter plasmid (NF-κB-Luc) was a kind gift from Dr. Peter Johnson (National Cancer Institute, Frederick, MD). The amount of DNA was normalized in all transfection experiments with pcDNA3. Expression of proteins was verified by Western blotting.
Cell Culture
Both human renal epithelial 293T cells and Jurkat T-lymphocyte cells were obtained from the American Type Culture Collection (Manassas, VA). 293T cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% fetal bovine serum, 200mM L-glutamine, 100U/ml penicillin, 100μg/ml streptomycin and 5% CO2 in plates pre-coated with 0.02% gelatin prepared in phosphate buffered saline (PBS). Twenty-four hours prior to transfection, cells were seeded into the required format (12- or 48-well) on plates pre-coated with 10μg/ml collagen IV (rat tail) in 10% DMEM medium without antibiotics. Jurkat suspensions were maintained in RPMI medium supplemented with 10% fetal bovine serum, 400mM L-glutamine, 100U/ml penicillin, 100μg/ml streptomycin and maintained in 5% CO2. On the day of experiment, cells were spun down, split and resuspended in appropriate medium depending on the specific assay.
ROS Detection
Intracellular peroxide and ·O2−− production was determined by fluorometric analysis using specific probes DCF and DHE, respectively. Following appropriate treatments, cells (1×105/ml) were incubated with the fluorescent probes (5μM) for 30min at 37°C, after which the cells were washed, re-suspended in PBS, and analyzed for DCF (485/535 nm) and DHE fluorescence (535/610 nm) intensity using a multi-well plate reader (FLUOstar OPTIMA, BMG LABTECH Inc., Durham, NC).
Western Blotting
After specific treatments, 293T cells were incubated in lysis buffer containing 20mM Tris-HCl (pH 7.5), 1% Triton X-100, 150mM NaCl, 10% glycerol, 1mM sodium orthovanadate, 50mM sodium fluoride, 100mM phenylmethylsulfonyl fluoride (PMSF), and a protease inhibitor mixture (Roche Molecular Biochemicals, Basel, Switzerland) for 20 min on ice. Alternatively, for Jurkats, cells were first collected and spun down at 2000 rpm for 5 min, and then incubated in lysis buffer. After insoluble debris was precipitated for both 293Ts and Jurkats by centrifugation at 14,000g for 15 min at 4°C, the supernatants were collected and assayed for protein content using bicinchoninic acid (BCA) method (Pierce Biotechnology, Rockford, IL). Equal amount of protein per sample (30μg) were resolved on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a 0.45-μm nitrocellulose membrane (Pierce). The transferred membranes were blocked for 1 h in 5% non-fat dry milk in TBST (25mM Tris-HCl (pH 7.4), 125mM NaCl, 0.05% Tween-20) and incubated with the appropriate primary antibodies and horseradish peroxidase-conjugated isotype specific secondary antibodies. The immune complexes were detected by chemiluminescence (Supersignal® West Pico, Pierce) and quantified by imaging densitometry, using UN-SCAN-IT automated digitizing software (Silk Scientific Corp., Orem, UT). Mean densitometry data from independent experiments were normalized to the control where indicated. The data were presented as mean ± S.D. and analyzed by Student’s t test.
NF-κB Reporter Gene Assays
Cells were seeded in 48-well plates and cultured to 80–90% confluence. Cells were co-transfected with 100ng/well of NF-κB reporter (NF-κB-Luc) plasmid, 10ng/well of the renilla luciferase vector (pRL-tk) and differing amounts of c-FLIP plasmid (either full length or domain mutants). Empty pcDNA-3 control vector was used either as a control or added to c-FLIP such that the total DNA in each well was equal. In other experiments, plasmid constructs for Akt and IKK (described above) were co-transfected (1μg/well) as described, and equal concentration of empty pcDNA3 vector was used as control. Cell were transfected using the LipofectAMINE 2000 transfection reagent (Invitrogen, Carlsbad, CA) for 293T cells or Metafectene Pro for Jurkats respectively, according to the manufacturer’s protocol. After a 24 h recovery period, transfected cells were treated with CD95/Fas ligand (FasL) with or without pre-treatment with inhibitors for 12 h. Cell extracts were prepared and analyzed for luciferase activity using the Promega dual-luciferase assay kit.
Luciferase Activity Assay
Luciferase activity was measured by enzyme-dependent light production using a luciferase assay kit (Promega, Madison, WI). After each experiment, cells were washed and incubated at roomtemperature for 10 min in 250μl of lysis buffer (Promega). For analysis, 10μl of each sample was loaded in an automated luminometer (Bio-Rad, Hercules, CA). At the time of measurement, 100μl of luciferase assay reagent (LAR) was automatically injected in each sample, and total firefly luminescence was measured over a 10 sec time interval. This was immediately followed by the injection of 100μl of stop-and-glow (SNG) reagent to measure the control renilla luminescence. The output was quantified as total firefly light units relative to the control renilla luminescence for each sample.
Apoptosis measurements
Sub-confluent (80%) densities of cells were treated with FasL and incubated with 10μg/ml Hoechst 33342 nuclear stain for 30 min at 37°C. The percentage of cells having intensely condensed chromatin and/or fragmented nuclei was scored by fluorescence microscopy (Axiovert 100; Carl Zeiss) using Pixera software. Data from at least ten separate fields were recorded and plotted.
Statistical Analysis
The data represent mean ± SD from three or more independent experiments. Statistical analysis was performed byStudent’s t test at a significance level of p < 0.05 for all experiments for the indicated data sets.
RESULTS
FasL-dependent NF-κB activity is mediated by hydrogen peroxide
The activation of NF-κB upon FasL stimulation was first ascertained in human renal epithelial cells (293Ts) using a NF-κB luciferase reporter system, which has been shown to be a reliable indicator of NF-κB promoter activity (39). FasL induced NF-κB activation in a dose-dependent manner; a dose-dependent increase in overall apoptosis was also observed (Fig. 1A). Since the role of ROS in mediating the pro-apoptotic effect of death ligands such as FasL tumor necrosis factor-alpha (TNF-α) is well established (40, 41), we assessed for ROS involvement in mediating NF-κB activity observed with FasL stimulation. Therefore, cells were pre-treated with the general anti-oxidant N-acetyl cysteine (NAC) prior to assaying for NF-κB. NAC inhibited FasL-dependent NF-κB activation, suggesting that ROS may play a positive role in mediating FasL-induced NF-κB activation (Fig. 1B). Both superoxide (·O2−−) and hydrogen peroxide (H2O2) are important second messengers generated upon FasL treatment in mouse macrophages (39). In order to identify the dominant ROS involved in FasL-induced NF-κB activation, we used specific fluorescent probes for H2O2 (DCF) and ·O2−− (DHE). FasL induced H2O2 in a dose-dependent manner, and NAC significantly inhibited this effect (Fig. 1C); however, ·O2−− levels remained largely unchanged with increasing doses of FasL, even in the presence of NAC (Fig. 1D). Pre-treatment of cells with catalase, a specific H2O2 scavenger was sufficient to cause inhibition of both FasL-induced NF-κB activity and overall H2O2 levels (Figs. 1, E and F). Thus, H2O2 seemed to be the specific ROS that seemed to play a predominant role in FasL signaling and subsequent NF-κB activity.
Fig. 1. FasL-dependent NF-κB activity is mediated by hydrogen peroxide.

A. Cells were co-transfected with 100ng/well of NF-κB-Luc and 10ng/well of pRL-tk normalizing luciferase plasmid for 24 h. Transfected cells were treated with increasing concentrations of FasL (0–200ng/ml) for 12 h and assayed for NF-κB luciferase activity. Plots show relative NF-κB activity over non-treated control (*P < 0.05 for each FasL data point as compared to control without FasL). Concurrently, cells that were treated with increasing concentrations of FasL were assayed for apoptosis using the Hoechst assay and graphed. B. Cells co-transfected with the NF-κB-Luc (100ng/well) and pRL-tk vectors (10ng/well) for 24 h. Transfected cells were pre-treated with 10mM NAC for 1 h followed by treatment with increasing doses (0–200ng/ml) of FasL for 12 h. NF-κB activity was measured by luciferase assay. C and D. Cells were either left untreated or were pre-treated with 10mM NAC for 1 h followed by treatment with various concentrations (0–200ng/ml) of FasL. Cells were then analyzed for either (B) H2O2 or (C) ·O2− production by measuring DCF and DHE fluorescence intensity, respectively. Plots show relative fluorescence intensity over non-treated control at the peak response time of 1 h. E. Cells were pre-treated with 1000U/ml catalase for 1 h, followed by FasL treatment (0–200ng/ml) for 12 h and were analyzed for H2O2 production by measuring DCF fluorescence intensity. Plots show relative fluorescence intensity over non-treated control at the peak response time of 1 h. F. Cells co-transfected with the NF-κB-Luc (100ng/well) and pRL-tk vectors (10ng/well) were pretreated with 1000U/ml catalase for 1 h prior to treatment with FasL (0–200ng/ml) for 12 h. FasL-dependent NF-κB activity was measured by luciferase assay.
FasL activates PI3K/Akt in a dose-dependent manner – Akt negatively regulates NF-κB
In conformity with previous studies that demonstrated PI3K/Akt regulation by FasL (7, 39, 42), we observed that increasing doses of FasL induced phosphorylation of PI3K/Akt in 293T cells without affecting total Akt levels (Fig. 2A). Since FasL had a positive effect on both PI3K/Akt and NF-κB, we assessed for potential cross talk between the two molecules in the context of FasL signaling. 293T cells were pretreated with PI3K inhibitors LY294002 and wortmannin, and assayed for NF-κB activity using the luciferase reporter assay. Surprisingly, inhibition of PI3K/Akt led to a marked increase in NF-κB activity in the presence of FasL (Fig. 2B). A similar effect was observed in Jurkats as well (Supplemental Fig. 2). In order to ascertain whether Akt was directly involved in NF-κB inhibition, or acted downstream of other molecules in the PI3K/Akt pathway, cells were co-transfected with a plasmid encoding constitutively active Akt (Akt-WT) along with the NF-κB reporter plasmid in 293T cells, and luciferase activity was measured in the absence or presence of FasL. We observed a decrease in NF-κB luciferase activity with over-expression of Akt plasmid, and Akt-mediated inhibition was observed even in the face of LY294002, suggesting a direct role for Akt in down-regulation of FasL-dependent NF-κB signaling (Fig. 2C). Thus, although FasL activates both PI3K/Akt as well as NF-κB, Akt inhibits NF-κB upon FasL stimulation.
Fig. 2. FasL activates PI3K/Akt in a dose-dependent manner – Akt negatively regulates NF-κB.

A. 293T cells were treated with increasing doses of FasL (25–200ng/ml) or left in serum free medium (SFM) alone without FasL (referred to as “Ntx” here and henceforth in the figures) for 1 h and assayed for total Akt and phosphorylated Akt-473 levels by immunoblot analysis. Blots were re-probed with GAPDH antibody to confirm equal loading of the samples. The immunoblot signals for phosphorylated Akt were quantified by densitometry. B. Cells co-transfected with the NF-κB-Luc and pRL-tk vectors were either left untreated or pre-treated with 20μM LY294002 or 10μM wortmannin for 1 h. Following pre-treatment, cells were treated with FasL (0–200ng/ml) for 12 h, and assayed for NF-κB activity. Concurrently, cells were lysed and assayed for phosphorylated Akt to confirm modulation of Akt using western blotting. Blots were reprobed with GAPDH antibody to confirm equal loading of samples. C. In addition to the luciferase vectors described in A, cells were co-transfected with either 1μg of control pcDNA3 plasmid or constitutively active Akt plasmid (Akt-WT). Post transfection, cells were treated with 200ng/ml FasL for 12 h in the presence or absence of 20μM LY294002 (1 h pre-treatment) and assayed for NF-κB activity by luciferase assay. Further, FasL-treated lysates were probed for phosphorylated Akt to confirm effect of Akt-WT plasmid. Plots show relative NF-κB levels over non-treated control. Values are mean ± SD (n = 4). *P < 0.05 versus non-treated control. **P < 0.05, #P < 0.05 versus FasL-treated datasets.
c-FLIP is a negative regulator of ROS generation and FasL-induced NF-κB activity
Since several studies have implicated c-FLIP in the regulation of NF-κB (13, 43), we investigated its role in mediating the effect of FasL on NF-κB. Transfection of 293T cells with increasing doses of plasmid encoding c-FLIP at low concentrations (0–4ng) alone caused a small albeit significant increase in basal NF-κB activity; however, c-FLIP strongly inhibited NF-κB levels in the presence of FasL in a dose-dependent manner (Fig. 3A and Supplemental Figure 2). In order to assess whether the observed changes in NF-κB reporter levels were reflected in modulation of protein expression of NF-κB-regulatory proteins, we measured protein expression levels of total IκB subunit, an inhibitory protein that remains bound to inactive NF-κB and sequesters it in the cytoplasm (44), in the presence or absence of FasL. As with NF-κB, c-FLIP alone down-regulated IκB protein levels in a dose-dependent manner (Fig. 3B); however, in the presence of FasL, c-FLIP up-regulated IκB (Fig. 3C). This suggested a dichotomy in c-FLIP signaling depending upon the absence or presence of FasL stimulation. Also, in addition to NF-κB, c-FLIP down-regulated H2O2 levels in the presence of FasL (Fig. 3D). Given that inhibition of H2O2 leads to direct down-regulation of NF-κB (Figs 1B and 1F), the inhibitory effects of c-FLIP on NF-κB may also be driven by its anti-oxidant effect in the presence of FasL. In order to assess whether modulation of H2O2 by c-FLIP led to an effect on NF-κB activity, cells co-transfected with c-FLIP and NF-κB reporter plasmid were assayed for luciferase activity in the presence of both catalase and NAC. As shown previously, pre-incubation with NAC and catalase caused a decrease in NF-κB activity in the presence of FasL. Interestingly, transfection with c-FLIP caused a further decrease in NF-κB activity as compared to ROS inhibitors alone (Fig. 3E).
Fig. 3. c-FLIP is a negative regulator of ROS generation and FasL-induced NF-κB activity.

In addition to transfection with 100ng/well NF-κB-Luc and 10ng/well pRL-tk normalization luciferase construct, cells were transfected with either empty pcDNA3 vector or increasing concentrations (0–4ng/ml) of c-FLIP. Total DNA in each cell was kept equal by addition of empty control vector. Further, cells were lysed and assayed for total c-FLIP protein using immunoblot analysis. Blots were re-probed for GAPDH to ensure equal loading. A. Transfected cells were treated with (0–200ng/ml) FasL for 12 h and analyzed for NF-κB activity by luciferase assay. Plots show relative luciferase activity over pcDNA3 control. B, C. Cells transfected with either control pcDNA3 or c-FLIP plasmids (0–4ng/ml) were assessed for total IκB levels in the absence (B) or presence (C) of FasL (200ng/ml) by immunoblotting. Blots were re-probed with GAPDH antibody to confirm equal loading of the samples. The immunoblot signals were quantified by densitometry. D. Cells transfected with either control pcDNA3 or c-FLIP plasmids (0–4ng/ml) were treated with 200ng/ml FasL (12 h) with or without 1000U/ml catalase (1 h pre-treatment) and analyzed for H2O2 by measuring DCF fluorescence. Plots show relative fluorescence intensity over non-treated control at the peak response time of 1 h. Values are mean ± SD (n = 4). *P < 0.05 versus non-treated control; *P < 0.05 versus pcDNA controls. **P < 0.05 and ##P < 0.05 as shown. E. Cells were co-transfected with 100ng/well of NF-κB-Luc plasmid, 10ng/well of pRL-tk plasmid and 4ng/ml of either pcDNA3 or c-FLIP plasmids. 24 h post transfection, cells were treated with 200ng/ml FasL (12 h), with or without 1 h pre-treatment with 10mM NAC and 1000U/ml catalase. Treated cells were lysed and assayed for NF-κB luciferase activity, and plotted.
Akt-dependent NF-κB inhibition occurs through c-FLIP
Thus far, the data suggests that both c-FLIP and Akt repress FasL-induced NF-κB activity. Furthermore, this inhibitory action of both proteins is mediated at least in part by ROS, suggesting that cross-talk may exist between Akt and c-FLIP in the context of FasL signaling. In order to assess whether such an interaction exists, 293T cells were transfected with either empty pcDNA3 vector or 4ng c-FLIP, and assayed for activated Akt. Transfection with c-FLIP led to increased levels of phosphorylated Akt in the presence of FasL, and this increase was sustained even in cells pretreated with LY294002 and NAC (Fig. 4A). Pre-treatment with NAC also upregulated basal level of FasL-induced Akt in the absence of c-FLIP (Fig. 4A – lane 5), which suggests that the positive effect of c-FLIP on Akt could at least partially be due to its anti-oxidant role. Conversely, modulation of Akt activity also led to a concurrent change in levels of c-FLIP (Fig. 4B). Inhibition of Akt by pre-treatment with LY294002 decreased c-FLIP protein expression, whereas transfection with the constitutively active Akt-WT plasmid increased c-FLIP protein levels.
Fig. 4. Akt-dependent NF-κB inhibition occurs through c-FLIP.
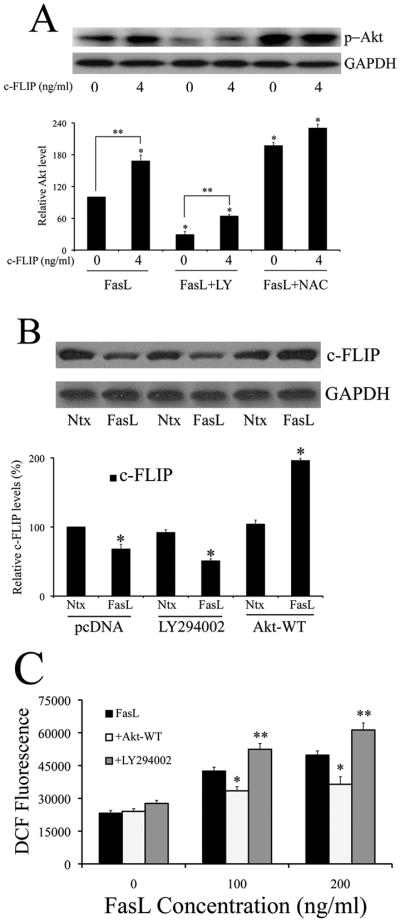
A. Cells transfected with either 4ng/ml of either pcDNA3 or c-FLIP was treated with 200ng/ml FasL (12 h) in the presence or absence of 20μM LY294002 or 10mM NAC (1 h pre-treatment). Treated cells were analyzed for phospho-Akt levels by immunoblotting. Blots were re-probed with GAPDH antibody to confirm equal loading of the samples. The immunoblot signals were quantified by densitometry (*P < 0.05 for all data points as compared to FasL treatment alone without c-FLIP (data point 1). **P < 0.05 for c-FLIP versus pcDNA-transfected datasets for both FasL treatment and FasL treated with LY294002). B. Cells transfected with 4ng/ml c-FLIP, in addition to 1mg of either Akt-WT or pcDNA control plasmid, were exposed to 200ng/ml FasL in the absence or presence of 20μM LY294002 (1 h pre-treatment) and probed for c-FLIP levels using western blotting. Blots were re-probed with GAPDH antibody to confirm equal loading of the samples. The immunoblot signals were quantified by densitometry. C. Cells were either transfected with 1μg Akt-WT plasmid or pcDNA3 control plasmid for 24 h. pcDNA3-transfected cells were then either pre-treated or with SFM with 20μM LY294002 for 1 h, followed by FasL treatment for 12 h. Akt-WT transfected cells were directly treated with FasL for 12 h. The sampled were then analyzed for H2O2 production by measuring DCF fluorescence intensity. Plots show relative fluorescence intensity over non-treated control at the peak response time of 1 h. Values are mean ± SD (n = 4). *P < 0.05, **P < 0.05 versus FasL-treated controls.
Since Akt has a positive effect on c-FLIP, this suggests that Akt may also indirectly possess anti-oxidant activity in the context of FasL signaling, which was ascertained by measuring the effect of modulation of Akt on H2O2 levels. Indeed, pre-treatment with LY294002 led to an increase in H2O2 levels, whereas transfection with Akt-WT plasmid had an inhibitory effect (Fig. 4C).
Regulation of NF-κB is mediated by positive cross talk between c-FLIP and Akt
Since we had independently demonstrated that Akt and c-FLIP exerted inhibitory effect on NF-κB in the presence of FasL, we wanted to assess for cross talk between these two molecules in the context of NF-κB inhibition, and the putative role of ROS in such an interaction. We first used PI3K/Akt modulators in the absence or presence of low levels of c-FLIP, and assayed for FasL-induced NF-κB activity. Cells transfected with c-FLIP significantly inhibited LY294002- and wortmannin-mediated increase in FasL-dependent NF-κB activity (Fig. 5A). This was reflected even in phosphorylated IκB, with c-FLIP inhibiting phospho-IκB levels in a dose dependent manner in the presence of LY294002 and NAC (Fig. 5B). Therefore, regulation of FasL-induced NF-κB by Akt seems to be mediated by c-FLIP. This was further confirmed by assaying for NF-κB luciferase activity in cells transfected with constitutively active Akt-WT plasmid. c-FLIP inhibited NF-κB in cells transfected with Akt-WT plasmid, and this effect was sustained even in the presence of LY294002 and NAC (Fig. 5C). Finally, assaying for H2O2 levels with c-FLIP-transfected cells in the presence of PI3K/Akt modulators showed an inhibition of DCF fluorescence with c-FLIP, which confirmed a role for ROS in c-FLIP-mediated down-regulation of FasL-induced NF-κB levels (Fig. 5D). Taken together, the data suggests that both Akt and c-FLIP play a co-regulatory role in inhibiting FasL-induced NF-κB activity. Furthermore, the data shows that the anti-oxidant role of c-FLIP is important in driving its cross talk with Akt, and also suggests an important overall role for ROS in FasL-induced NF-κB activity.
Fig. 5. Regulation of NF-κB is mediated by positive cross-talk between c-FLIP and Akt.

A. Cells were co-transfected with 100ng/well of NF-κB-Luc plasmid, 10ng/well of pRL-tk plasmid and 4ng/ml of either pcDNA3 or c-FLIP plasmids. 24 h post transfection, cells were treated with 200ng/ml FasL (12 h), with or without 1 h pre-treatment with 20μM LY294002 and 10μM wortmannin. Treated cells were lysed and assayed for NF-κB luciferase activity. B. Levels of phosphorylated IκB were assessed for cells transfected with increasing doses of c-FLIP (0–4ng/ml) by western blotting. Transfected cells were treated with 200ng/ml FasL (12 h) alone or in the presence of 20μM LY294002 or 10mM NAC (1 h pre-treatment). Blots were re-probed with GAPDH antibody to confirm equal loading of the samples. The immunoblot signals were quantified by densitometry. C. Cells were co-transfected with 1μg of Akt-Wt and 4ng/ml c-FLIP, and assessed for NF-κB activity in the presence of 200ng/ml FasL (12 h) with our without 20μM LY294002 or 10mM NAC (1 h pre-treatment). D. Cells were either transfected with 1μg Akt-WT plasmid or pcDNA3 control plasmid for 24 h, in addition to 4ng/ml c-FLIP plasmid. pcDNA3-transfected cells were pre-treated with 20μM LY294002 for 1 h, followed by FasL treatment (0–200ng/ml) for 12 h. Akt-WT transfected cells were directly treated with FasL (0–200ng/ml) for 12 h. The samples were then analyzed for H2O2 production by measuring DCF fluorescence intensity. Plots show relative fluorescence intensity over non-treated control at the peak response time of 1 h. Values are mean ± SD (n = 4). *P < 0.05, **P < 0.05 and ##P < 0.05 versus control datasets as shown in the respective figures.
Regulation of NF-κB by c-FLIP is upstream of IKK
Since both c-FLIP and Akt can regulate NF-κB, we wanted to assess which regulatory components of the NF-κB pathway could mediate this inhibitory effect. The IKK complex lies upstream of NF-κB and is composed of three subunits – α, β and γ. Previous studies have shown that the IKK-γ subunit plays a regulatory role in NF-κB activity, and can be directly modulated by c-FLIP (13). Therefore, we co-transfected cells with either a constitutively active (IKK-WT) or dominant negative (IKK-DN) form of the IKK-γ subunit, along with 4ng/ml c-FLIP, and assessed the effect on NF-κB luciferase activity. In the absence of c-FLIP, IKK-WT caused a basal increase in NF-κB activity (graph middle), whereas IKK-DN decreased it both in the presence or absence of FasL (graph right) (Fig. 6, A and B). Transfection with c-FLIP down-regulated both IKK-WT- and IKK-DN-mediated NF-κB activity (Fig. 6, B and C). This suggested that c-FLIP acted upstream of the IKK complex. Further, over-expression of IKKγ by transfection of the IKK-WT plasmid was not sufficient to overcome c-FLIP-mediated down-regulation of FasL-induced NF-κB activity. This effect was at least partially mediated by ROS and PI3K/Akt, since c-FLIP could further inhibit FasL-dependent NF-κB levels in the presence of NAC (Fig. 6B) and LY294002 (Fig. 6C). Overall, the data suggested that the inhibitory effect of c-FLIP on NF-κB occurs upstream in the IKK pathway, possibly by modulation of the IKK complex.
Fig. 6. Regulation of NF-κB by c-FLIP is upstream of IKK.

A. Cells transfected with either pcDNA control plasmid or 4ng/ml c-FLIP and exposed to 200ng/ml FasL in the absence or presence of 10mM NAC were probed for phosphorylated IKKα/β using western blotting. Blots were re-probed with GAPDH antibody to confirm equal loading of the samples. B, C. Cells were co-transfected with 100ng/well of NF-κB-Luc plasmid, 10ng/well of pRL-tk plasmid, 4ng/ml of either pcDNA3 or c-FLIP plasmids and 1 μg pcDNA3, IKK-WT or IKK-DN plasmids. 24 h post transfection, cells were treated with 200ng/ml FasL along with (B) 10mM NAC or (C) 20μM LY294002. Treated cells were analyzed for NF-κB levels by luciferase assay. Plots show relative NF-κB levels over non-treated control. Values are mean ± SD (n = 4). *P < 0.05 versus non-treated control. **P < 0.05 versus pcDNA-transfected control datasets.
Role of c-FLIP processing and S-nitrosylation in FasL-induced NF-κB activation
Previous studies have shown that FLIPS is mainly responsible for the activation of NF-κB by the regulation of the IKK complex (44). Upon activation, full-length c-FLIP is processed into shorter forms such as p43-FLIP and p22-FLIP (45). Since the processing of c-FLIP may also be important in the down-regulation of FasL-induced NF-κB activity, we wanted to identify and assess individual domains in c-FLIP that may be responsible for NF-κB inhibition. We found that c-FLIP was mainly undetectable in non-transfected cells (left lanes in both blot panels in Fig. 7A), but was processed predominantly into p43-FLIP upon FasL stimulation in cells transfected with 4ng/ml c-FLIP (Fig. 7A – second lane in right panel). Therefore, we generated relevant domain deletion mutants of c-FLIP, and co-transfected cells with either full-length c-FLIP or its deletion constructs along with NF-κB luciferase construct (Fig. 7B). In the presence of FasL, full-length c-FLIP inhibited NF-κB as expected. However, the shorter forms of c-FLIP (p22-FLIP and p43-FLIP), could also lead to inhibition of NF-κB activity, with p22-FLIP driving inhibition of FasL-dependent NF-κB activity the most as compared to the other c-FLIP constructs (Fig. 7C). On the other hand, NF-κB inhibitory activity was lost when transfected with c-FLIP construct comprising only of death effector domain 1 (DED1), suggesting that both DED1 and DED2 were required for NF-κB inhibition. Previous studies by our group had suggested an important role for S-nitrosylation in mediating c-FLIP expression and stability in and FasL signaling (46, 47). Therefore, we assessed the importance of c-FLIP S-nitrosylation in mediating NF-κB activity. Cells were transfected with a non-nitrosylable c-FLIP mutant (46), and assessed for FasL-dependent NF-κB activity. The non-nitrosylable mutant further down-regulated FasL-dependent NF-κB activity, suggesting that S-nitrosylation of c-FLIP may lead to a decrease in its effectiveness in inhibition of FasL-dependent NF-κB activity. (Fig. 7D).
Fig. 7. Role of c-FLIP processing and S-nitrosylation in FasL-induced NF-κB activation.

A. Cells transfected with either pcDNA3 control plasmid or 4ng/ml c-FLIP were exposed to 200ng/ml FasL, and probed for c-FLIP levels using western blotting. Blots were re-probed with GAPDH antibody to confirm equal loading of the samples. B. Domain mutants were generated using standard techniques as mentioned in the material and methods section and were cloned into the pcDNA3 vector. The physiologically relevant processed forms include p43-FLIP (D1), p22-FLIP (D2) and domain mutant expressing only the death effector domain 1 (D3). In addition, full-length c-FLIP with the two cysteines in its caspase-like domain substituted by alanines (D4) was generated such that the mutated form cannot undergo S-nitrosylation. Western blots probed with c-myc antibody show the expression of the c-FLIP domain mutants. C. Cells co-transfected with 100ng/well NF-κB-Luc, 10ng/well pRL-tk and domain mutants of c-FLIP (4ng/ml) were treated with 200ng/ml FasL (12 h) in the absence or presence of 20μM LY294002 and 10mM NAC (1 h pre-treatment), and then assayed for NF-κB activity by luciferase assay. D. Cells were co-transfected with 100ng/well NF-κB-Luc, 10ng/well pRL-tk and non-nitrosylable c-FLIP mutant (D4). Transfected cells were either left untreated or pretreated with 20μM LY294002 and 10mM NAC for 1 h followed by 200ng/ml FasL treatment for 12 h. Treated cells were analyzed for NF-κB activity by luciferase assay. Values are mean ± SD (n = 4). *P < 0.05, **P < 0.05 for respective data versus pcDNA-transfected control datasets as indicated.
DISCUSSION
Homeostasis is a tightly regulated phenomenon, which requires the continuous interplay of both cell death and cell survival pathways. NF-κB and PI3K/Akt are important regulators of cellular homeostasis. In the context of FasL-mediated apoptosis, NF-κB primarily exerts a pro-survival signal, probably as a defense mechanism in order to protect cells from superfluous death ligand-induced apoptosis (48). We found that FasL-induced NF-κB activity was ROS dependent in 293T cells, with H2O2 being the predominant species responsible for NF-κB activation (Fig. 1, E and F). This is consistent with previous studies showing that transcriptional inhibition of FasL using antioxidants also leads to the inhibition of NF-κB and a net inhibitory effect on FasL (49–52).
Both PI3K/Akt and NF-κB play a role in protecting cells from undergoing apoptosis (53–56). However, this is the first study that demonstrates an inhibitory effect of PI3K/Akt on FasL-induced NF-κB activity. This result is certainly surprising, given that FasL positively stimulates both PI3K/Akt and NF-κB activity (Fig. 1A and 2A). However, given that PI3K/Akt has also been shown to play a pro-apoptotic role in response to FasL (42), inhibition of NF-κB fits well with the model of PI3K/Akt-induced apoptosis, since NF-κB is a strong anti-apoptotic signaling molecule and its inhibition may be required for potent induction of cell death (57, 58).
Furthermore, this is the first study that demonstrates that c-FLIP can inhibit FasL-induced NF-κB activity (Fig. 3). c-FLIP is a known antioxidant that is regulated by the NF-κB signaling pathway (59), and c-FLIP in turn can lead to a direct and potent increase in NF-κB activity (10, 60–62). The role of c-FLIP as an oncogenic factor in cancer progression and chemo-resistance has been well documented (60, 63). However, studies assessing its role in tumors predominantly involve high levels of c-FLIP expression (more than 100ng/ml). The ‘physiological’ expression levels of c-FLIP vary depending on cell type, and are typically extremely low, which has hindered the delineation of c-FLIP function under normal cellular conditions (16). In fact, basal c-FLIP levels were undetectable even in 293T cells in our study (Fig. 7A), which has also been corroborated by others (61). However, there have been a few important studies assessing the role of c-FLIP under physiological conditions, the overall results of which are reaffirmed in our study. For example, Kreuz et al. suggested that physiological levels of c-FLIP exerted an inhibitory effect on FasL-induced NF-κB-driven interleukin-8 activity in a caspase-dependent manner (16). Also, Chang et al. demonstrated that c-FLIP may play a dual role depending upon its expression levels – at low levels that mimic physiological conditions, c-FLIP is pro-apoptotic, whereas at higher protein levels of c-FLIP such as that observed in tumors, c-FLIP promotes survival (23). Two recent studies investigating the role of c-FLIP and its processed forms in FasL signaling by Neumann et al. and Fricker et al. from the German Cancer Research Center in Germany have suggested a similar pro-apoptotic effect for c-FLIP at physiological concentrations using a systems biology approach (64–66). Overall, our results effectively complement the data presented by the aforementioned studies, while demonstrating a novel role for PI3K/Akt in regulating c-FLIP-mediated inhibition of NF-κB upon FasL treatment.
We demonstrate that at low levels, c-FLIP alone activates NF-κB as expected. However, in the presence of FasL, NF-κB activity was inhibited by c-FLIP in a dose-dependent manner (Fig. 3, A and C). Furthermore, low levels of c-FLIP also inhibited H2O2 levels in the presence of FasL (Fig. 3D), which suggested that the inhibition of NF-κB by c-FLIP might be due at least partially to its antioxidant properties, which has been shown to be important in FasL signaling (67). However, c-FLIP can further inhibit NF-κB even in the presence of NAC, suggesting that a ROS-independent mechanism of NF-κB regulation by c-FLIP may also exist. We now know from corroborating evidence provided by the studies performed in Germany that c-FLIP-driven inhibition of NF-κB may be due to direct interaction of the p43 form of c-FLIP with the IKKγ subunit. The increased processing of c-FLIP upon FasL stimulation observed in our study supports this idea (Fig. 7A). Secondly, c-FLIP is capable of inhibiting NF-κB activity even when IKK is upregulated using constitutively active IKKγ construct (Fig. 6). Finally, much higher inhibition of NF-κB is observed when transfected with the p43-FLIP construct as compared to c-FLIPL. However, the important aspect of this result is the fact that c-FLIP seems to play a regulatory role depending upon its expression levels, with higher levels leading to an exactly opposite effect as proposed by the recent studies (64–66). Such concentration-dependent duality in function for c-FLIP is very significant in that it suggests an extremely critical function for c-FLIP in regulation of the cell death. Some of the important findings in 293T cells have also been confirmed using Jurkat T-lymphocytes, which offers further credibility to this study, and makes these findings translatable to physiological conditions (Supplemental Fig. 2).
We also observe that both PI3K/Akt and c-FLIP positively regulate each other in the presence of FasL (Fig. 4, A and B), and can both inhibit NF-κB, which has not been reported previously. A feedback loop is observed between c-FLIP and Akt, leading to an overall decrease in NF-κB levels, which is corroborated by data observed with co-expression of c-FLIP and Akt-WT (Fig. 4F). The modulation of c-FLIP by PI3K/Akt was observed only in c-FLIP-transfected cells (Fig. 6A), which suggests that either any increase in endogenous c-FLIP expression with Akt stimulation is undetectable, or that there might be a high turnover of c-FLIP protein, which has also been observed by others (16). Our results also show that c-FLIP promotes PI3K/Akt-mediated down-regulation of NF-κB, which may be due to the antioxidant properties of c-FLIP, as suggested by recapitulation of this effect upon co-treatment with NAC. Also, we modulated levels of the regulatory IKKγ subunit (Fig. 6) and not other subunits of the IKK complex because both c-FLIP and v-FLIP have been previously shown to directly act on IKKγ, thereby exerting effects on overall NF-κB levels (68, 69).
As mentioned earlier, processing of c-FLIP was important for inhibition of NF-κB. This is supported by the fact that c-FLIP is found to be processed in the presence of FasL and not in its absence at least in 293T cells (compare lanes 2 and 4 in Fig. 6A), and opposite effects are seen with c-FLIP modulation of NF-κB depending upon either the presence or absence of FasL (Fig. 2 – A–C). Assessment of NF-κB activity using deletion mutants of c-FLIP that lack one or more important domains indicate that p22-FLIP was particularly effective in inhibiting NF-κB, leading to a much higher decrease in NF-κB activity as compared to full-length c-FLIP (Fig. 6C). Although previous studies show that p22-FLIP promotes NF-κB activity, the seeming inconsistency can be explained by the fact that only low levels of c-FLIP (up to 4ng/ml) were used in our study as compared to the much higher levels in previous work. This provides further validation to the dichotomy observed with NF-κB regulation in the context of c-FLIP. Interestingly, the non-S-nitrosylable mutant of c-FLIP caused further inhibition of NF-κB activity, with levels even lower than that observed with p22-FLIP. This suggests that not only is the processing of c-FLIP important for its inhibitory effect on NF-κB, but post-translational modifications such as S-nitrosylation may impede this down-regulation under physiological conditions, and may have an important role to play in c-FLIP associated pathophysiological effects.
In addition to the effects of Akt on c-FLIP, previous studies have also shown that the stress-activated protein kinase/Jun-amino-terminal kinase (SAPK/JNK) pathway may also regulate c-FLIP, and can potentially play a regulatory role in the inhibition of NF-κB. However, our data indicates that modulation of JNK activity (using the JNK inhibitor SP600125) did not lead to any significant changes in Akt levels, nor affected downstream proteins at the time-points included in this study (see Supplemental Fig. 1). However, treatment with SP600125 was able to counter the increases in NF-κB activity when Akt was repressed (see Supplemental Fig. 1D), suggesting that Akt and JNK may have some form of cross-talk, which needs to be investigated further. In addition, no significant changes in FasR levels were observed for the time-points assayed, suggesting that inhibitory effects of c-FLIP on NF-κB may be independent of FasR levels (Supplemental Fig. 1).
FasL is an important stimulator of apoptosis, and we observe a decrease in NF-κB levels with an increase in c-FLIP levels in our system. Since NF-κB is an important pro-survival factor, cells transfected with low levels of c-FLIP show a higher level of FasL-dependent apoptosis as compared to non-transfected cells (Fig. 8A). Thus, our study purports the importance of c-FLIP mediated down-regulation of NF-κB as a novel and significant mechanism in induction of FasL-induced apoptosis. Also, in our system, we observe an increase in apoptosis but a decrease in c-FLIP-mediated H2O2 production. Although this may be counter-intuitive, given that a positive co-relation exists between ROS levels and degree of apoptosis, several studies also show that ROS may also have anti-apoptotic effects (70). On the other hand, this may suggest that inhibition of NF-κB by physiological levels of c-FLIP may have a more significant impact on overall apoptosis as compared to the putative protective mechanism as a result of the anti-oxidant effects of c-FLIP. Therefore, our model may serve as an alternate and updated representation of the role of c-FLIP signaling in the context of FasL-induced inflammatory response under physiological conditions (Fig. 8B). The data presented in this article recapitulates some of the important conclusions drawn by recent studies investigating the physiological role of c-FLIP, and highlights the duality of c-FLIP signaling based upon levels of both c-FLIP itself and of FasL, and the resulting effect on overall apoptosis (Fig. 8A). In addition, our data also hints at the importance of other factors such as NO (which contributes to S-nitrosylation of c-FLIP), which may have an influence on the function of c-FLIP, and will be pursued in the future.
Fig. 8. Model for c-FLIP-induced apoptosis at low levels of c-FLIP expression.

A. Cells transfected with increasing levels of c-FLIP (0–4ng/ml) were either treated with 200ng/ml FasL and assayed for apoptosis, or co-transfected with 100ng NF-κB-Luc and 10ng pRL-tk vectors and assayed for luciferase levels. In the presence of FasL, increasing c-FLIP levels caused a dose-dependent increase in apoptosis, but a corresponding decrease in NF-κB activity. B. FasL binding causes the formation of the DISC, composed of Fas, FADD, pro-caspase 8, and RIP (not shown). Formation of the DISC recruits c-FLIP, where it is activated and cleaved into its shorter forms such as p43-FLIP (45). In this study, we show that FasL can activate Akt through activation of c-FLIP protein, and vice-versa. Increased c-FLIP levels at the physiological range leads to decreased H2O2 (primary ROS involved), which occurs upstream of the IKK complex, leads to a decrease in NF-κB activity. Further, Akt may also cause NF-κB down-regulation in a c-FLIP independent manner. Down-regulation of NF-κB by either mechanism leads to decrease in apoptosis.
We believe that this study would help pave the way for further understanding of FasL-mediated inflammatory response, and shed new light on the role of PI3K/Akt and c-FLIP in apoptosis, which are traditionally understood to be pro-survival factors. Particularly, such studies are important to extend the understanding of c-FLIP under normal biological conditions including growth and immune response, and may lay the foundation for detailed mechanistic studies on c-FLIP and its role in apoptosis.
Supplementary Material
Acknowledgments
This work was supported by the NIH grant R01HL76340.
References
- 1.Fulda S. Tumor resistance to apoptosis. Int J Cancer. 2009;124:511–515. doi: 10.1002/ijc.24064. [DOI] [PubMed] [Google Scholar]
- 2.Nagata S. Fas ligand-induced apoptosis. Annu Rev Genet. 1999;33:29–55. doi: 10.1146/annurev.genet.33.1.29. [DOI] [PubMed] [Google Scholar]
- 3.Debatin KM, Beltinger C, Bohler T, Fellenberg J, Friesen C, Fulda S, Herr I, Los M, Scheuerpflug C, Sieverts H, Stahnke K. Regulation of apoptosis through CD95 (APO-I/Fas) receptor-ligand interaction. Biochem Soc Trans. 1997;25:405–410. doi: 10.1042/bst0250405. [DOI] [PubMed] [Google Scholar]
- 4.Suda T, Nagata S. Purification and characterization of the Fas-ligand that induces apoptosis. J Exp Med. 1994;179:873–879. doi: 10.1084/jem.179.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- 6.Xu G, Shi Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007;17:759–771. doi: 10.1038/cr.2007.52. [DOI] [PubMed] [Google Scholar]
- 7.Medan D, Wang L, Toledo D, Lu B, Stehlik C, Jiang BH, Shi X, Rojanasakul Y. Regulation of Fas (CD95)-induced apoptotic and necrotic cell death by reactive oxygen species in macrophages. J Cell Physiol. 2005;203:78–84. doi: 10.1002/jcp.20201. [DOI] [PubMed] [Google Scholar]
- 8.Devadas S, Hinshaw JA, Zaritskaya L, Williams MS. Fas-stimulated generation of reactive oxygen species or exogenous oxidative stress sensitize cells to Fas-mediated apoptosis. Free Radic Biol Med. 2003;35:648–661. doi: 10.1016/s0891-5849(03)00391-5. [DOI] [PubMed] [Google Scholar]
- 9.Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 10.Kataoka T. The caspase-8 modulator c-FLIP. Crit Rev Immunol. 2005;25:31–58. doi: 10.1615/critrevimmunol.v25.i1.30. [DOI] [PubMed] [Google Scholar]
- 11.Peter ME. The flip side of FLIP. Biochem J. 2004;382:e1–3. doi: 10.1042/BJ20041143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274:1541–1548. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- 13.Golks A, Brenner D, Krammer PH, Lavrik IN. The c-FLIP-NH2 terminus (p22-FLIP) induces NF-kappaB activation. J Exp Med. 2006;203:1295–1305. doi: 10.1084/jem.20051556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirchhoff S, Muller WW, Li-Weber M, Krammer PH. Up-regulation of c-FLIPshort and reduction of activation-induced cell death in CD28-costimulated human T cells. Eur J Immunol. 2000;30:2765–2774. doi: 10.1002/1521-4141(200010)30:10<2765::AID-IMMU2765>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 15.Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN. c-FLIPR, a new regulator of death receptor-induced apoptosis. J Biol Chem. 2005;280:14507–14513. doi: 10.1074/jbc.M414425200. [DOI] [PubMed] [Google Scholar]
- 16.Kreuz S, Siegmund D, Rumpf JJ, Samel D, Leverkus M, Janssen O, Hacker G, Dittrich-Breiholz O, Kracht M, Scheurich P, Wajant H. NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J Cell Biol. 2004;166:369–380. doi: 10.1083/jcb.200401036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han DK, Chaudhary PM, Wright ME, Friedman C, Trask BJ, Riedel RT, Baskin DG, Schwartz SM, Hood L. MRIT, a novel death-effector domain-containing protein, interacts with caspases and BclXL and initiates cell death. Proc Natl Acad Sci U S A. 1997;94:11333–11338. doi: 10.1073/pnas.94.21.11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu S, Vincenz C, Ni J, Gentz R, Dixit VM. I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD-95-induced apoptosis. J Biol Chem. 1997;272:17255–17257. doi: 10.1074/jbc.272.28.17255. [DOI] [PubMed] [Google Scholar]
- 19.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 20.Krueger A, Baumann S, Krammer PH, Kirchhoff S. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol. 2001;21:8247–8254. doi: 10.1128/MCB.21.24.8247-8254.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thome M, Tschopp J. Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol. 2001;1:50–58. doi: 10.1038/35095508. [DOI] [PubMed] [Google Scholar]
- 22.Micheau O, Thome M, Schneider P, Holler N, Tschopp J, Nicholson DW, Briand C, Grutter MG. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem. 2002;277:45162–45171. doi: 10.1074/jbc.M206882200. [DOI] [PubMed] [Google Scholar]
- 23.Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME, Yang X. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. Embo J. 2002;21:3704–3714. doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponton A, Clement MV, Stamenkovic I. The CD95 (APO-1/Fas) receptor activates NF-kappaB independently of its cytotoxic function. J Biol Chem. 1996;271:8991–8995. doi: 10.1074/jbc.271.15.8991. [DOI] [PubMed] [Google Scholar]
- 25.Tada K, Okazaki T, Sakon S, Kobarai T, Kurosawa K, Yamaoka S, Hashimoto H, Mak TW, Yagita H, Okumura K, Yeh WC, Nakano H. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-kappa B activation and protection from cell death. J Biol Chem. 2001;276:36530–36534. doi: 10.1074/jbc.M104837200. [DOI] [PubMed] [Google Scholar]
- 26.Borset M, Hjorth-Hansen H, Johnsen AC, Seidel C, Waage A, Espevik T, Sundan A. Apoptosis, proliferation and NF-kappaB activation induced by agonistic Fas antibodies in the human myeloma cell line OH-2: amplification of Fas-mediated apoptosis by tumor necrosis factor. Eur J Haematol. 1999;63:345–353. doi: 10.1111/j.1600-0609.1999.tb01138.x. [DOI] [PubMed] [Google Scholar]
- 27.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 28.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 29.Brasier AR. The NF-kappaB regulatory network. Cardiovasc Toxicol. 2006;6:111–130. doi: 10.1385/ct:6:2:111. [DOI] [PubMed] [Google Scholar]
- 30.Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- 31.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 32.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 33.Sale EM, Sale GJ. Protein kinase B: signalling roles and therapeutic targeting. Cell Mol Life Sci. 2008;65:113–127. doi: 10.1007/s00018-007-7274-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toker A. Protein kinases as mediators of phosphoinositide 3-kinase signaling. Mol Pharmacol. 2000;57:652–658. [PubMed] [Google Scholar]
- 35.Gulbins E, Hermisson M, Brenner B, Grassme HU, Linderkamp O, Dichgans J, Weller M, Lang F. Cellular stimulation via CD95 involves activation of phospho-inositide-3-kinase. Pflugers Arch. 1998;435:546–554. doi: 10.1007/s004240050551. [DOI] [PubMed] [Google Scholar]
- 36.Suhara T, Mano T, Oliveira BE, Walsh K. Phosphatidylinositol 3-kinase/Akt signaling controls endothelial cell sensitivity to Fas-mediated apoptosis via regulation of FLICE-inhibitory protein (FLIP) Circ Res. 2001;89:13–19. doi: 10.1161/hh1301.092506. [DOI] [PubMed] [Google Scholar]
- 37.Plas DR, Talapatra S, Edinger AL, Rathmell JC, Thompson CB. Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J Biol Chem. 2001;276:12041–12048. doi: 10.1074/jbc.M010551200. [DOI] [PubMed] [Google Scholar]
- 38.Seol JW, Lee YJ, Kang HS, Kim IS, Kim NS, Kwak YG, Kim TH, Seol DW, Park SY. Wortmannin elevates tumor necrosis factor-related apoptosis-inducing ligand sensitivity in LNCaP cells through down-regulation of IAP-2 protein. Exp Oncol. 2005;27:120–124. [PubMed] [Google Scholar]
- 39.Lu B, Wang L, Medan D, Toledo D, Huang C, Chen F, Shi X, Rojanasakul Y. Regulation of Fas (CD95)-induced apoptosis by nuclear factor-kappaB and tumor necrosis factor-alpha in macrophages. Am J Physiol Cell Physiol. 2002;283:C831–838. doi: 10.1152/ajpcell.00045.2002. [DOI] [PubMed] [Google Scholar]
- 40.Chen JJ, Sun Y, Nabel GJ. Regulation of the proinflammatory effects of Fas ligand (CD95L) Science. 1998;282:1714–1717. doi: 10.1126/science.282.5394.1714. [DOI] [PubMed] [Google Scholar]
- 41.Woo CH, Eom YW, Yoo MH, You HJ, Han HJ, Song WK, Yoo YJ, Chun JS, Kim JH. Tumor necrosis factor-alpha generates reactive oxygen species via a cytosolic phospholipase A2-linked cascade. J Biol Chem. 2000;275:32357–32362. doi: 10.1074/jbc.M005638200. [DOI] [PubMed] [Google Scholar]
- 42.Lu B, Wang L, Stehlik C, Medan D, Huang C, Hu S, Chen F, Shi X, Rojanasakul Y. Phosphatidylinositol 3-kinase/Akt positively regulates Fas (CD95)-mediated apoptosis in epidermal Cl41 cells. J Immunol. 2006;176:6785–6793. doi: 10.4049/jimmunol.176.11.6785. [DOI] [PubMed] [Google Scholar]
- 43.Yu JW, Shi Y. FLIP and the death effector domain family. Oncogene. 2008;27:6216–6227. doi: 10.1038/onc.2008.299. [DOI] [PubMed] [Google Scholar]
- 44.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 45.Ueffing N, Singh KK, Christians A, Thorns C, Feller AC, Nagl F, Fend F, Heikaus S, Marx A, Zotz RB, Brade J, Schulz WA, Schulze-Osthoff K, Schmitz I, Schwerk C. A single nucleotide polymorphism determines protein isoform production of the human c-FLIP protein. Blood. 2009 doi: 10.1182/blood-2009-02-204230. [DOI] [PubMed] [Google Scholar]
- 46.Chanvorachote P, Nimmannit U, Wang L, Stehlik C, Lu B, Azad N, Rojanasakul Y. Nitric oxide negatively regulates Fas CD95-induced apoptosis through inhibition of ubiquitin-proteasome-mediated degradation of FLICE inhibitory protein. J Biol Chem. 2005;280:42044–42050. doi: 10.1074/jbc.M510080200. [DOI] [PubMed] [Google Scholar]
- 47.Iyer AK, Azad N, Wang L, Rojanasakul Y. Role of S-nitrosylation in apoptosis resistance and carcinogenesis. Nitric Oxide. 2008;19:146–151. doi: 10.1016/j.niox.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. 1999;17:331–367. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- 49.Bauer MK, Vogt M, Los M, Siegel J, Wesselborg S, Schulze-Osthoff K. Role of reactive oxygen intermediates in activation-induced CD95 (APO-1/Fas) ligand expression. J Biol Chem. 1998;273:8048–8055. doi: 10.1074/jbc.273.14.8048. [DOI] [PubMed] [Google Scholar]
- 50.Dumont A, Hehner SP, Hofmann TG, Ueffing M, Droge W, Schmitz ML. Hydrogen peroxide-induced apoptosis is CD95-independent, requires the release of mitochondria-derived reactive oxygen species and the activation of NF-kappaB. Oncogene. 1999;18:747–757. doi: 10.1038/sj.onc.1202325. [DOI] [PubMed] [Google Scholar]
- 51.Vogt M, Bauer MK, Ferrari D, Schulze-Osthoff K. Oxidative stress and hypoxia/reoxygenation trigger CD95 (APO-1/Fas) ligand expression in microglial cells. FEBS Lett. 1998;429:67–72. doi: 10.1016/s0014-5793(98)00562-6. [DOI] [PubMed] [Google Scholar]
- 52.Meyer M, Schreck R, Baeuerle PA. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. Embo J. 1993;12:2005–2015. doi: 10.1002/j.1460-2075.1993.tb05850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burow ME, Weldon CB, Melnik LI, Duong BN, Collins-Burow BM, Beckman BS, McLachlan JA. PI3-K/AKT regulation of NF-kappaB signaling events in suppression of TNF-induced apoptosis. Biochem Biophys Res Commun. 2000;271:342–345. doi: 10.1006/bbrc.2000.2626. [DOI] [PubMed] [Google Scholar]
- 54.Jones RG, Saibil SD, Pun JM, Elford AR, Bonnard M, Pellegrini M, Arya S, Parsons ME, Krawczyk CM, Gerondakis S, Yeh WC, Woodgett JR, Boothby MR, Ohashi PS. NF-kappaB couples protein kinase B/Akt signaling to distinct survival pathways and the regulation of lymphocyte homeostasis in vivo. J Immunol. 2005;175:3790–3799. doi: 10.4049/jimmunol.175.6.3790. [DOI] [PubMed] [Google Scholar]
- 55.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 56.Beneteau M, Pizon M, Chaigne-Delalande B, Daburon S, Moreau P, De Giorgi F, Ichas F, Rebillard A, Dimanche-Boitrel MT, Taupin JL, Moreau JF, Legembre P. Localization of Fas/CD95 into the lipid rafts on down-modulation of the phosphatidylinositol 3-kinase signaling pathway. Mol Cancer Res. 2008;6:604–613. doi: 10.1158/1541-7786.MCR-07-0331. [DOI] [PubMed] [Google Scholar]
- 57.Guicciardi ME, Gores GJ. Life and death by death receptors. Faseb J. 2009;23:1625–1637. doi: 10.1096/fj.08-111005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 59.Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21:5299–5305. doi: 10.1128/MCB.21.16.5299-5305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chaudhary PM, Jasmin A, Eby MT, Hood L. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene. 1999;18:5738–5746. doi: 10.1038/sj.onc.1202976. [DOI] [PubMed] [Google Scholar]
- 61.Kataoka T, Budd RC, Holler N, Thome M, Martinon F, Irmler M, Burns K, Hahne M, Kennedy N, Kovacsovics M, Tschopp J. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr Biol. 2000;10:640–648. doi: 10.1016/s0960-9822(00)00512-1. [DOI] [PubMed] [Google Scholar]
- 62.Kataoka T, Tschopp J. N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol Cell Biol. 2004;24:2627–2636. doi: 10.1128/MCB.24.7.2627-2636.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Safa AR, Day TW, Wu CH. Cellular FLICE-like inhibitory protein (C-FLIP): a novel target for cancer therapy. Curr Cancer Drug Targets. 2008;8:37–46. doi: 10.2174/156800908783497087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neumann L, Pforr C, Beaudouin J, Pappa A, Fricker N, Krammer PH, Lavrik IN, Eils R. Dynamics within the CD95 death-inducing signaling complex decide life and death of cells. Mol Syst Biol. 2010;6:352. doi: 10.1038/msb.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lavrik IN. Systems biology of apoptosis signaling networks. Curr Opin Biotechnol. 2010 doi: 10.1016/j.copbio.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 66.Fricker N, Beaudouin J, Richter P, Eils R, Krammer PH, Lavrik IN. Model-based dissection of CD95 signaling dynamics reveals both a pro-and antiapoptotic role of c-FLIPL. J Cell Biol. 2010;190:377–389. doi: 10.1083/jcb.201002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang L, Azad N, Kongkaneramit L, Chen F, Lu Y, Jiang BH, Rojanasakul Y. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J Immunol. 2008;180:3072–3080. doi: 10.4049/jimmunol.180.5.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Field N, Low W, Daniels M, Howell S, Daviet L, Boshoff C, Collins M. KSHV vFLIP binds to IKK-gamma to activate IKK. J Cell Sci. 2003;116:3721–3728. doi: 10.1242/jcs.00691. [DOI] [PubMed] [Google Scholar]
- 69.Liu L, Eby MT, Rathore N, Sinha SK, Kumar A, Chaudhary PM. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J Biol Chem. 2002;277:13745–13751. doi: 10.1074/jbc.M110480200. [DOI] [PubMed] [Google Scholar]
- 70.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.