

Abstract
Narcolepsy is a rare sleep disorder characterized by excessive daytime sleepiness and cataplexy. Familial narcolepsy accounts for less than 10% of all narcolepsy cases. However, documented multiplex families are very rare and causative mutations have not been identified to date. To identify a causative mutation in familial narcolepsy, we performed linkage analysis in the largest ever reported family, which has 12 affected members, and sequenced coding regions of the genome (exome sequencing) of three affected members with narcolepsy and cataplexy. We successfully mapped a candidate locus on chromosomal region 6p22.1 (LOD score = 3.85) by linkage analysis. Exome sequencing identified a missense mutation in the second exon of MOG within the linkage region. A c.398C>G mutation was present in all affected family members but absent in unaffected members and 775 unrelated control subjects. Transient expression of mutant myelin oligodendrocyte glycoprotein (MOG) in mouse oligodendrocytes showed abnormal subcellular localization, suggesting an altered function of the mutant MOG. MOG has recently been linked to various neuropsychiatric disorders and is considered as a key autoantigen in multiple sclerosis and in its animal model, experimental autoimmune encephalitis. Our finding of a pathogenic MOG mutation highlights a major role for myelin and oligodendrocytes in narcolepsy and further emphasizes glial involvement in neurodegeneration and neurobehavioral disorders.
Main Text
Narcolepsy (NARCLP1 [MIM 161400]) is a disabling sleep disorder characterized by irresistible excessive daytime sleepiness (EDS) and cataplexy, a condition triggered by strong emotions leading to a sudden loss of muscle tone.1 Narcolepsy is a rare and mainly sporadic disorder. Genetic studies revealed a tight association with the HLA haplotype DRB5∗01:01-DRB1∗15:01-DQA1∗01:02-DQB1∗06:02 (MIM 604305) on chromosome 6 in more than 95% of individuals with narcolepsy with cataplexy.2 This HLA haplotype represents an almost necessary but not sufficient risk factor for narcolepsy because approximately 20% of the general healthy population carries the same haplotype. The discovery of hypocretin-1 (HCRT [MIM 602358]) deficiency shed light on the underlying pathophysiology of the disease.3 The hypocretin neurotransmission system, which was identified in the lateral hypothalamus,4,5 was shown to play a major role in controlling vigilance states by its diffuse projections to major wakefulness centers.6 Hypocretin deficiency, defined as dramatically decreased if not undetectable levels of hypocretin-1 in the cerebrospinal fluid (CSF), as found in narcolepsy individuals, is thought to induce an imbalance between waking neurotransmitter systems resulting in abnormal transitions between vigilance states.3 Because of the strong HLA association, hypocretin-1 deficiency is believed to be caused by an autoimmune attack. Only recently, circulating TRIB2-specific antibodies reactive to hypocretin neurons were detected in individuals with narcolepsy, confirming the autoimmune nature of narcolepsy.7 Population-based genome-wide association studies (GWAS) of sporadic narcolepsy in individuals of European descent also revealed association with variants in the immune-related genes (TCRA [MIM 186880] and P2RY11 [MIM 602697]).8,9 However, in contrast to the well-established HLA contribution (odds ratio > 40), the identified associations have a small effect size (odds ratio < 2). We recently suggested a causal involvement of the HLA class 2 region in the pathogenesis of sporadic narcolepsy and identified a HLA DQB1 allele (06:03) that confers a 50-fold increase in protection against narcolepsy.10 So far, except in an atypical and unique sporadic case of narcolepsy that had a mutation in HCRT,11 no mutation in either hypocretin ligands or receptors has been reported.
Estimates of prevalence of familial forms of narcolepsy vary from 1% to 10% but documented multiplex families are extremely rare (1%–2%).12 Because there are limited numbers of affected individuals in families, only two linkage studies are available, and they suggest candidate loci on chromosome 21 (21q22.3)13 and 4 (4p13-q21).14 However, no pathogenic mutation has been identified so far in any of the reported candidate regions.
We describe here the characterization of a large Spanish family with 12 affected members diagnosed with narcolepsy and cataplexy. We used both genome-wide linkage analysis and exome capturing with subsequent next-generation sequencing to identify the causative mutation in this family.
All participants provided written informed consent and the study protocol was approved by the ethics committee of the Gregorio Marañón University Hospital (Madrid, Spain). The family was identified through a dizygotic twin pair concordant for narcolepsy (Figure 1, Table S1, available online). The twins are part of a four-generation pedigree that was referred to the Sleep and Epilepsy Unit of the Department of Clinical Neurophysiology in Madrid. Both twins as well as all other affected family members met the diagnostic criteria for narcolepsy and cataplexy according to the International Classification of Sleep Disorders.15 The diagnosis was additionally confirmed by undetectable levels of hypocretin-1 in the cerebrospinal fluid of both twins. The twin's mother was diagnosed with narcolepsy-cataplexy but passed away 10 years ago. One niece and three of four offspring of the twins are diagnosed with narcolepsy-cataplexy as well (left branch). Narcolepsy-cataplexy is also present in two cousins of the twins and in four of their offspring (right branch, Table S2). Fifteen additional family members were interviewed or investigated in the sleep laboratory and three of them underwent whole-night polysomnography followed by a multiple sleep latency test. In 14 of these family members, narcolepsy was excluded. In one (III-3) narcolepsy could not be definitely excluded because EDS (although without clear cataplexy) was present, but this person did not completely fulfill the diagnostic criteria for narcolepsy and her EDS was not improved by continuous positive airway pressure (CPAP) treatment for sleep apnea. Several family members are obese (or morbidly obese), and others are suffering from type 2 diabetes (Figure 1). In total, 12 living members of the family are affected with narcolepsy-cataplexy, seven by obesity, and four by type 2 diabetes. One of the twins (III-5) and one of their cousins (III-10) were diagnosed with all three conditions.
Figure 1.
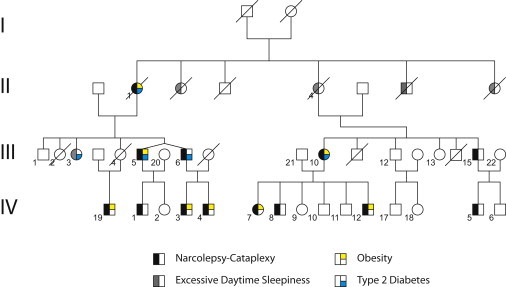
Family Pedigree
Four-generation pedigree of the Spanish family including the dizygotic twin pair concordant for narcolepsy-cataplexy in the third generation; the distribution of the disorder indicates an autosomal-dominant transmission of the disease-causing gene. The family consists of 13 (12 living) family members affected with narcolepsy-cataplexy. Note that individual III-3 did not fulfill the full diagnostic criteria though the clinical characteristics of this person could not exclude the possibility of narcolepsy-cataplexy. Seven family members were also affected with obesity and four with type 2 diabetes. Individuals III-5 and III-10 (as well as II-1) were diagnosed for all three conditions.
DNA samples of 11 affected and 15 nonaffected family members were genotyped on Affymetrix Genome-Wide Human 5.0 SNP arrays. The affected individual IV-19 was not included in the linkage analysis because this person was diagnosed very recently. A SNP data set equivalent to the Affymetrix Mapping 100K genotyping array was extracted for each individual. With this data set, we performed a parametric linkage analysis by using the Merlin 1.1.2 program.16 Eleven family members were considered as affected, whereas III-3 was considered as unknown. Parametric linkage analysis was performed with a dominant model, a disease allele frequency of 0.00025, and a 90% penetrance.
The four-generation pedigree was clearly compatible with an autosomal-dominant transmission of a gene responsible for narcolepsy with cataplexy (Figure 1). HLA genotyping revealed that all affected family members of the right branch carried the known associated DQB1∗06:02 allele, whereas all affected members of the left branch carried a different DQB1 allele indicating that DQB1∗06:02 is neither necessary nor sufficient in this family. Because the dizygotic twins are DQB1∗06:02 negative and the only known mutation was found in HCRT in a DQB1∗06:02 negative individual, we first sequenced the coding region of HCRT and its two receptors but found no pathogenic mutation (Table S1). These findings excluded the HLA class II and the hypocretin system. Our genome-wide linkage analysis revealed significant linkage to a region on chromosome 6p22.1-6p22.3, encompassing a chromosomal region of 6.08 Mb (3.2 cM) flanked by the SNPs rs9295612 and rs2517592, with a maximum LOD score of 3.85 (Figure S1). No additional loci with suggestive evidence of linkage were detected in the genome. The linkage region contains 54 genes in addition to clusters of butyrophilin, histone, zinc finger, and olfactory receptors. Because the maximum LOD score was observed within a region previously linked to reading disability17 and containing genes with potential function in the brain, we then sequenced the coding region of candidate genes GPLD1 (MIM 602515), ALDH5A1 (MIM 610045), KIAA0319 (MIM 609269), TTRAP (MIM 605764), ACOT13, and C6orf62 but did not find any pathogenic mutation.
We next performed exome sequencing in three affected family members (IV-3, IV-12, and IV-5) by means of exome capturing with the Agilent SureSelect Human All Exon Kit (38 Mb) according to manufacturer's protocols. Paired-end sequencing was performed on an Illumina Genome Analyzer IIx and the short reads (75 bp) were aligned to the Human Genome (UCSC hg19) with Bowtie (v0.12.7).18 Single nucleotide variants (SNVs) were subsequently called with SAMTools (v0.1.12). Because narcolepsy is a rare disease (general population prevalence = 0.025%), known variants according to the SNP databases of dbSNP (Build 132) and the 1000 Genomes Project were filtered out with the SeattleSeq's annotation tool. This lead to the identification of 148 unknown, heterozygous, and mainly missense SNVs, which were present in all three exomes (Table S3). Two of those variants were found on chromosome 6 (in MOG and C4B), but only one fell into our candidate linkage region. The missense variant was located in the second exon of MOG (MIM 159465) and represented a C to G substitution (c.398C>G in NM_206809.3, Figure 2A; nucleotide 29627405 on chromosome 6, [GRCh37/hg19]) resulting in serine to cysteine substitution (p.Ser133Cys in Q16653). Sanger sequencing confirmed the presence of this mutation in all affected family members. None of the 15 nonaffected family members carried the mutation. One sister of the twins (III-3), in whom the presence of narcolepsy could not be excluded, also carried the mutation. We next genotyped the putative mutation in 775 unrelated control subjects, 370 individuals with sporadic narcolepsy-cataplexy, and 41 individuals with narcolepsy without cataplexy. None of them carried the mutation. Similarly, resequencing of the coding region of MOG (Table S4) in 35 unrelated familial cases revealed, except for known SNPs, neither the same nor additional causative mutation(s). A search of the 1000 Genomes database identified many rare variants, but none was considered as pathogenic (Table S5). These findings strongly suggest that the identified mutation is unique to our family.
Figure 2.
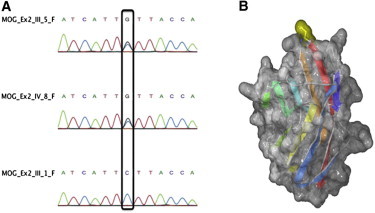
MOG Mutation
Chromatograph example displaying the identified heterozygous mutation (A) in the second exon of MOG (C398G) in two affected (III-5, IV-8) and one healthy family member (III-1). A model of the Ig-like domain of human p.Ser133Cys MOG mutant (B). Secondary structure elements are colored from blue (N terminal) to red (C terminal), and the location of the p.Ser133Cys mutation is highlighted on the molecular surface in yellow, at the top of the image. The image was prepared with Swiss-PdbViewer and rendered with POV-Ray.
PolyPhen-2 annotating tool19 was then used to predict the functional effect of the c.398C>G mutation, and the analysis indicated that the mutation is pathogenic with the maximum score of 1. We next used Swiss-PdbViewer20 to align the human MOG (Q16653) onto the mouse MOG (1PY9 crystal structure) and introduced the p.Ser133Cys mutation. The analysis revealed that this residue is located at the tip of the solvent exposed FG-loop of the Ig-like domain of MOG (Figure 2B), which has been previously described as a critical epitope region interacting with the demyelinating monoclonal antibody 8-18C5.21 The Ig-like domain contains two cysteines (Cys53 and Cys127), which form a disulfide bond characteristic of the Ig-like fold. Because the additional cysteine present in the p.Ser133Cys mutant is spatially very distant from either of them in the native fold (over 19 Å), the creation of a wrong disulfide bond during protein folding would result in a misfolded domain. Cysteine misbonding in the Ig-like domain has been previously reported to result in the genetic disease hyaline fibromatosis.22 On the other hand, even if the domain folds correctly, the presence of a surface-exposed cysteine is likely to result in the formation of protein dimers or aggregates resulting in a nonfunctional MOG. To assess this possibility, the identified MOG mutation was introduced in a mammalian expression vector containing full-length human MOG cDNA (pcDNA6.2/C-EmGFP-Topo-hMOG, subsequently called MOG-GFP)23 by means of site-directed in-vitro mutagenesis. Both wild-type (MOG-GFP) and mutated vectors (c.398C>G-MOG-GFP) were transiently transfected in murine oligodendroglial precursor cells (Oli-neu)24 and the effect of the mutation on the subcellular localization of MOG investigated (Figure 3). Cells transfected with MOG-GFP showed mainly a perinuclear and membrane localization as previously described.24 In contrast, transfection with c.398C>G-MOG-GFP revealed an ectopically clustered localization of the mutated protein in the cytoplasm of both the precursor and differentiated cells although its membrane localization did not seem affected. These findings support the idea that the c.398C>G mutation must be pathogenic.
Figure 3.
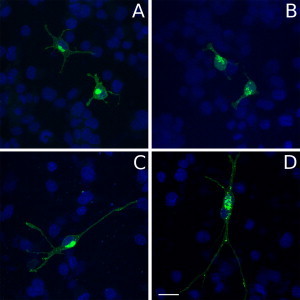
In Vitro Expression of the Mutant MOG
Localization of wild-type and mutated forms of MOG in Oli-neu cells. Oli-neu cells kept at nondifferentiated (A and B) or differentiated (C and D) stages were used to localize wild-type (MOG-GFP [A and C]) or mutated (C398G-MOG-GFP; [B and D]) forms of myelin oligodendrocyte glycoprotein (MOG: green; nuclear dye DAPI: blue). Wild-type MOG mainly shows a perinuclear and membrane localization (A and C). In contrast, the mutated C398G-MOG-GFP shows ectopically clustered localization in cytoplasm although its membrane localization seems to be preserved (B and D). The scale bar represents 20 μm.
The myelin oligodendrocyte protein is a member of the immunoglobulin superfamily and a minor component of the myelin sheath in the CNS, where it is exclusively expressed at the outmost layer of the myelin sheath as well as in oligodendrocytes.25 MOG is a target myelin antigen, and the mature protein has an extracellular IgV-like domain that contains three encephalitogenic epitopes. Thus, MOG serves as a target for autoimmune attacks in CNS-directed immune responses, and its implication in the pathogenesis of MS, especially in experimental autoimmune encephalomyelitis (EAE), the animal model of MS, is well established.26 Because young individuals with MS (who share the same HLA DRB1-DQB1 haplotype as narcolepsy), and particularly individuals diagnosed with acute disseminated encephalomyelitis, display serum MOG antibodies23,27,28 and because our identified mutation is located in the third encephalitogenic epitope of MOG (RDHSYQEE epitope), we performed an ELISA test for MOG antibodies in six affected family members (III-5, III-6, III-15, IV-3, IV-4, and IV-5) and found no evidence that the mutation induces autoimmunity against MOG. Also, TRIB2 antibodies recently reported to target hypocretin neurons7 were not detected in any family members. Considering the possibility that unknown antibodies targeting hypocretin neurons might play a role in the pathogenesis of the disease, sections of human hypothalamus were costained with the sera of affected family members and an antibody against hypocretin-1. Again, no antibodies were detected in the sera of affected family members, excluding a potential autoimmune attack targeting hypocretin neurons. We also performed a MRI scan of the brain in two affected family members (III-15, IV-12) that revealed no structural lesions, and especially no white matter lesions in the hypothalamic region. This finding, together with a lack of clinical symptoms suggestive of MS, excluded the possibility that the presence of narcolepsy is a consequence of MS lesions in the CNS.
Recent studies suggested that myelin and oligodendroyctes can play an important role in neuronal signaling and are also involved in the dopaminergic and glutaminergic circuitry of the brain.29 In this context, myelin and oligodendrocytes were linked to the pathogenesis of neurodevelopmental disorders such as schizophrenia. Genetic association studies in neuropsychiatric disorders revealed evidence for association of myelin and oligodendrocyte-related genes, such as NRG1 (MIM 142445) and ERBB4 (MIM 600543), with schizophrenia.29 However, causative mutations in MOG have not been reported and SNVs in MOG have not been shown to be significantly associated with MS, schizophrenia, or other neuropsychiatric disorders. Nevertheless, we showed both by structural modeling and our transfection experiments that the identified mutation leads to an ectopically clustered expression pattern in the cytoplasm. The observed abnormal pattern of expression might result from two nonexclusive possibilities. First, the mutation changes a serine to cysteine (Ser133Cys), six amino acids downstream of another cysteine that participates in the unique disulfuide bond (Cys53-Cys127).30 The presence of this new cysteine might create the wrong disulfide bond and therefore disrupt the folding and the stability of the protein. Second, the RDHSYQEE epitope of MOG, which harbors the identified mutation, is known to interact with the mouse monoclonal antibody 8-18C5. Binding of 8-18C5 to MOG on the surface of oligodendrocytes leads to microtubule depolymerization in vitro, suggesting a role for MOG in the organization of the cytoskeleton that is compromised by binding of 8-18C5.31 The altered expression pattern we found could be a consequence of an inactivation of the MOG-cytoskeleton interaction. Accordingly, mutating Ser133 to Glu was shown to abolish the binding of 8-18C5 antibody to MOG.21 Alternatively, it could also be a consequence of a stronger interaction(s) between MOG and its potential interactors. The identification of such interactors might be one of the major goals in follow-up studies to identify and explain the possible link between myelin, oligodendrocytes, and hypocretin deficiency.
It was previously shown that taiep myelin mutant rats develop REM sleep abnormalities and cataplexy-like episodes and share pharmacological alterations similar to the canine model of narcolepsy; this suggests that abnormalities in myelination can lead to a narcolepsy-like phenotype.32 Interestingly, the phenotype of taiep rats was linked to microtubule alterations in the cytoplasm of oligodendrocytes that lead to deficits in myelin membrane formation.33 In this context it should be noted that MOG deficient mice (MOG−/−) have been extensively investigated for CNS-directed immune responses34 but have not been examined for possible sleep phenotypes. In a pilot study we have video-recorded four MOG−/− mice but did not observe any cataplexy-like behavior (sleep recordings in these mice are in progress). The absence of narcolepsy-like behavior in MOG−/− mice further suggests that the identified mutation might be a dominant-negative mutation (gain of function). Generation of transgenic mice carrying c.398C>G-MOG will be an appropriate model to identify a narcolepsy-like phenotype in mice and could possibly help to find the missing link with hypocretin deficiency.
Gene expression studies in major depression, bipolar disorder, schizophrenia, and MS indicates that genes expressed in oligodendrocytes are downregulated,35 corroborating the hypothesis that loss of trophic support by oligodendrocytes might be causal to neurodegeneration and neurodevelopmental disorders. The identification of a mutation in MOG, so far unique to our family, not only provides an insight into pathogenesis of narcolepsy, but also highlights the role of myelin and oligodendrocytes in disease susceptibility in other complex neuropsychiatric disorders.
Acknowledgments
Supported by the University of Lausanne, Swiss National Science Foundation (grant PP00P3_124833 to R.C.), the European Narcolepsy Network (EU-NN), and an unrestricted research grant from UCB Pharma (Belgium). Thanks to Markus Reindl for providing the MOG plasmids, Petra Zavadakova for assistance in linkage analysis, the Genomic Technologies Facility of the University of Lausanne for exome sequencing, María-José Domínguez, Antonia García, and Nunci Pereda for sleep studies at the Sleep Unit of Gregorio Marañón University Hospital in Madrid, and Ioannis Xenarios from Vital-It- Swiss Institute of Bioinformatics for protein modeling analysis. We thank especially Dr. Michel Billiard for advice and suggestions and all the individuals and family members who participated in this research. MT and GJL received honorarium as invited speakers and members of the narcolepsy advisory board and unrestricted research grants from UCB Pharma (Belgium).
Contributor Information
Mehdi Tafti, Email: mehdi.tafti@unil.ch.
Rosa Peraita-Adrados, Email: mperaita.hgugm@salud.madrid.org.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
SeattleSeq Annotation, http://gvs.gs.washington.edu/SeattleSeqAnnotation/
References
- 1.Dauvilliers Y., Arnulf I., Mignot E. Narcolepsy with cataplexy. Lancet. 2007;369:499–511. doi: 10.1016/S0140-6736(07)60237-2. [DOI] [PubMed] [Google Scholar]
- 2.Mignot E., Lin L., Rogers W., Honda Y., Qiu X., Lin X., Okun M., Hohjoh H., Miki T., Hsu S. Complex HLA-DR and -DQ interactions confer risk of narcolepsy-cataplexy in three ethnic groups. Am. J. Hum. Genet. 2001;68:686–699. doi: 10.1086/318799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nishino S., Ripley B., Overeem S., Lammers G.J., Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355:39–40. doi: 10.1016/S0140-6736(99)05582-8. [DOI] [PubMed] [Google Scholar]
- 4.Sakurai T., Amemiya A., Ishii M., Matsuzaki I., Chemelli R.M., Tanaka H., Williams S.C., Richardson J.A., Kozlowski G.P., Wilson S. Orexins and orexin receptors: A family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 5.de Lecea L., Kilduff T.S., Peyron C., Gao X., Foye P.E., Danielson P.E., Fukuhara C., Battenberg E.L., Gautvik V.T., Bartlett F.S., 2nd The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. USA. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peyron C., Tighe D.K., van den Pol A.N., de Lecea L., Heller H.C., Sutcliffe J.G., Kilduff T.S. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J. Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cvetkovic-Lopes V., Bayer L., Dorsaz S., Maret S., Pradervand S., Dauvilliers Y., Lecendreux M., Lammers G.J., Donjacour C.E., Du Pasquier R.A. Elevated Tribbles homolog 2-specific antibody levels in narcolepsy patients. J. Clin. Invest. 2010;120:713–719. doi: 10.1172/JCI41366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hallmayer J., Faraco J., Lin L., Hesselson S., Winkelmann J., Kawashima M., Mayer G., Plazzi G., Nevsimalova S., Bourgin P. Narcolepsy is strongly associated with the T-cell receptor alpha locus. Nat. Genet. 2009;41:708–711. doi: 10.1038/ng.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kornum B.R., Kawashima M., Faraco J., Lin L., Rico T.J., Hesselson S., Axtell R.C., Kuipers H., Weiner K., Hamacher A. Common variants in P2RY11 are associated with narcolepsy. Nat. Genet. 2011;43:66–71. doi: 10.1038/ng.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hor H., Kutalik Z., Dauvilliers Y., Valsesia A., Lammers G.J., Donjacour C.E., Iranzo A., Santamaria J., Peraita Adrados R., Vicario J.L. Genome-wide association study identifies new HLA class II haplotypes strongly protective against narcolepsy. Nat. Genet. 2010;42:786–789. doi: 10.1038/ng.647. [DOI] [PubMed] [Google Scholar]
- 11.Peyron C., Faraco J., Rogers W., Ripley B., Overeem S., Charnay Y., Nevsimalova S., Aldrich M., Reynolds D., Albin R. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- 12.Mignot E. Genetic and familial aspects of narcolepsy. Neurology. 1998;50(2, Suppl 1):S16–S22. doi: 10.1212/wnl.50.2_suppl_1.s16. [DOI] [PubMed] [Google Scholar]
- 13.Dauvilliers Y., Blouin J.L., Neidhart E., Carlander B., Eliaou J.F., Antonarakis S.E., Billiard M., Tafti M. A narcolepsy susceptibility locus maps to a 5 Mb region of chromosome 21q. Ann. Neurol. 2004;56:382–388. doi: 10.1002/ana.20208. [DOI] [PubMed] [Google Scholar]
- 14.Nakayama J., Miura M., Honda M., Miki T., Honda Y., Arinami T. Linkage of human narcolepsy with HLA association to chromosome 4p13-q21. Genomics. 2000;65:84–86. doi: 10.1006/geno.2000.6143. [DOI] [PubMed] [Google Scholar]
- 15.American Academy of Sleep Medicine . Second Edition. American Academy of Sleep Medicine; Westchester, IL: 2005. International Classification of Sleep Disorders. Diagnostic and coding manual. [Google Scholar]
- 16.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 17.Francks C., Paracchini S., Smith S.D., Richardson A.J., Scerri T.S., Cardon L.R., Marlow A.J., MacPhie I.L., Walter J., Pennington B.F. A 77-kilobase region of chromosome 6p22.2 is associated with dyslexia in families from the United Kingdom and from the United States. Am. J. Hum. Genet. 2004;75:1046–1058. doi: 10.1086/426404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 21.Breithaupt C., Schäfer B., Pellkofer H., Huber R., Linington C., Jacob U. Demyelinating myelin oligodendrocyte glycoprotein-specific autoantibody response is focused on one dominant conformational epitope region in rodents. J. Immunol. 2008;181:1255–1263. doi: 10.4049/jimmunol.181.2.1255. [DOI] [PubMed] [Google Scholar]
- 22.Deuquet J., Lausch E., Guex N., Abrami L., Salvi S., Lakkaraju A., Ramirez M.C., Martignetti J.A., Rokicki D., Bonafe L. Hyaline fibromatosis syndrome inducing mutations in the ectodomain of anthrax toxin receptor 2 can be rescued by proteasome inhibitors. EMBO Mol. Med. 2011;3:208–221. doi: 10.1002/emmm.201100124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Pauli F., Mader S., Rostasy K., Schanda K., Bajer-Kornek B., Ehling R., Deisenhammer F., Reindl M., Berger T. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin. Immunol. 2011;138:247–254. doi: 10.1016/j.clim.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 24.Winterstein C., Trotter J., Krämer-Albers E.M. Distinct endocytic recycling of myelin proteins promotes oligodendroglial membrane remodeling. J. Cell Sci. 2008;121:834–842. doi: 10.1242/jcs.022731. [DOI] [PubMed] [Google Scholar]
- 25.Breithaupt C., Schubart A., Zander H., Skerra A., Huber R., Linington C., Jacob U. Structural insights into the antigenicity of myelin oligodendrocyte glycoprotein. Proc. Natl. Acad. Sci. USA. 2003;100:9446–9451. doi: 10.1073/pnas.1133443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linington C., Bradl M., Lassmann H., Brunner C., Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am. J. Pathol. 1988;130:443–454. [PMC free article] [PubMed] [Google Scholar]
- 27.Reindl M., Linington C., Brehm U., Egg R., Dilitz E., Deisenhammer F., Poewe W., Berger T. Antibodies against the myelin oligodendrocyte glycoprotein and the myelin basic protein in multiple sclerosis and other neurological diseases: A comparative study. Brain. 1999;122:2047–2056. doi: 10.1093/brain/122.11.2047. [DOI] [PubMed] [Google Scholar]
- 28.Lalive P.H., Häusler M.G., Maurey H., Mikaeloff Y., Tardieu M., Wiendl H., Schroeter M., Hartung H.P., Kieseier B.C., Menge T. Highly reactive anti-myelin oligodendrocyte glycoprotein antibodies differentiate demyelinating diseases from viral encephalitis in children. Mult. Scler. 2011;17:297–302. doi: 10.1177/1352458510389220. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi N., Sakurai T., Davis K.L., Buxbaum J.D. Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog. Neurobiol. 2011;93:13–24. doi: 10.1016/j.pneurobio.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clements C.S., Reid H.H., Beddoe T., Tynan F.E., Perugini M.A., Johns T.G., Bernard C.C., Rossjohn J. The crystal structure of myelin oligodendrocyte glycoprotein, a key autoantigen in multiple sclerosis. Proc. Natl. Acad. Sci. USA. 2003;100:11059–11064. doi: 10.1073/pnas.1833158100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dyer C.A., Matthieu J.M. Antibodies to myelin/oligodendrocyte-specific protein and myelin/oligodendrocyte glycoprotein signal distinct changes in the organization of cultured oligodendroglial membrane sheets. J. Neurochem. 1994;62:777–787. doi: 10.1046/j.1471-4159.1994.62020777.x. [DOI] [PubMed] [Google Scholar]
- 32.Eguibar J.R., Cortés Mdel.C., Lara-Lozano M. Presynaptic dopaminergic agonists increased gripping-generated immobility episodes in the myelin-mutant taiep rat. Neurosci. Lett. 2010;483:189–192. doi: 10.1016/j.neulet.2010.07.086. [DOI] [PubMed] [Google Scholar]
- 33.Song J., O'connor L.T., Yu W., Baas P.W., Duncan I.D. Microtubule alterations in cultured taiep rat oligodendrocytes lead to deficits in myelin membrane formation. J. Neurocytol. 1999;28:671–683. doi: 10.1023/a:1007060832459. [DOI] [PubMed] [Google Scholar]
- 34.Liñares D., Mañá P., Goodyear M., Chow A.M., Clavarino C., Huntington N.D., Barnett L., Koentgen F., Tomioka R., Bernard C.C. The magnitude and encephalogenic potential of autoimmune response to MOG is enhanced in MOG deficient mice. J. Autoimmun. 2003;21:339–351. doi: 10.1016/j.jaut.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 35.Konradi C., Sillivan S.E., Clay H.B. Mitochondria, oligodendrocytes and inflammation in bipolar disorder: Evidence from transcriptome studies points to intriguing parallels with multiple sclerosis. Neurobiol. Dis. 2011 doi: 10.1016/j.nbd.2011.01.025. in press. Published online February 17, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.