

Abstract
Generalized pustular psoriasis (GPP) is a rare and yet potentially lethal clinical variant of psoriasis, characterized by the formation of sterile cutaneous pustules, neutrophilia, fever and features of systemic inflammation. We sequenced the exomes of five unrelated individuals diagnosed with GPP. Nonsynonymous, splice-site, insertion, and deletion variants with an estimated population frequency of <0.01 were considered as candidate pathogenic mutations. A homozygous c.338C>T (p.Ser113Leu) missense substitution of IL36RN was identified in two individuals, with a third subject found to be a compound heterozygote for c.338C>T (p.Ser113Leu) and a c.142C>T (p.Arg48Trp) missense substitution. IL36RN (previously known as IL1F5) encodes an IL-1 family receptor antagonist, which opposes the activity of the IL-36A and IL-36G innate cytokines. Homology searches revealed that GPP mutations alter evolutionarily conserved residues. Homozygosity for the c.338C>T (p.Ser113Leu) variant is associated with an elevated proinflammatory response following ex vivo stimulation with IL36A. These findings suggest loss of function of IL36RN as the genetic basis of GPP and implicate innate immune dysregulation in this severe episodic inflammatory disease, thereby highlighting IL-1 signaling as a potential target for therapeutic intervention.
Main Text
Generalized pustular psoriasis (GPP) is a rare and potentially life-threatening disease characterized by episodic, widespread skin inflammation with pustule development associated with marked systemic features (Figure 1). Acute attacks are associated with pregnancy and may be triggered by infection and exposure to drugs. GPP may be the only manifestation of disease or occur with other forms of psoriasis, including psoriasis vulgaris (PV) and palmar-plantar pustulosis (PPP).1 Although GPP is classified as a variant of psoriasis, the striking clinical and histological differences indicate that it is likely to be a disease of distinct etiology.
Figure 1.
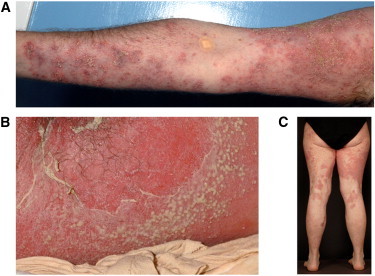
Clinical Features of Generalized Pustular Psoriasis
(A) Individual GPP-02 II:1. Note extensive plaques of erythema with superimposed pustules on the arm.
(B) Individual GPP-01 II:5. Note erythematous, intensely inflamed skin with active pustulation and desquamation on the buttocks.
(C) Individual GPP-03 II:2. Note widespread, healing erythematous plaques following active pustulation on the legs.
Here, we sought to identify disease-causal alleles for GPP by using an established exome-sequencing strategy.2 We selected five unrelated affected individuals with GPP (Table 1) on the basis of the following criteria: (1) recurrent, severe GPP requiring hospital admission; (2) absence of associated PV; and (3) absence of the HLA-Cw∗0602 allele, the major genetic determinant of PV, at the HLA-C locus.3 The clinical features of each of the five subjects are presented in Table 1. None had a family history of GPP. All subjects provided full written consent, and ethical approval for this study was obtained from St Thomas' Hospital Research Ethics Committee (reference 06/Q0702/7).
Table 1.
Clinical and Genetic Data for the Five GPP Cases in Whom Exome Sequencing Was Performed
| Individual | Gender | Ethnicity | Age at Onset (years) | GPP | PPP | CPP | Inflammatory Arthritis | Triggers of Exacerbations | Past Treatments / Responsea |
Systemic Features during Flares |
IL36RN Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GPP-01 II:5 | female | European | 51 | yes | no | no | no | stress | acitretin / no methotrexate / no ciclosporin / yes fumaderm / no etanercept / initial infliximab / initial adalimumab / partial |
fever > 38°C: yes malaise: yes CRP > 100 mg/l: yes ESR > 50 mm/hr: yes neutrophilia > 15 × 109/l: yes |
c.338C>T (p.Ser113Leu) |
| GPP-02 II:1 | male | European | 5 | yes | no | no | no | stress, medication (penicillin, codeine), viral infection | prednisolone / yes acitretin / no methotrexate / yes ciclosporin / yes |
fever > 38°C: yes malaise: yes CRP > 100 mg/l: yes ESR > 50 mm/hr: yes neutrophilia > 15 × 109/l: yes |
c.338C>T (p.Ser113Leu) |
| GPP-03 II:2 | female | European | 7 | yes | no | no | no | stress, medication (paracetamol), viral infection, pregnancy | prednisolone / yes acitretin / no isotretinoin / no methotrexate / yes ciclosporin / yes infliximab / yes |
fever > 38°C: yes malaise: yes CRP > 100 mg/l: yes ESR > 50 mm/hr: yes neutrophilia > 15 × 109/l: yes |
c.142C>T (p.Arg48Trp) and c.338C>T (p.Ser113Leu) |
| GPP-04 I:1 | female | European | 10 | yes | yes | no | no | stress, pregnancy | prednisolone / no methotrexate / yes ciclosporin / no infliximab / yes |
fever > 38°C: yes malaise: yes CRP > 100 mg/l: no ESR > 50 mm/hr: yes neutrophilia > 15 × 109/l: yes |
no mutation identified |
| GPP-05 I:1 | female | European | 45 | yes | yes | no | no | - | acitretin / yes ciclosporin / yes |
fever > 38°C: no malaise: no CRP > 100 mg/l: no ESR > 50 mm/hr: no neutrophilia > 15 × 109/l: no |
no mutation identified |
Abbreviations are as follows: CRP, C-reactive protein; ESR, erythrocyte sedimentation rate.
Response defined as treatment resulting in disease remission for > 1 month.
DNA was extracted from blood, and whole-exome capture was performed by in-solution hybridization followed by massively parallel sequencing. Three micrograms of genomic DNA was sheared via focused acoustic technology (Covaris), yielding a mean fragment size of 150 bp. Fragment ends were repaired and sequencing adaptors were ligated. The sequence library was hybridized for 24 hr with biotinylated 120 bp RNA probes designed against coding regions of the genome (Agilent). Streptavidin-coated magnetic beads were utilized to retain DNA bound to the RNA probes, while unbound DNA was washed off. The exome-enriched pool of DNA was then eluted and amplified with a low-cycle PCR. Finally, the enriched DNA fragments were sequenced with 76 bp paired-end reads on the Illumina GAIIx platform. The resulting reads were aligned to the reference human genome (hg18, NCBI build 36) with Novoalign (Novocraft Technologies). Over 4.7 Gb of sequence was generated for each subject, such that > 75% of the coding bases of the GENCODE-defined exome were represented by at least 20 reads (Table S1 available online). Single-nucleotide substitutions and small insertion/deletion variants were identified with Samtools4 and annotated with respect to coding genes with the Annovar software package5 (Table S2).
Because vertical transmission has not been reported for GPP, and the proband GPP-01 II-5 is the offspring of consanguineous parents (Figure 2), we prioritized analysis of the exome variant profiles with a model of a rare autosomal-recessive inheritance. This analysis required the presence of at least one homozygous or two heterozygous nonconservative sequence changes (nonsynonymous/splice-site substitution or coding insertion/deletion occurring with an estimated population frequency of < 0.01) in the same gene, in all five individuals. Population-frequency estimates were derived from the March 2011 data release of the 1000 Genomes Project and from 250 control exome variant profiles sequenced via the same methodology in our laboratory. The analysis failed to identify a single gene fitting a rare recessive model in all five individuals. However, evaluation of the data with a prior expectation of genetic heterogeneity revealed IL36RN (previously known as IL1F5, MIM 605507, RefSeq NM_173170.1) as the only gene harboring low-frequency variants compatible with recessive inheritance in three of the five subjects (Table 2). Two (GPP-01 II:5 and GPP-02 II:1) were homozygous for a c.338C>T (p.Ser113Leu) missense substitution, whereas the third subject (GPP-03 II:2) was found to be compound heterozygous for c.338C>T (p.Ser113Leu) and a c.142C>T (p.Arg48Trp) missense substitution.
Figure 2.
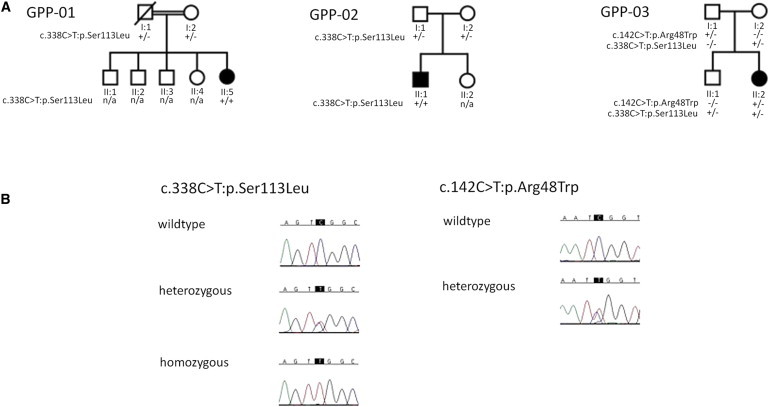
Mutation and Segregation Analysis
(A) Pedigrees and mutation status of three GPP kindreds in whom mutations were identified in IL36RN.
(B) Sequence chromatograms of the c.338C>T (p.Ser113Leu) and c.142C>T (p.Arg48Trp) mutations.
Table 2.
Numbers of Candidate Genes Highlighted by Exome Sequencing
| Any x of five individuals | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Genes with homozygous or compound-heterozygous nonsynonymous, splice-site, or insertion/deletion variants | 6721 | 5141 | 4030 | 3108 | 2162 |
| With an estimated frequency < 0.01 | 80 | 5 | 1 | 0 | 0 |
The identified variants were each confirmed by Sanger sequencing (Figure 2). Genotyping of these substitutions in available unaffected family members confirmed that the segregation of the variants was consistent with a recessive inheritance pattern for IL36RN-associated GPP (Figure 2). Neither variant has been observed by the 1000 Genomes Project or observed in the homozygous state in 250 ethnically matched control individuals. A single carrier of the c.338C>T (p.Ser113Leu) missense substitution was observed in our control cohort; no other coding variation in IL36RN was observed in this individual. Sanger sequencing of the two individuals in whom no mutation was identified by exome sequencing confirmed the wild-type sequence. It is of note that PPP was observed in these two cases. PPP is a clinical feature not present in the three individuals in whom we identified IL36RN mutations, these findings are therefore consistent with the notion that mutation of IL36RN is responsible for a specific subtype of GPP not associated with PPP. This specificity was further supported by Sanger sequencing of the four IL36RN coding exons and their associated splice sites in a cohort of 30 individuals with a range of pustular forms of psoriasis, including GPP, in individuals with preexisting PV and PPP. Wild-type coding sequence was observed in all subjects. In addition, no significant association of common variation at the IL36RN locus with PV was observed under recessive or additive models in data available from a large genome-wide association study of psoriasis previously undertaken by our group (data not shown).3 IL36RN has also been previously investigated in psoriatic arthritis; although association was reported with variants in other genes located within the IL1 gene cluster, no direct association was observed with a variant located in intron 1 of IL36RN (rs990524).6 More recently, a genome-wide association study of psoriatic arthritis did not report association to IL36RN locus.7
IL36RN belongs to the IL-1 cytokine family, which comprises a group of evolutionary ancient cytokines with potent and critical roles in innate immunity; a system of cells, receptors, and mediators that provides a rapid and nonspecific response to pathogens. There are 11 IL-1 family members, nine of which are clustered in a 400 kb region on chromosome 2. Like many members of the IL-1 family, IL36RN and its paralogs IL36A (MIM 605509), IL36B (MIM 605508), and IL36G (MIM 605542) (previously known as IL1F6, IL1F8, and IL1F9, respectively) are abundantly expressed in skin. The proteins encoded by these genes modulate NF-κB signaling, through their interaction with the IL-1RL2 receptor (previously known as IL-1Rrp2).8,9 IL36RN in particular antagonizes the activity of IL-36A and IL-36G, by blocking the recruitment of the IL-1RL2 receptor complex.10 IL36RN itself consists of a putative signal peptide followed by a large IL-1 homology motif, thus reflecting the typical domain architecture of IL-1 family cytokines.10 In this context, homology searches and three-dimensional structural modeling demonstrated that both the p.R48W and the p.S113L mutation affect evolutionary conserved IL36RN residues. which are located in close proximity to critical binding residues (Figure 3).11 Both missense substitutions are predicted by SIFT to be damaging.12 To assess the functional impact of the p.Ser113Leu mutation, we obtained peripheral-blood mononuclear cells (PBMCs) from a homozygous affected individual (GPP-02 II:1) and an unrelated healthy volunteer. We examined the effect of IL-36A stimulation on the production of four NF-κB-induced proinflammatory cytokines (IL-1α, IL-6, IL-8, and TNF). After 7 hr of treatment with 1 μg/ml IL-36A (R&D Systems), both intracellular transcript levels (Applied Biosystems TaqMan Gene Expression assays) and extracellular protein secretion (Millipore MILLIPLEX MAP Human Cytokine/Chemokine panel and a Luminex FLEXMAP 3D flow-based analyzer) were measured. The induction of TNF mRNA was not analyzed because the 7 hr time point exceeds the time span of the gene transcriptional response. Both sets of experiments demonstrated a marked increased in cytokine production in the affected individual PBMCs (Figure 4), in keeping with the notion that the p.Ser113Leu missense substitution affects the ability of IL36RN to antagonize IL36A-induced NF-κB signaling.
Figure 3.
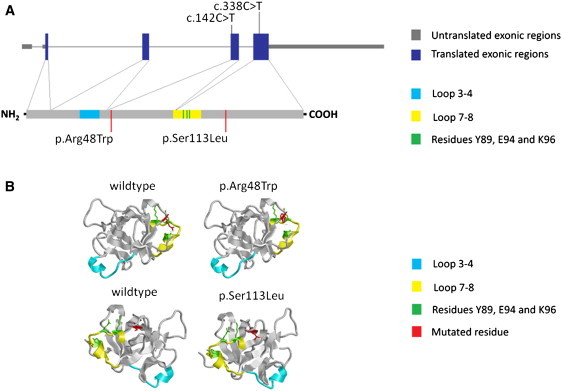
Location of the Two Identified Mutations
(A) Location of the two identified mutations with respect to the genomic organization of IL36RN (upper panel) and IL36RN (lower panel).
(B) Locations of the c.142C>T (p.Arg48Trp) and c.338C>T (p.Ser113Leu) mutations with respect to the three-dimensional structure of IL36RN, including the loop domains and critical residues that mediate receptor interaction.9
Figure 4.
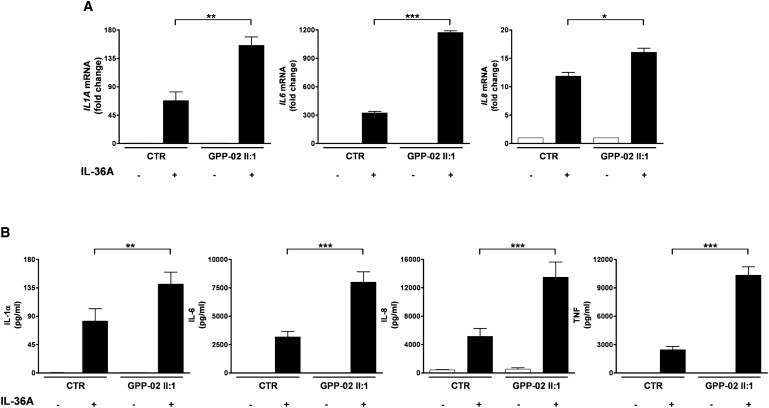
Analysis of Case and Control PBMCs, after Ex Vivo Stimulation with IL-36A
(A) The induction of IL1A, IL6, and IL8 mRNA was assessed by real-time PCR, with the RPLPO transcript used as an endogenous control. Data are plotted as the mean, and error bars represent the SEM of duplicate stimulations with the expression level of the unstimulated sample set as a baseline value. CTR, control.
(B) The concentration of IL-1α, IL-6, IL-8, and TNF was assayed in the PBMC culture medium. Data are plotted as the mean, and error bars represent the SEM of duplicate stimulations. All significance values were calculated via a one-way ANOVA test, followed by a Bonferroni posttest. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. CTR, control.
In mice, abnormal Il-36 signaling due to Il1f6 (the mouse ortholog of IL-36A) overexpression results in transient skin inflammation characterized by acanthosis, hyperkeratosis, and a mixed-cell infiltrate rich in neutrophils,13 features observed in both PV and GPP. When crossed with Il1f5 (the mouse ortholog of IL36RN)-deficient mice, the skin phenotype is strikingly enhanced and persistent with extensive pustule formation.13 Taken together, these data provide the platform to postulate that GPP is caused by abnormal IL-36A signaling resulting from IL36RN loss-of-function mutations. The variable age at onset of clinical manifestations of GPP in the three individuals in whom we have identified mutations in IL36RN suggests a complex interaction among this major genetic determinant, potential environmental triggers, and/or additional genetic modifiers. Interestingly mutations in another IL-1 family receptor antagonist, IL1RN (MIM 147679), have recently been reported to cause an autoinflammatory disease characterized in part by pustular skin eruptions (MIM 612852).14,15 However, there are notable differences between this disease and GPP, including neonatal onset and major skeletal abnormalities. Finally, it is also of interest that anakinra, a recombinant IL-1 receptor antagonist (IL-1RA), has been reported to be effective in two individuals with GPP,16 providing evidence for the clinical benefit of treatment targeting IL1 family pathways.
GPP is a severe, debilitating, and potentially life-threatening disease. Our data indicate that mutations in IL36RN underlie this distinct form of pustular disease, separate from other forms of psoriasis. They implicate defects of the innate immune system and highlight IL-1 signaling as a potential target for therapeutic intervention.
Acknowledgments
The authors express their gratitude to the families for participating in this study. This work was supported by an MRC Programme grant to R.C.T., F.O.N., and J.N.B. (G0601387), a Wellcome Trust Programme grant to F.O.N. (GR078173MA), and a British Skin Foundation award to F.C. (1006). A.O. is supported by a studentship funded by The Generation Trust. A.E.P. is supported by an MRC/BAD/BSF Clinical Research Training Fellowship. The authors acknowledge the use of BRC Core Facilities provided by the financial support from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy's & St Thomas' NHS Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust. We acknowledge technical assistance from Anna Bertoni and Christian Hundhausen.
Contributor Information
Richard C. Trembath, Email: richard.trembath@kcl.ac.uk.
Jonathan N. Barker, Email: jonathan.barker@kcl.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org
The GENCODE Project, http://www.gencodegenes.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Griffiths C.E.M., Barker J.N.W.N. Psoriasis. In: Burns T., Breathnach S., Cox N., Griffiths C.E.M., editors. Rook's Textbook of Dermatology. Wiley-Blackwell; Chichester, England: 2010. pp. 20.1–20.60. [Google Scholar]
- 2.Simpson M.A., Irving M.D., Asilmaz E., Gray M.J., Dafou D., Elmslie F.V., Mansour S., Holder S.E., Brain C.E., Burton B.K. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 2011;43:303–305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- 3.Strange A., Capon F., Spencer C.C., Knight J., Weale M.E., Allen M.H., Barton A., Band G., Bellenguez C., Bergboer J.G., Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2 A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010;38:164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rahman P., Sun S., Peddle L., Snelgrove T., Melay W., Greenwood C., Gladman D. Association between the interleukin-1 family gene cluster and psoriatic arthritis. Arthritis Rheum. 2006;54:2321–2325. doi: 10.1002/art.21928. [DOI] [PubMed] [Google Scholar]
- 7.Hüffmeier U., Uebe S., Ekici A.B., Bowes J., Giardina E., Korendowych E., Juneblad K., Apel M., McManus R., Ho P. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat. Genet. 2010;42:996–999. doi: 10.1038/ng.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnston A., Xing X., Guzman A.M., Riblett M., Loyd C.M., Ward N.L., Wohn C., Prens E.P., Wang F., Maier L.E. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J. Immunol. 2011;186:2613–2622. doi: 10.4049/jimmunol.1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Towne J.E., Garka K.E., Renshaw B.R., Virca G.D., Sims J.E. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 2004;279:13677–13688. doi: 10.1074/jbc.M400117200. [DOI] [PubMed] [Google Scholar]
- 10.Sims J.E., Smith D.E. The IL-1 family: regulators of immunity. Nat. Rev. Immunol. 2010;10:89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- 11.Dunn E.F., Gay N.J., Bristow A.F., Gearing D.P., O'Neill L.A., Pei X.Y. High-resolution structure of murine interleukin 1 homologue IL-1F5 reveals unique loop conformations for receptor binding specificity. Biochemistry. 2003;42:10938–10944. doi: 10.1021/bi0341197. [DOI] [PubMed] [Google Scholar]
- 12.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 13.Blumberg H., Dinh H., Trueblood E.S., Pretorius J., Kugler D., Weng N., Kanaly S.T., Towne J.E., Willis C.R., Kuechle M.K. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J. Exp. Med. 2007;204:2603–2614. doi: 10.1084/jem.20070157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aksentijevich I., Masters S.L., Ferguson P.J., Dancey P., Frenkel J., van Royen-Kerkhoff A., Laxer R., Tedgård U., Cowen E.W., Pham T.H. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N. Engl. J. Med. 2009;360:2426–2437. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddy S., Jia S., Geoffrey R., Lorier R., Suchi M., Broeckel U., Hessner M.J., Verbsky J. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N. Engl. J. Med. 2009;360:2438–2444. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viguier M., Guigue P., Pagès C., Smahi A., Bachelez H. Successful treatment of generalized pustular psoriasis with the interleukin-1-receptor antagonist Anakinra: lack of correlation with IL1RN mutations. Ann. Intern. Med. 2010;153:66–67. doi: 10.7326/0003-4819-153-1-201007060-00030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.