

Abstract
The genetic variants leading to impairment of intellectual performance are highly diverse and are still poorly understood. ST3GAL3 encodes the Golgi enzyme β-galactoside-α2,3-sialyltransferase-III that in humans predominantly forms the sialyl Lewis a epitope on proteins. ST3GAL3 resides on chromosome 1 within the MRT4 locus previously identified to associate with nonsyndromic autosomal recessive intellectual disability. We searched for the disease-causing mutations in the MRT4 family and a second independent consanguineous Iranian family by using a combination of chromosome sorting and next-generation sequencing. Two different missense changes in ST3GAL3 cosegregate with the disease but were absent in more than 1000 control chromosomes. In cellular and biochemical test systems, these mutations were shown to cause ER retention of the Golgi enzyme and drastically impair ST3Gal-III functionality. Our data provide conclusive evidence that glycotopes formed by ST3Gal-III are prerequisite for attaining and/or maintaining higher cognitive functions.
Introduction
Intellectual disability (ID) is a cognitive impairment disorder characterized by an intelligence quotient (IQ) below 70.1 Patients are persistently slow learners of basic language and motor skills during childhood and show significant subnormal global intellectual capacity as adults. ID is thought to occur with a prevalence between 1% and 3% and frequently results from genetic aberrations (see e.g., Ropers2). In developed countries the occurrence of ID with (syndromic ID) or without (nonsyndromic ID) other congenital anomalies is the most frequent reason for seeking clinical advice. However, the cases where definitive molecular alterations can be linked with ID represent a minority (see e.g., Ropers3), per se reflecting the extreme heterogeneity of genetic and environmental components that are involved in the etiology of this disorder. In the current study, we identified mutations in the sialyltransferase ST3GAL-III4 that cause nonsyndromic autosomal recessive ID (NSARID), and with this discovery move the glycocalyx, the outermost structure of each animal cell,5 into the focus of cognitive research.
Although largely neglected, the glycocalyx is without doubt the essential information carrier in all living systems. Only sugars, the molecular building blocks of the glycocalyx, possess the chemical potential to create a biological communication system that is rich enough to store and translate the myriads of details for steering and fine-tuning developmental processes in general6 and neurodevelopment in particular.7 Indeed, glycosylation defects in general were previously reported to play a role in the etiology of disorders with ID features including NSARID (for reviews see e.g., Grunewald et al.8 and Marquadt and Denecke9).10,11
In contrast to RNA, DNA, and protein biosynthesis, glycans are built without guiding templates and thus are prone to dynamic- and context-dependent alterations reflecting the state of the respective individual cell (for review see Cohen and Varki12).
Positioned at the outermost sites of the glycocalyx, the nine-carbon-sugar sialic acid (Sia) plays a prominent role in this scenario.13 The sialome, defined here as the total complement of sialo-glycotopes, is built by twenty sialyltransferases (STs) that vary in acceptor and linkage specificity.14 Complementing this, modern organisms have developed a large family of Sia-binding lectins to decipher the information encrypted in the sialome (for review see Crocker et al.15). Moreover, the recent evolution of the sialylation pathway in humans has been leading to the generation of ten uniquely human variants within the ∼60 genes directly involved in the biosynthesis of the sialome.16 This burst of genetic changes coincides with the separation of the modern humans from their closest hominide relatives, the great apes.17
ST3GAL3 located on chr1p34.1 encodes the β-galactoside-α2,3-sialyltransferase-III (ST3Gal-III), which in humans predominantly forms the sialyl Lewis a (sLea) epitope on glycoproteins (see Figure 1).18 The gene maps to overlapping linkage intervals recently identified in two families with members suffering from nonsyndromic autosomal recessive intellectual disability (NSARID).19,20 The identified intervals were 10.1 Mbp and 9.3 Mbp in size and showed highly significant LOD scores of 4.2 and 7.1, respectively. The shared common region was 7.9 Mbp and contained a total of 118 protein coding genes, including ST3GAL3.19,20 In each interval we identified a different mutation in ST3GAL3, both of which cosegregate with NSARID and have a deleterious impact upon the functionality of the gene product.
Figure 1.
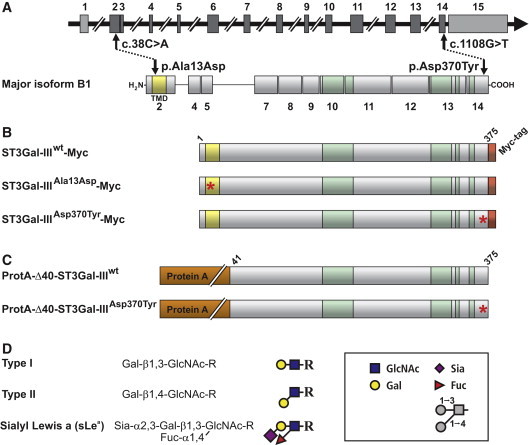
Schematic Representation of ST3GAL3 and the Major Translation Product Isoform B1
(A) ST3GAL3 is a complex transcriptional unit comprising 15 exons, 12 of which (dark gray boxes) contain protein coding sequence. The major translation product is isoform B1. As a typical Golgi-localized sialyltransferase ST3Gal-III contains a short, N-terminally localized cytoplasmic tail followed by a TMD (shaded in yellow). Positions of identified homozygous mutations and resulting amino acid exchanges are indicated by arrows. The highly conserved sialylmotifs are shaded in green.
(B and C) Illustration of ST3Gal-III expression constructs used in this study to analyze the impact of mutations. All constructs are based on isoform B1. Full-length sequences were extended by a C-terminal Myc-epitope to enable protein identification in the cellular context and in parallel to compartmental markers. For the generation of soluble proteins the first 40 amino acids were replaced by protein A. Construct names are indicated and mutations are highlighted by asterisks.
(D) ST3Gal-III is involved in the formation of the sialyl Lewis a (sLea) epitope on proteins. The transfer of sialic acid (Sia) onto galactose (Gal) present in type I and II structures precedes the transfer of fucose (Fuc) to the core sugar N-acetylglucosamine (GlcNAc).
Subjects and Methods
Patients
This study was performed in accordance with the ethical standards of the responsible institutional and national committees on human experimentation. The patients and their families were recruited in collaboration with local genetic counselors from Iran who obtained informed consent of the parents from both families in writing. Probands were examined by experienced clinical geneticists who assessed their physical and mental status by using a standard clinical evaluation form that had been designed as described earlier.20 The family pedigrees are shown in Figure 2 and clinical features of the patients from both families are listed in Table S2, available online.
Figure 2.
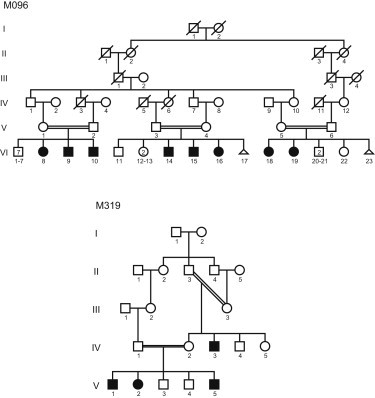
Family Pedigrees
Filled symbols indicate individuals suffering from intellectual disability.
Genome Partitioning and Mutation Screening
We obtained a blood sample from one patient per family (1721 of M096 and 381of M319) and used it to generate Epstein-Barr-Virus-transformed lymphoblastoid cell lines (LCLs); we used these LCLs to enrich chromosome 1 containing the genomic region previously identified to cosegregate with the disease.19,20 The chromosome sorting was performed by Chrombios (Raubling, Germany) and resulted in about 300,000 metaphase copies of chromosome 1s per patient. DNA was prepared and sequenced with an Illumina Genome Analyzer IIx. Original sequencing data were deposited in the Sequence Read Archive SRA (see Web Resources), accession number SRA026713.
We next aligned the 36 bp paired-end reads obtained from each sequencing run onto the human reference genome (hg18) and focused for mutation screening on the target regions defined by the linkage intervals of each family by using SOAP2.2021 and the parameter settings –a –b –D –o –m 200 –x 600 −2 –g 3 –e. Reads that could be unambiguously aligned to the target regions were then used for variant calling, that is, the detection of nonsynonymous mutations located in coding regions and changes in canonical splice sites (based on the RefSeq gene model). We excluded variants with a potential effect on splice sites (i.e., destroying an existing splice site or introducing a cryptic splice site) by using NNSplice (see Web Resources). Putatively causative mutations located in untranslated regions or introns were excluded by comparison with known regulatory elements from the ORegAnno and TargetScan miRNA regulatory site databases (see Web Resources). Variant calls had to be supported by at least three nonidentical reads, a phred-like quality score ≥ 20, and an allelic percentage ≥ 70. To remove neutral polymorphisms, variants were filtered with dbSNP130 as well as the published genomes from 185 individuals22 and 200 Danish exomes.23
Expression Plasmids
We amplified the cDNA encoding the B1 isoform of ST3Gal-III (accession number NM_006279.2) by PCR on cDNA purchased from Origene (RC208261) and cloned it into pcDNA3 (Invitrogen) to generate untagged full-length or into pcDNA3.1-Myc/His A (Invitrogen) to generate C-terminally Myc-His6 tagged translation products. Mutations were introduced by site-directed mutagenesis. Details on the used primer sequences are given in Table S1.
For the construction of soluble ProtA-tagged enzyme variants, the sequence encoding amino acids 41–375 of ST3Gal-III was amplified by PCR with Origene RC208261 as a template and the resulting PCR product subcloned into pPROTA vector.24 The p.Asp370Tyr mutant enzyme was generated by site-directed mutagenesis. Detailed information on the cloning strategies is given in Table S3.
Cell Culture and Transfection
The mouse fibroblast cell line LMTK− and Chinese hamster ovary (CHO) cells were cultured in DMEM/Ham's F-12 (1:1) medium (Biochrom) containing 5% FCS and 2 mM sodium pyruvate. Transient transfections with plasmid DNAs were carried out with Lipofectamine (Invitrogen) in OpitMEM (GIBCO BRL). EBV-transformed lymphoblasts were cultured in RPMI 1640 medium (Biochrom) containing 10% FCS, 2 mM sodium pyruvate, 100 U/ml penicillin and 0.1 mg/ml streptomycin.
Immunocytochemistry
Transiently transfected LMTK− cells were fixed 72 hr after transfection with 4% PFA (AppliChem) in PBS for 30 min at room temperature, permeabilised with 0.2% Triton X-100 in PBS for 30 min, and incubated with primary antibodies for 1 hr. Rabbit α-Mannosidase II antiserum (kindly provided by Kelley Moremen, University of Georgia, Athens, USA) was used in a dilution of 1:8570, rabbit IRE1 polyclonal antibody (Abcam) in a dilution of 1:40, Myc antibody 9E10 (Roche) in a concentration of 5 μg/ml, and ST3Gal-III antibody (Sigma, av46706) in a concentration of 2 μg/ml. Cells were stained with a combination of 1:2000 diluted Cy3 conjugated sheep anti-mouse IgG (whole molecule) F(ab)2 fragments (Sigma) and 1 μg/ml Alexa Fluor 488 donkey anti-rabbit IgG (H+L; Invitrogen Molecular Probes) or 1:1000 diluted Cy3 conjugated sheep anti-rabbit IgG (whole molecule) F(ab)2 fragments (Sigma) for 1 hr at room temperature in the dark. Slides were mounted in Vectashield mounting medium with 4′-6-Diamidino-2-phenylindole (DAPI) (Vector laboratories) and analyzed with a Plan Apochromat 63x/1.40 Oil DIC M27 differential interference contrast objective with an Axiovert 200M microscope in combination with an ApoTome, an AxioCAM MRm digital camera and AxioVision 4.5 software (Zeiss). Images were taken in an automatic exposure setting with filter sets for DAPI, Alexa Fluor 488, and Cy3 and converted in blue, green, and red, respectively. Intensities were adapted to be equal for the three colors.
Immunoprecipitation
Soluble ProtA fusion constructs were extracted from cell culture supernatants of transiently transfected CHO cells by the use of IgG Sepharose Fast Flow 6 (GE Healthcare). Myc-tagged full-length constructs were immunoprecipitated from CHO cell lysates by the use of anti-Myc mAb 9E10 coupled to Protein G Sepharose Fast Flow 4 (GE Healthcare). Protein concentrations were determined by immunoblotting against the Myc-epitope with serial dilutions of a purified Myc-tagged protein.
Immunoblot Analysis
Following a 5 min boiling step, samples were subjected to 10% SDS-PAGE according to Laemmli25 under reducing conditions with 2.5% (v/v) β-mercaptoethanol, and transferred onto nitrocellulose membranes (Schleicher and Schuell) by immunoblotting. For expression analysis, membranes were developed as described in Mühlenhoff et al.26 For the activity assay, membranes were blocked with Odyssey Blocking Buffer (LI-COR,1:2 diluted in PBS), incubated with penta-His antibody (QIAGEN) or mouse IgG (Pierce) as described above, followed by incubation with 1:10,000 diluted anti-mouse IRDye680 (LI-COR) for 1 hr, and analyzed with an Odyssey Imaging System (LI-COR). Bands were quantified with the Odyssey 2.1 software.
Enzymatic Testing of ST3Gal-III Variants
ST3Gal-III variants immunoprecipitated as described above were equilibrated with reaction buffer (50 mM 2-(N-morpholino)ethanesulfonic acid (MES) [pH 6.5] and 10 mM MnCl2) and subsequently, incubated with the synthetic acceptor Gal-β1,3-GlcNAc-β-para-nitrophenol (1mM) and 50 μM (0.9 kBq) CMP-[14C]Neu5Ac at 37°C. After 1h, the reaction mix was loaded onto methanol-activated SepPak Plus C18-columns (Waters). The columns were washed three times with PBS, and reaction products were eluted with 4.5 ml MeOH. Eluates were air-dried, resolved in 3 ml Filtersafe scintillation cocktail (Zinsser Analytic), and measured in an LS 6500 Multi-Purpose Scintillation Counter (Beckman Coulter).
Results
To identify the disease-causing mutations in the two families with overlapping linkage intervals on chr1p34.1, we used flow sorting to isolate in excess of 300,000 copies of chromosome 1 from LCLs of each index patient. DNA preparations from these chromosomes were submitted to high-throughput sequencing with a Genome Analyzer IIx (Illumina), resulting in a sequence coverage of more than 99% with a mean depth between 17 and 24 reads per bp position on chromosome 1 for the each patient (Table 1). Alignment (with SOAP21) of the mappable paired-end reads from the each linkage interval with the human reference genome revealed 6384 (M096) and 4874 (M319) homozygous sequence changes.
Table 1.
Deep Sequencing Data
| Patient/Family | Total Accumulative Length of Obtained Sequence Fragments [Gb] | Total Accumulative Length of Mappable Fragments [Gb] | Physical Coordinates of Linkage Intervala[bp] | Total Accumulative Length of Mappable Fragments in Linkage Interval [Mb] | Relative Sequence Coverage in Linkage Interval | Fold Coverage in Linkage Interval |
|---|---|---|---|---|---|---|
| M096 | 7.43 | 6.81 | chr1:37058812-46775726 | 166 | 99.7% | 17 |
| M319 | 10.66 | 9.02 | chr1:38895940-48981455 | 246 | 99.4% | 24 |
Based on NCBI genome build 36.1 (hg18).
After in silico filtering of these data, 197 (M096) and 187 (M319) previously unreported DNA variants remained; however, all but one mutation per family lay outside the protein coding sequences as well as canonical splice donor and acceptor sites. The single remaining change in each patient affected the same gene, ST3GAL3, at positions either in exon 2 (NM_006279.2: c.38C>A, p.Ala13Asp) or exon 14 (NM_006279.2: c.1108G>T, p.Asp370Tyr; see also Figure 1, Table S1), this change affected the transmembrane domain (TMD) and the catalytic domain (CD), respectively. In silico analyses carried out with different bioinformatic tools (Pmut,27 SIFT,28 and MutationTaster29) suggested a disease-causing potential for both changes. The mutations were confirmed by Sanger sequencing and shown to be present in patients in a homozygous state and heterozygous or absent in all tested healthy individuals from each pedigree. In contrast, the newly identified ST3GAL3 mutations appeared neither in the 278 copies of chromosome 1 from ethnically matched controls nor in the published genomes of 185 individuals from the 1000 Genomes Project22 and were also absent in the exomes of 200 Danish individuals,23 in whom no other deleterious mutation within the whole coding region of ST3GAL3 was also not observed.
Like all other members of the ST family, ST3Gal-III is a Golgi-resident type II membrane protein30,31 containing four highly conserved sialylmotifs that form part of the active site32 (Figure 1). The ST3GAL3 mutations identified in NSARID patients introduce primary sequence changes in the TMD (ST3Gal-IIIAla13Asp) and the CD (ST3Gal-IIIAsp370Tyr) outside of the conserved motifs (Figure 1). We first investigated the presence of ST3GAL3 transcripts in normal human lymphoblastoid cell lines by RT-PCR and found extremely low expression in this cell type (data not shown). Thus, facing a lack of suitable patient specimens to study the consequences of these mutations directly, we used cellular and in vitro test systems.
Because the TMDs of several glycosyltransferases have previously been demonstrated to be involved in correct subcellular localization of the enzymes (reviewed in Tu and Banfield33), we suspected that the p.Ala13Asp mutation affected ST3Gal-III localization. To test this, we expressed full-length forms of ST3Gal-IIIwt and the mutants ST3Gal-IIIAla13Asp and ST3Gal-IIIAsp370Tyr in the murine fibroblast cell line LMTK− and evaluated the subcellular localization of the enzymes by indirect immunofluorescence by using a ST3Gal-III-specific antibody. Notably, the perinuclear Golgi signal obtained for the wild-type enzyme was replaced by bright signals highly reminiscent of ER staining in cells expressing one of the mutant enzymes (Figure 3, left panel). To further explore this aberrant localization, we used C-terminally Myc-tagged ST3Gal-III variants (ST3Gal-III-Myc), enabling the parallel staining of compartmental markers (α-Mannosidase II [α-Man II] for Golgi and IRE1 for ER). In keeping with the direct detection of ST3Gal-III, the antibody recognizing the Myc-tag demonstrated strict Golgi localization for ST3Gal-IIIwt-Myc (overlay with anti-α-Man II), whereas both mutants colocalized with the ER marker IRE1. Remarkably, the segregation between α-Man II and ST3Gal-IIIAsp370Tyr-Myc was complete, whereas in rare cases ST3Gal-IIIAla13Asp-Myc reached the Golgi apparatus in this cellular assay system (Figure 3, middle panel). In both cases, ER retention prevents the encounter of ST3Gal-III with its Golgi-localized substrates and thus causes loss of function.
Figure 3.
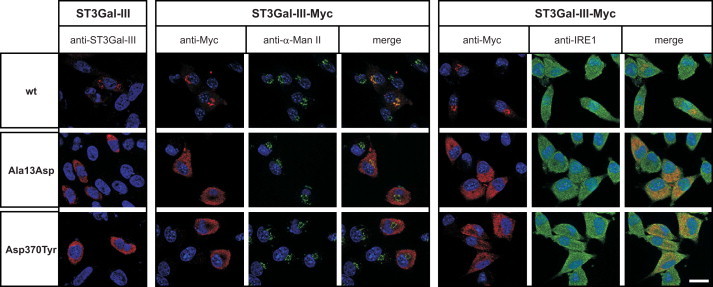
Impaired Golgi Transport of Mutant ST3Gal-III
The subcellular localization of wild-type (wt) and mutant forms of ST3Gal-III (p.Ala13Asp and p.Asp370Tyr) was analyzed in transiently transfected LMTK− cells. Proteins were expressed without (first column; ST3Gal-III) or with C-terminally added Myc-epitope (ST3Gal-III-Myc). A rabbit polyclonal antibody (anti-ST3Gal-III) was used to visualize the untagged protein (first column). Cells transfected with Myc-tagged ST3Gal-III variants enabled the parallel display of recombinant proteins (anti-Myc; red) and of ER (anti-IRE1; green) and Golgi (anti-α-Mannosidase II, α-Man II; green) markers, respectively. ST3Gal-III proteins of wild-type sequence showed the typical Golgi pattern and signals obtained for the recombinant protein fully overlapped with the Golgi marker α-Man II. In contrast, the mutant proteins (p.Ala13Asp and p.Asp370Tyr) displayed an ER-like distribution and an extensive overlap with the ER marker IRE1. Remarkably, the segregation from the Golgi marker was complete for the p.Asp370Tyr mutant, whereas a significant overlap with the Golgi marker α-Man II was observed for the p.Ala13Asp mutant. Nuclei were stained with 4,6′diamidino-2-phenylindole (DAPI; blue). Scale bar: 20 μm.
To address the question of a direct effect of the identified mutations on ST3Gal-III activity, we determined the specific activity of the enzymes. Myc-tagged constructs (see Figure 1B) were immunoisolated from transiently transfected cells and tested in a radioactive incorporation assay. The specific activities calculated from five independent experiments are shown in Figure 4A. Under these in vitro conditions, in which compartmental borders are destroyed and enzymes and substrates come into direct contact, the TMD mutant behaved like the wild-type. In contrast, the CD mutant (p.Asp370Tyr) was found to be completely inactive.
Figure 4.
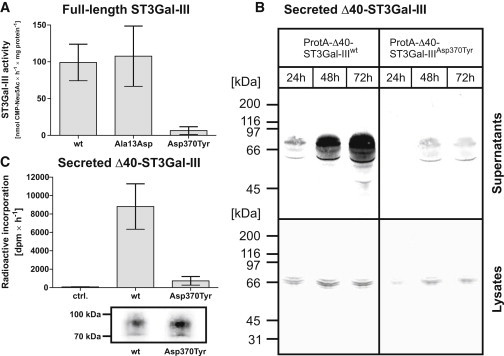
Expression and Functional Testing of Wild-Type and Mutant ST3Gal-III
(A) Sialyltransferase activity of Myc-tagged full-length enzyme variants. Wild-type and mutant enzymes were immunoprecipitated from cell lysates of transiently transfected CHO cells and tested for enzymatic activity with the radioactively labeled substrate CMP-[14C]Neu5Ac and the type I acceptor Gal-β1,3-GlcNAc-β-para-nitrophenol (see Figure 1D). Incorporated radioactivity was measured by scintillation counting after purification with C18 columns. Activities were calculated from five independent experiments and are shown as mean values ± standard error of the mean.
(B) Expression analysis of secreted ProtA-Δ40-ST3Gal-III variants. CHO cells were transiently transfected and the appearance of recombinant proteins in the cell culture supernatants (upper panel) and cell lysates (lower panel) was monitored over 72 hr by immunoblotting.
(C) Sialyltransferase activity of secreted ProtA-Δ40-ST3Gal-III variants. Wild-type and mutant enzymes were immunoprecipitated from cell culture supernatants of transiently transfected CHO cells and the activity incorporation assay was performed as described in (A). One third of the immunoprecipitated samples was analyzed by immunoblotting against the Protein A-part to control the amounts of protein incorporated into the activity assay. Data obtained from three independent experiments are shown as mean values +/− standard error of the mean.
Although STs are extensively studied enzymes, the existing information on structure-function relationships (for review see Audry et al.14) does not provide an explanation for the deleterious effect caused by p.Asp370Tyr. The amino acid exchange lies outside of the functionally important sialylmotifs in an area not known to be involved in protein folding or function. Soluble variants for wild-type and CD mutant were therefore generated to enable enzyme testing outside the cellular environment. The N-terminal 40 amino acids (encompassing the cytoplamic tail and TMD of ST3Gal-III; see Figure 1C) were replaced with Staphylococcus aureus protein A. Fusion constructs (ProtA-Δ40-ST3Gal-IIIwt and ProtA-Δ40-ST3Gal-IIIAsp370Tyr) were expressed in CHO cells, and the time course of the appearance of recombinant proteins in the supernatant was monitored by immunoblotting (Figure 4B). Compared to ProtA-Δ40-ST3Gal-IIIwt, the secretion of ProtA-Δ40-ST3Gal-IIIAsp370Tyr was drastically reduced (Figure 4B). Notably, however, this lack of secretion did not correspond with an intracellular protein accumulation. In contrast, similar intracellular expression levels observed for mutant- and wt-protein argue for equal translation rates, which, in the case of the mutant, might be followed by a rapid clearance of the defective protein by the ER-associated quality control system (for review see e.g., Hoseki et al.34 and Jarosch et al.35). Still, sufficient protein could be isolated and used for in vitro testing. A representative experiment is shown in Figure 4C and confirms that p.Asp370Tyr dramatically affects the enzymatic function of ST3Gal-III. Although the lack of structural information prevents the molecular interpretation, this naturally occurring mutation highlights another aspect of structure-function-relationships in the family of sialyltransferases.
Discussion
Sialic acids are key elements in cellular communication processes because of their exposed position and unique physiochemical properties. Consequently, genetic alterations that disturb or prevent the formation of sialo-glycoconjugates are embryonic lethal (E 6.5 in mouse36) or cause the development of diseases.37
In this study, we demonstrate a link between ST3Gal-III activity and the establishment of higher brain functions. By means of linkage analysis, chromosome sorting and next-generation sequencing, we were able to identify two mutations in ST3GAL3 cosegregating with NSARID, which affect cellular localization and the activity and abundance of the gene product ST3Gal-III. Although this sialyltransferase can act on both type I and type II acceptor structures (Figure 1D), the human ST3Gal-III has been demonstrated to exhibit a marked preference for type I structures on glycoproteins.18,38 The resulting major product, sLea (Figure 1D), is well described in the context of immune reactions39 but has never been linked to neural development and/or function. However, high isotranscript numbers of ST3GAL3 with patterns distinctly different from transcript patterns in all other tissues have been observed in human fetal neural tissue40 and indicate a role of ST3Gal-III activity in brain function.
The N-terminal part of glycosyltransferases, and in particular the transmembrane domain, have been demonstrated to be important for correct Golgi localization of the enzymes.33 Because the biosynthesis of glycotopes is a sequential process strictly dependent on the vectorial organization of enzymes in the ER and Golgi, the ST3Gal-III glycan acceptor structures (see Figure 1D) are formed by glycosyltransferases that are localized in the Golgi.30,31 Moreover, provision of metabolically activated sialic acid, the sugar donor substrate used by sialyltransferases, is restricted to the late Golgi compartments.41 Accordingly, correct localization in the Golgi apparatus is a prerequisite for ST3Gal-III activity. The charged amino acid residue introduced into the hydrophobic TMD in ST3Gal-IIIAla13Asp is likely to disturb the transport information eventually leading to ER retention. The partial rescue of Golgi localization of ST3Gal-IIIAla13Asp-Myc relates to the massive overexpression of the mutant enzyme in transiently transfected cells. However, in the natural situation where glycosyltransferases are generally low expressed (for review see Roth42), the complete loss of ST3Gal-III activity is highly likely.
Fully unexpected was the finding that the very C-terminally located p.Asp370Tyr mutation abolished forward transport of ST3Gal-IIIAsp370Tyr to the Golgi apparatus and prevented secretion of the construct ProtA-Δ40-ST3Gal-IIIAsp370Tyr. This mutation that concerns a protein segment outside the sialylmotifs14 seems to impact protein folding and/or stability. As no accumulation in the ER could be observed by immunoblot analysis, we assume that the severe misfolding of the enzyme triggers degradation by the ERAD system.35 It is important to emphasize that this naturally occurring mutation has revealed a so far unknown function of the C terminus in the folding of functional ST3Gal-III.
Interestingly, no behavioral abnormalities were reported for a previously investigated ST3Gal-III knockout mouse model.43 This promotes the conclusion that highly dynamic combinatorial functions, which are characteristic for human cognitive processes and a priori absent in the mouse are affected. Moreover, in contrast to the known congenital glycosylation disorders,8,9,44 patients with ST3Gal-III mutations do not show any clinical features apart from ID. Thus, it is tempting to speculate that peripheral functions of ST3Gal-III, as for instance described in the mouse,45 are compensated in humans by other members of the ST3Gal family.14 However, higher brain functions have a complex molecular basis and particularly the cognitive aspects thereof represent an evolutionarily young feature. It can thus be assumed that functional redundancy to secure the system against the loss of individual components is not as developed as in older features. This might explain why we find ST3Gal-III and products of ST3Gal-III catalysis indispensable for higher cognition in humans. In this context, it is also of note that recent findings implicate sialylation in the modulation of voltage-gated neuronal potassium channels,46 which seem to play a role in the etiology of psychiatric symptoms and autism as well.47–49 We therefore conclude that our results constitute an important contribution to the understanding of the molecular basis of ID and other autism spectrum disorders. What is more, by implicating glycobiological aspects that were so far not considered to play a role in the etiology of cognitive dysfunction, our findings open up interesting new avenues of research into the function of the human central nervous system in health and disease.
Acknowledgments
We are grateful to the patients and their families for their participation in the study. We thank Herbert Hildebrandt for helpful discussion and Corinna Jensen, Marianne Schlicht, Anna-Maria Baumann, and Maike Hartmann for expert technical assistance. This work was supported financially by a grant from the Iranian National Science Foundation. Support was also received from the Max Planck Innovation Fund, a grant from the German Federal Ministry of Education and Research to H.H.R. (MRNET 01GS08161-2), and a grant from the German Research Foundation to R.G.-S. (GE 801/10-1). H.H.R., H.N., and K.K. participate in the GENCODYS Consortium.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://omim.org
ORegAnno and TargetScan miRNA regulatory site databases, http://genome.ucsc.edu/cgi-bin/hgTables
Sequence Read Archive (SRA), http://www.ncbi.nlm.nih.gov/sra
Accession Numbers
The Sequence Read Archive (SRA) accession number for the sequencing data reported in this paper is SRA026713.
References
- 1.Ropers H.H., Hamel B.C. X-linked mental retardation. Nat. Rev. Genet. 2005;6:46–57. doi: 10.1038/nrg1501. [DOI] [PubMed] [Google Scholar]
- 2.Ropers H.H. Genetics of intellectual disability. Curr. Opin. Genet. Dev. 2008;18:241–250. doi: 10.1016/j.gde.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 3.Ropers H.H. Genetics of early onset cognitive impairment. Annu. Rev. Genomics Hum. Genet. 2010;11:161–187. doi: 10.1146/annurev-genom-082509-141640. [DOI] [PubMed] [Google Scholar]
- 4.Kitagawa H., Paulson J.C. Cloning and expression of human Gal beta 1,3(4)GlcNAc alpha 2,3-sialyltransferase. Biochem. Biophys. Res. Commun. 1993;194:375–382. doi: 10.1006/bbrc.1993.1830. [DOI] [PubMed] [Google Scholar]
- 5.Varki A., Cummings R., Esko J., Freeze H., Hart G., Marth J.D. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1999. Essentials of Glycobiology. [PubMed] [Google Scholar]
- 6.Haltiwanger R.S., Lowe J.B. Role of glycosylation in development. Annu. Rev. Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- 7.Kleene R., Schachner M. Glycans and neural cell interactions. Nat. Rev. Neurosci. 2004;5:195–208. doi: 10.1038/nrn1349. [DOI] [PubMed] [Google Scholar]
- 8.Grunewald S., Matthijs G., Jaeken J. Congenital disorders of glycosylation: A review. Pediatr. Res. 2002;52:618–624. doi: 10.1203/00006450-200211000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Marquardt T., Denecke J. Congenital disorders of glycosylation: Review of their molecular bases, clinical presentations and specific therapies. Eur. J. Pediatr. 2003;162:359–379. doi: 10.1007/s00431-002-1136-0. [DOI] [PubMed] [Google Scholar]
- 10.Garshasbi M., Hadavi V., Habibi H., Kahrizi K., Kariminejad R., Behjati F., Tzschach A., Najmabadi H., Ropers H.H., Kuss A.W. A defect in the TUSC3 gene is associated with autosomal recessive mental retardation. Am. J. Hum. Genet. 2008;82:1158–1164. doi: 10.1016/j.ajhg.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molinari F., Foulquier F., Tarpey P.S., Morelle W., Boissel S., Teague J., Edkins S., Futreal P.A., Stratton M.R., Turner G. Oligosaccharyltransferase-subunit mutations in nonsyndromic mental retardation. Am. J. Hum. Genet. 2008;82:1150–1157. doi: 10.1016/j.ajhg.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen M., Varki A. The sialome—far more than the sum of its parts. OMICS. 2010;14:455–464. doi: 10.1089/omi.2009.0148. [DOI] [PubMed] [Google Scholar]
- 13.Schauer R. Sialic acids as regulators of molecular and cellular interactions. Curr. Opin. Struct. Biol. 2009;19:507–514. doi: 10.1016/j.sbi.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Audry M., Jeanneau C., Imberty A., Harduin-Lepers A., Delannoy P., Breton C. Current trends in the structure-activity relationships of sialyltransferases. Glycobiology. 2011;21:716–726. doi: 10.1093/glycob/cwq189. [DOI] [PubMed] [Google Scholar]
- 15.Crocker P.R., Paulson J.C., Varki A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 16.Varki N.M., Strobert E., Dick E.J., Jr., Benirschke K., Varki A. Biomedical differences between human and nonhuman hominids: Potential roles for uniquely human aspects of sialic acid biology. Annu. Rev. Pathol. 2011;6:365–393. doi: 10.1146/annurev-pathol-011110-130315. [DOI] [PubMed] [Google Scholar]
- 17.Varki A. Colloquium paper: Uniquely human evolution of sialic acid genetics and biology. Proc. Natl. Acad. Sci. USA. 2010;107(Suppl 2):8939–8946. doi: 10.1073/pnas.0914634107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinstein J., de Souza-e-Silva U., Paulson J.C. Sialylation of glycoprotein oligosaccharides N-linked to asparagine. Enzymatic characterization of a Gal beta 1 to 3(4)GlcNAc alpha 2 to 3 sialyltransferase and a Gal beta 1 to 4GlcNAc alpha 2 to 6 sialyltransferase from rat liver. J. Biol. Chem. 1982;257:13845–13853. [PubMed] [Google Scholar]
- 19.Kuss A.W., Garshasbi M., Kahrizi K., Tzschach A., Behjati F., Darvish H., Abbasi-Moheb L., Puettmann L., Zecha A., Weissmann R. Autosomal recessive mental retardation: homozygosity mapping identifies 27 single linkage intervals, at least 14 novel loci and several mutation hotspots. Hum. Genet. 2011;129:141–148. doi: 10.1007/s00439-010-0907-3. [DOI] [PubMed] [Google Scholar]
- 20.Najmabadi H., Motazacker M.M., Garshasbi M., Kahrizi K., Tzschach A., Chen W., Behjati F., Hadavi V., Nieh S.E., Abedini S.S. Homozygosity mapping in consanguineous families reveals extreme heterogeneity of non-syndromic autosomal recessive mental retardation and identifies 8 novel gene loci. Hum. Genet. 2007;121:43–48. doi: 10.1007/s00439-006-0292-0. [DOI] [PubMed] [Google Scholar]
- 21.Li R., Yu C., Li Y., Lam T.W., Yiu S.M., Kristiansen K., Wang J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 22.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y., Vinckenbosch N., Tian G., Huerta-Sanchez E., Jiang T., Jiang H., Albrechtsen A., Andersen G., Cao H., Korneliussen T. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 2010;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez-Lopez R., Nicholson R., Gesnel M.C., Matrisian L.M., Breathnach R. Structure-function relationships in the collagenase family member transin. J. Biol. Chem. 1988;263:11892–11899. [PubMed] [Google Scholar]
- 25.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Mühlenhoff M., Eckhardt M., Bethe A., Frosch M., Gerardy-Schahn R. Autocatalytic polysialylation of polysialyltransferase-1. EMBO J. 1996;15:6943–6950. [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrer-Costa C., Gelpí J.L., Zamakola L., Parraga I., de la Cruz X., Orozco M. PMUT: A web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- 28.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 29.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 30.Roth J., Berger E.G. Immunocytochemical localization of galactosyltransferase in HeLa cells: Codistribution with thiamine pyrophosphatase in trans-Golgi cisternae. J. Cell Biol. 1982;93:223–229. doi: 10.1083/jcb.93.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burger P.C., Lötscher M., Streiff M., Kleene R., Kaissling B., Berger E.G. Immunocytochemical localization of alpha2,3(N)-sialyltransferase (ST3Gal III) in cell lines and rat kidney tissue sections: Evidence for golgi and post-golgi localization. Glycobiology. 1998;8:245–257. doi: 10.1093/glycob/8.3.245. [DOI] [PubMed] [Google Scholar]
- 32.Datta A.K., Chammas R., Paulson J.C. Conserved cysteines in the sialyltransferase sialylmotifs form an essential disulfide bond. J. Biol. Chem. 2001;276:15200–15207. doi: 10.1074/jbc.M010542200. [DOI] [PubMed] [Google Scholar]
- 33.Tu L., Banfield D.K. Localization of Golgi-resident glycosyltransferases. Cell. Mol. Life Sci. 2010;67:29–41. doi: 10.1007/s00018-009-0126-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoseki J., Ushioda R., Nagata K. Mechanism and components of endoplasmic reticulum-associated degradation. J. Biochem. 2010;147:19–25. doi: 10.1093/jb/mvp194. [DOI] [PubMed] [Google Scholar]
- 35.Jarosch E., Lenk U., Sommer T. Endoplasmic reticulum-associated protein degradation. Int. Rev. Cytol. 2003;223:39–81. doi: 10.1016/s0074-7696(05)23002-4. [DOI] [PubMed] [Google Scholar]
- 36.Schwarzkopf M., Knobeloch K.P., Rohde E., Hinderlich S., Wiechens N., Lucka L., Horak I., Reutter W., Horstkorte R. Sialylation is essential for early development in mice. Proc. Natl. Acad. Sci. USA. 2002;99:5267–5270. doi: 10.1073/pnas.072066199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reinke S.O., Lehmer G., Hinderlich S., Reutter W. Regulation and pathophysiological implications of UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE) as the key enzyme of sialic acid biosynthesis. Biol. Chem. 2009;390:591–599. doi: 10.1515/BC.2009.073. [DOI] [PubMed] [Google Scholar]
- 38.Kono M., Ohyama Y., Lee Y.C., Hamamoto T., Kojima N., Tsuji S. Mouse beta-galactoside alpha 2,3-sialyltransferases: Comparison of in vitro substrate specificities and tissue specific expression. Glycobiology. 1997;7:469–479. doi: 10.1093/glycob/7.4.469. [DOI] [PubMed] [Google Scholar]
- 39.Kannagi R., Ohmori K., Kimura N. Anti-oligosaccharide antibodies as tools for studying sulfated sialoglycoconjugate ligands for siglecs and selectins. Glycoconj. J. 2009;26:923–928. doi: 10.1007/s10719-008-9122-z. [DOI] [PubMed] [Google Scholar]
- 40.Grahn A., Barkhordar G.S., Larson G. Cloning and sequencing of nineteen transcript isoforms of the human alpha2,3-sialyltransferase gene, ST3Gal III; its genomic organisation and expression in human tissues. Glycoconj. J. 2002;19:197–210. doi: 10.1023/A:1024253808424. [DOI] [PubMed] [Google Scholar]
- 41.Eckhardt M., Mühlenhoff M., Bethe A., Gerardy-Schahn R. Expression cloning of the Golgi CMP-sialic acid transporter. Proc. Natl. Acad. Sci. USA. 1996;93:7572–7576. doi: 10.1073/pnas.93.15.7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roth J. Localization of glycosylation sites in the Golgi apparatus using immunolabeling and cytochemistry. J. Electron Microsc. Tech. 1991;17:121–131. doi: 10.1002/jemt.1060170202. [DOI] [PubMed] [Google Scholar]
- 43.Ellies L.G., Sperandio M., Underhill G.H., Yousif J., Smith M., Priatel J.J., Kansas G.S., Ley K., Marth J.D. Sialyltransferase specificity in selectin ligand formation. Blood. 2002;100:3618–3625. doi: 10.1182/blood-2002-04-1007. [DOI] [PubMed] [Google Scholar]
- 44.Krasnewich D., O'Brien K., Sparks S. Clinical features in adults with congenital disorders of glycosylation type Ia (CDG-Ia) Am. J. Med. Genet. C. Semin. Med. Genet. 2007;145C:302–306. doi: 10.1002/ajmg.c.30143. [DOI] [PubMed] [Google Scholar]
- 45.Guo J.P., Brummet M.E., Myers A.C., Na H.J., Rowland E., Schnaar R.L., Zheng T., Zhu Z., Bochner B.S. Characterization of expression of glycan ligands for Siglec-F in normal mouse lungs. Am. J. Respir. Cell Mol. Biol. 2011;44:238–243. doi: 10.1165/rcmb.2010-0007OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwetz T.A., Norring S.A., Ednie A.R., Bennett E.S. Sialic acids attached to O-glycans modulate voltage-gated potassium channel gating. J. Biol. Chem. 2011;286:4123–4132. doi: 10.1074/jbc.M110.171322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dhamija R., Renaud D.L., Pittock S.J., McKeon A., Lachance D.H., Nickels K.C., Wirrell E.C., Kuntz N.L., King M.D., Lennon V.A. Neuronal voltage-gated potassium channel complex autoimmunity in children. Pediatr. Neurol. 2011;44:275–281. doi: 10.1016/j.pediatrneurol.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 48.Parthasarathi U.D., Harrower T., Tempest M., Hodges J.R., Walsh C., McKenna P.J., Fletcher P.C. Psychiatric presentation of voltage-gated potassium channel antibody-associated encephalopathy. Case report. Br. J. Psychiatry. 2006;189:182–183. doi: 10.1192/bjp.bp.105.012864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitten S., Gupta N., Bloch R.M., Dunham C.K. Voltage-gated potassium channel antibody associated mood disorder without paraneoplastic disease. Biol. Psychiatry. 2011;70:e15–e17. doi: 10.1016/j.biopsych.2011.03.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.