

Abstract
Interaction (nonadditive effects) between genetic variants has been highlighted as an important mechanism underlying phenotypic variation, but the discovery of genetic interactions in humans has proved difficult. In this study, we show that the spectrum of variation in the human genome has been shaped by modifier effects of cis-regulatory variation on the functional impact of putatively deleterious protein-coding variants. We analyzed 1000 Genomes population-scale resequencing data from Europe (CEU [Utah residents with Northern and Western European ancestry from the CEPH collection]) and Africa (YRI [Yoruba in Ibadan, Nigeria]) together with gene expression data from arrays and RNA sequencing for the same samples. We observed an underrepresentation of derived putatively functional coding variation on the more highly expressed regulatory haplotype, which suggests stronger purifying selection against deleterious coding variants that have increased penetrance because of their regulatory background. Furthermore, the frequency spectrum and impact size distribution of common regulatory polymorphisms (eQTLs) appear to be shaped in order to minimize the selective disadvantage of having deleterious coding mutations on the more highly expressed haplotype. Interestingly, eQTLs explaining common disease GWAS signals showed an enrichment of putative epistatic effects, suggesting that some disease associations might arise from interactions increasing the penetrance of rare coding variants. In conclusion, our results indicate that regulatory and coding variants often modify the functional impact of each other. This specific type of genetic interaction is detectable from sequencing data in a genome-wide manner, and characterizing these joint effects might help us understand functional mechanisms behind genetic associations to human phenotypes—including both Mendelian and common disease.
Main Text
Genetic variants can have joint, nonadditive functional effects,1–4 but characterizing such epistasis between common variants in humans has proven difficult, and undetected epistasis remains one potential reason for the low proportion of heritability of complex traits explained by common genetic variants.5–10 In addition to gene-gene interactions, linked loci can also have epistatic effects,11–14 one putative mechanism being the interaction between regulatory and coding variants of the same gene.15,16 Potential targets of these interactions are abundant: cis-regulatory variation is common17–19 and has been estimated to affect at least 20% of protein-coding variants within an individual in only a single tissue.16,20 In this study, we have analyzed population genetic signatures of a specific but probably abundant type of epistasis: common cis-regulatory variation modifying the penetrance of rare putatively deleterious coding variants. Our results indicate that such interactions are common and likely contribute to genetic predisposition to complex disease.
Regulatory variation in cis might affect the penetrance of a deleterious coding variant of the same gene through allelic imbalance: In individuals who are heterozygous for both a regulatory and a coding single nucleotide variant (rSNV and cSNV, respectively), the deleterious coding allele might have a much more severe phenotypic outcome if it is more highly expressed than the other allele. This might make the coding heterozygote functionally close to a deleterious homozygote (Figure 1A). In such a situation, regulatory variation modifies the functional impact—and selection coefficient—of a deleterious coding variant, which might have a secondary effect also on the selection coefficient of the regulatory variant even when the change of gene expression level itself does not affect fitness. Importantly, whether or not a rare deleterious cSNV allele resides on the more highly or less expressed haplotype in a gene with cis-regulatory variation is not fully random: the probability that a new coding mutation lands on a particular haplotype is equal to the haplotype frequency. Altogether, these phenomena can shape the patterns of both regulatory and coding variation, and in this paper we show that these specific patterns are common in the human genome.
Figure 1.
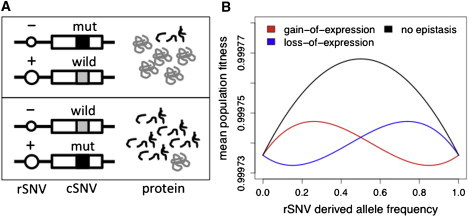
Model of Epistasis between Regulatory and Coding Variation
Within a gene, the functional effect of a coding variant wild-type (cSNVw) and mutant (cSNVm) alleles can be dramatically altered by linkage to a more highly or less expressed allele of a regulatory variant (rSNV+ and rSNV−) (A). Combining this epistatic effect with the probability that the cSNVm is on each rSNV haplotype gives us a model of epistasis in which the average population fitness varies as a function of rSNV frequency (B). In this model, epistatic selection alters the double-heterozygote fitnesses: w[cSNVwrSNV+/cSNVmrSNV−] = 1 − (1 − i)hs, and w[cSNVwrSNV−/cSNVmrSNV+] = 1 − [i + (1 − i)h]s, where i denotes the magnitude of allelic imbalance, s is the selection coefficient, and h is the dominance of the m allele. Allele frequencies are based on Hardy-Weinberg equilibrium and additional new mutations cSNVw → cSNVm hitting the rSNV+ and rSNV− haplotypes with a probability of their frequency in rate μ. Here, we have used parameters μ = 10−4, s = 0.8, h = 0.4, and i = 0.9 or i = 0. See Table S1 for details.
We analyzed genetic variation discovered in the low-coverage resequencing data of the 1000 Genomes Project pilot 1 and 2 (release March 2010), from 60 samples of European origin (CEU [Utah residents with ancestry from northern and western Europe]) and 58 Yoruba individuals from Nigeria (YRI [Yoruba in Ibadan, Nigeria]).21 The study was approved by the institutional review boards of the Coriell Institute for Medical Research and the University Hospitals of Geneva. To analyze common regulatory variation, we mapped expression quantitative trait loci (eQTLs) in cis by Spearman rank correlation by using gene expression array data from transformed lymphoblastoid cell lines of 57 CEU and 56 YRI individuals and SNPs with MAF > 5% and less than 1 Mb from transcription start site, by using a permutation threshold of 0.0120. This yielded a total of 433 eQTLs with ancestral allele information (provided by the 1000 Genomes Consortium) in CEU and 446 in YRI (false discovery rate 25%). We designate the more highly and less expressed rSNV alleles rSNV+ and rSNV−, respectively, and classify the eQTLs in gain-of-expression (GOE) and loss-of-expression (LOE) variants according to the effect of the derived allele. Instead of testing all variants against all across the genome for statistical epistasis, we analyzed our data for specific patterns of variation predicted by our model of local epistasis.
The model of epistasis predicts increased penetrance of deleterious cSNVs when the derived cSNV allele is on the more highly expressed haplotype in cSNV-rSNV double heterozygotes. These cases are most likely to arise when the rSNV has high heterozygosity, and novel putatively deleterious coding mutations hit the rSNV+ allele—that is, in common rSNVs with a high rSNV+ allele frequency. These rSNVs might be under increased purifying selection (Figure 1B). We observe a signal consistent with this in the frequency distribution of eQTLs: GOE eQTLs had significantly lower derived allele frequencies (DAF) than LOE eQTLs (Figure 2; DAFGOE versus DAFLOE Mann-Whitney p = 0.0092 in CEU and p = 0.026 in YRI), that is, the rSNV+ alleles tend to have lower frequencies among common regulatory variants, consistently with epistatic selection. An alternative explanation to this pattern would be increased gene-expression levels being more deleterious in general but then the proportion of GOE rSNVs should grow exponentially toward lower rSNV frequencies. Because eQTL analysis does not capture rare regulatory variants, we investigated whether such a pattern can be observed by analyzing allele-specific expression (ASE) from RNA sequencing data of 60 CEU individuals.22 By using the frequency of the coding variant with rare ASE to predict which cSNV allele is linked to the derived allele of the unknown putative rare rSNV (Figure S1, available online), we estimated that 78 ± 12% (linear regression p = 2.1 × 10−10) of rare rSNVs are loss-of-expression variants (Figure S2). Altogether, whereas common regulatory variants with DAF 5%–50% are predominantly GOE, rare rSNVs—as well as common variants of DAF > 50%—appear to be usually LOE. This is inconsistent with the gain of expression being more deleterious in general and follows the predictions of the epistasis model (Figure 1B).
Figure 2.
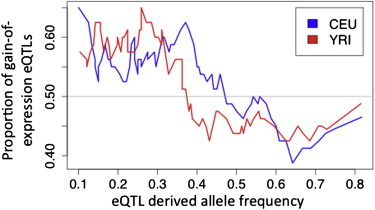
Frequency Distribution of eQTLs
The proportion of gain-of-expression eQTLs with respect to derived allele frequency in sliding windows of 80 SNPs with an overlap of five SNPs; this shows that the more highly expressed alleles tend to have low frequencies (p = 0.0092 in CEU and p = 0.026 in YRI).
Furthermore, we analyzed the distribution of fold change of eQTLs (calculated as the ratio of the median expression values of the major homozygote and the heterozygote eQTL genotype classes), which describes the magnitude of putative cSNV allelic imbalance and epistasis effect in eQTL heterozygotes. We observed that especially in CEU, common eQTLs with high frequency of the rSNV− allele tend to have higher fold changes (Figure S3). These eQTLs are likely to have most coding mutations occurring on the rSNV− haplotype and thus benefit from epistasis: the bigger the allelic imbalance is, the lower the penetrance of these cSNVs. Conversely, strong epistatic effects in eQTLs with high rSNV+ frequencies are more likely to be disadvantageous, which is consistent with their low fold changes. Thus, epistatic effects appear to shape not only the frequency spectrum of regulatory variants but also the distribution of the magnitude of their effect.
Patterns of coding variation are also expected to be affected by epistasis. Increased purification of deleterious cSNV alleles from the more highly expressed haplotype might lead to different distribution of putatively deleterious cSNVs and neutral cSNVs on the regulatory haplotypes. To investigate this, we compared nonsynonymous and synonymous SNVs (nsSNVs and sSNVs, respectively), expecting the former to show stronger signs of interaction with rSNVs. First, epistasis would favor some haplotype combinations over others, which is expected to increase the overall linkage disequilibrium (LD). Indeed, LD between eQTLs and nsSNVs was stronger than for sSNVs (Figures 3A and 3B and Figure S6; Mann-Whitney p values for CEU and YRI are 0.012 and 0.008 for D′, and 0.002 and 0.078, for r2, respectively). This is consistent with our previous results based on RNA sequencing data from 60 CEU individuals.20 Second, we investigated whether this LD pattern is likely caused by underrepresentation of putatively deleterious coding alleles on the more highly expressed haplotype. Analysis of allele-specific expression (ASE) data from the same RNA sequencing dataset22 showed that a larger proportion of nsSNVs than sSNVs showed decreased expression of the derived allele (Figure 3C; p = 0.0035 for overall sSNV-nsSNV difference according to a linear regression model, and Fisher's exact test p = 0.046 for cSNVs with DAF < 0.15, see also Figures S1 and S2). This analysis is unlikely to be biased because of nsSNVs being putatively more often causal regulatory variants themselves because such an autoregulatory mechanism would be unlikely to cause allelic imbalance, and we have observed that ASE occurrence in sSNVs and nsSNVs overall is almost equal.20 Furthermore, in haplotype-phased eQTL data (from the 1000 Genomes July 2010 release) in CEU, the more highly expressed haplotype carried significantly fewer derived alleles of nsSNVs than of sSNVs (Fisher's exact test p = 2 × 10−4, Table S2). These results suggest that epistatic selection leads to deficiency of deleterious coding variation on the more highly expressed regulatory haplotype. Additionally, we investigated whether epistasis can also affect the total number of coding variants in genes with regulatory variation, and we observed that the number of cSNVs was decreased in eQTL genes (Mann-Whitney p < 2.2 × 10−16 in CEU, p = 6.4 × 10−3 in YRI; Figure S3) and, importantly, was decreased even more in CEU when the rSNV+ allele was common; this suggests that epistasis might expose coding variants to selection and lead to increased purifying selection.
Figure 3.
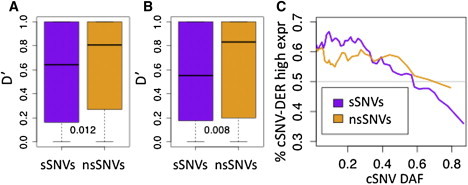
Signals of Epistasis in Coding Variation
Linkage disequilibrium (D′) between eQTLs and sSNVs or nsSNVs in CEU (A) and YRI (B). The sSNVs were sampled to the derived allele frequency distribution of nsSNVs, and the numbers denote the p values for sSNV − nsSNV comparisons. In (C), allele-specific expression data from CEU was used to analyze how frequently the derived allele of a coding variant (cSNV − DER) is more highly expressed; the plot shows medians in sliding window of 400 SNPs with an overlap of 50. The difference between sSNVs and nsSNVs variants (p = 0.0035) suggests selection against increased expression of the putatively deleterious derived allele of low-frequency nsSNVs. The overall decreasing trend is likely due to putative regulatory variants underlying the ASE effect being more often loss-of-expression variants (see Figures S1 and S2).
Finally, we asked whether epistatic effects might play a role in genetic predisposition to complex disease, with an enrichment of deleterious alleles on the more highly expressed haplotype potentially increasing disease risk. To this end, we used the Regulatory Trait Concordance score23 to define 98 disease-associated eQTLs in which the eQTL is likely to tag the same variant as a GWAS SNP (from NHGRI catalog24 accessed April 12, 2010) and compared them to 934 control eQTLs. This analysis was based on a dataset of 75 European individuals with genotypes imputed to HapMap2 and array expression data from fibroblasts, T cells, and LCLs.25 Figure 4A shows an enrichment of disease eQTLs in high frequencies of the rSNV+ allele, when random coding mutations are more likely to hit this haplotype and possibly have increased penetrance. This trend is opposite to that observed for eQTLs overall—suggesting that the variants not following the general pattern putatively optimized by evolution are more likely to contribute to disease. Furthermore, the disease risk allele is more often the eQTL+ allele or linked to it (21/30 cases with available data, χ2 test p = 0.023, Figure 4B)—although this pattern alone (but not the frequency pattern in Figure 4A) could be caused by the increased expression level itself being detrimental. Altogether, these results suggest that a proportion of disease associations due to regulatory variants might arise from the dysfunction of the rSNV+ allele because it increases the penetrance of linked cSNVs. However, we did not observe significant differences in the patterns of coding variation in the 60 CEU individuals between genes with disease-associated and control eQTLs (Figure S6). Future studies with case-control material will, we hope, clarify whether increased disease risk sometimes arises neither from changed gene-expression levels alone nor from an enrichment of rare cSNVs per se but from their interaction. Table S3 gives a list of disease-associated eQTLs with a high frequency of the more highly expressed haplotype; these would be the best candidates for searching for the epistatic effect.
Figure 4.
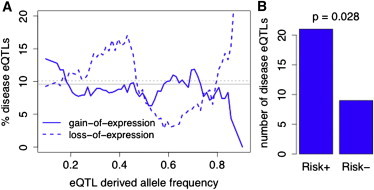
Properties of Disease-Associated eQTLs
Percentage of disease-associated eQTLs of control eQTLs with respect to derived allele frequency in sliding windows of 0.05 (A) suggests an enrichment of disease-associated eQTLs in the parts of the frequency spectrum in which epistasis might increase the penetrance of rare coding variants. The vertical lines denote the average percentages. (B) The eQTL alleles linked to the disease risk allele are shown; + and − denoting the more highly or less expressed alleles.
In conclusion, our study illustrates how epistasis between coding and cis-regulatory variants has shaped the spectrum of genetic variation in the human genome. The straightforward principle of interaction recapitulates many of the phenomena observed in the data and opens ground for future research of the role of epistasis in, for example, tissue specificity25 and genetic associations to complex disease.6,7,11 In future studies, more refined models characterizing the population genetic dynamics of epistasis will, we hope, shed light on differences between populations, genetic load caused by epistasis, and evolutionary equilibria.26 Additionally, this type of epistasis might contribute to varying penetrance of Mendelian disorders,27 in which the penetrance of a rare disease-causing allele could be modified by the individual's genotype of a common regulatory variant of that gene. In this study we focused on interactions between common rSNVs and rare cSNVs, but the accumulating genomic and RNA sequencing data will enable analysis of modifying effects that rare regulatory variation might have on both rare and common coding variation.
Altogether, our results show that the functional effects of regulatory variation often extend beyond gene-expression levels and that the impact of rare coding variants is frequently modified by regulatory variation. This might have important practical implications for understanding functional effects of genetic variants—and this specific type of genetic interaction can be relatively easily detected from sequencing data in a genome-wide manner, as outlined in this study. Phenotypic associations to regulatory variants have only rarely led to characterization of expression differences underlying the phenotype,18 and our results suggest that a proportion of these signals might actually be driven not by expression change itself but by increased penetrance of deleterious coding variants. Additionally, considerable effort is being directed to the discovery of loss-of-function coding variants from genome or exome sequencing data. Thus far, these data have rarely been complemented with RNA sequencing data from relevant tissues to understand how the predicted functional effect is actually manifested in the downstream pathways of the cell and in the phenotype. In the future, the integrated analysis of regulatory and coding variants will be important in characterizing the genetic sources of phenotypic variation in humans.
Acknowledgments
The funding for this study was provided by Louis Jeantet Foundation, Swiss National Science Foundation and National Centers of Competence in Research Frontiers in Genetics (Swiss National Science Foundation) to E.T.D. T.L. is funded by the Academy of Finland and the Emil Aaltonen foundation. We would like to thank Vital-IT.ch for managing computer resources, and Alfonso Buil and Eugenia Migliavacca for assistance with the analyses.
Contributor Information
Tuuli Lappalainen, Email: tuuli.lappalainen@unige.ch.
Emmanouil T. Dermitzakis, Email: emmanouil.dermitzakis@unige.ch.
Supplemental Data
References
- 1.Phillips P.C. Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 2008;9:855–867. doi: 10.1038/nrg2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carlborg O., Jacobsson L., Ahgren P., Siegel P., Andersson L. Epistasis and the release of genetic variation during long-term selection. Nat. Genet. 2006;38:418–420. doi: 10.1038/ng1761. [DOI] [PubMed] [Google Scholar]
- 3.Zhu J., Zhang B., Smith E.N., Drees B., Brem R.B., Kruglyak L., Bumgarner R.E., Schadt E.E. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 2008;40:854–861. doi: 10.1038/ng.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehner B., Crombie C., Tischler J., Fortunato A., Fraser A.G. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat. Genet. 2006;38:896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- 5.Moore J.H., Williams S.M. Epistasis and its implications for personal genetics. Am. J. Hum. Genet. 2009;85:309–320. doi: 10.1016/j.ajhg.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eichler E.E., Flint J., Gibson G., Kong A., Leal S.M., Moore J.H., Nadeau J.H. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clayton D.G. Prediction and interaction in complex disease genetics: experience in type 1 diabetes. PLoS Genet. 2009;5:e1000540. doi: 10.1371/journal.pgen.1000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cordell H.J. Detecting gene-gene interactions that underlie human diseases. Nat. Rev. Genet. 2009;10:392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marchini J., Donnelly P., Cardon L.R. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat. Genet. 2005;37:413–417. doi: 10.1038/ng1537. [DOI] [PubMed] [Google Scholar]
- 11.Haig D. Does heritability hide in epistasis between linked SNPs? Eur. J. Hum. Genet. 2011;19:123. doi: 10.1038/ejhg.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gregersen J.W., Kranc K.R., Ke X., Svendsen P., Madsen L.S., Thomsen A.R., Cardon L.R., Bell J.I., Fugger L. Functional epistasis on a common MHC haplotype associated with multiple sclerosis. Nature. 2006;443:574–577. doi: 10.1038/nature05133. [DOI] [PubMed] [Google Scholar]
- 13.Bickel R.D., Kopp A., Nuzhdin S.V. Composite effects of polymorphisms near multiple regulatory elements create a major-effect QTL. PLoS Genet. 2011;7:e1001275. doi: 10.1371/journal.pgen.1001275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stam L.F., Laurie C.C. Molecular dissection of a major gene effect on a quantitative trait: the level of alcohol dehydrogenase expression in Drosophila melanogaster. Genetics. 1996;144:1559–1564. doi: 10.1093/genetics/144.4.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emison E.S., Garcia-Barcelo M., Grice E.A., Lantieri F., Amiel J., Burzynski G., Fernandez R.M., Hao L., Kashuk C., West K. Differential contributions of rare and common, coding and noncoding Ret mutations to multifactorial Hirschsprung disease liability. Am. J. Hum. Genet. 2010;87:60–74. doi: 10.1016/j.ajhg.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimas A.S., Stranger B.E., Beazley C., Finn R.D., Ingle C.E., Forrest M.S., Ritchie M.E., Deloukas P., Tavaré S., Dermitzakis E.T. Modifier effects between regulatory and protein-coding variation. PLoS Genet. 2008;4:e1000244. doi: 10.1371/journal.pgen.1000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheung V.G., Spielman R.S. Genetics of human gene expression: mapping DNA variants that influence gene expression. Nat. Rev. Genet. 2009;10:595–604. doi: 10.1038/nrg2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Montgomery S.B., Dermitzakis E.T. From expression QTLs to personalized transcriptomics. Nat. Rev. Genet. 2011;12:277–282. doi: 10.1038/nrg2969. [DOI] [PubMed] [Google Scholar]
- 19.Majewski J., Pastinen T. The study of eQTL variations by RNA-seq: from SNPs to phenotypes. Trends Genet. 2011;27:72–79. doi: 10.1016/j.tig.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Montgomery S.B., Lappalainen T., Gutierrez-Arcelus M., Dermitzakis E.T. Rare and common regulatory variation in population-scale sequenced human genomes. PLoS Genet. 2011;7:e1002144. doi: 10.1371/journal.pgen.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durbin R.M., Abecasis G.R., Altshuler D.L., Auton A., Brooks L.D., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montgomery S.B., Sammeth M., Gutierrez-Arcelus M., Lach R.P., Ingle C., Nisbett J., Guigo R., Dermitzakis E.T. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nica A.C., Montgomery S.B., Dimas A.S., Stranger B.E., Beazley C., Barroso I., Dermitzakis E.T. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet. 2010;6:e1000895. doi: 10.1371/journal.pgen.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hindorff, L.A., Junkins, H.A., Hall, P.N., Mehta, J.P., and Manolio, T.A. (2010). A Catalog of Published Genome-Wide Association Studies. www.genome.gov/gwastudies.
- 25.Dimas A.S., Deutsch S., Stranger B.E., Montgomery S.B., Borel C., Attar-Cohen H., Ingle C., Beazley C., Gutierrez Arcelus M., Sekowska M. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinreich D.M., Watson R.A., Chao L. Perspective: Sign epistasis and genetic constraint on evolutionary trajectories. Evolution. 2005;59:1165–1174. [PubMed] [Google Scholar]
- 27.Van Heyningen V., Yeyati P.L. Mechanisms of non-Mendelian inheritance in genetic disease. Hum. Mol. Genet. 2004;13(Spec No 2):R225–R233. doi: 10.1093/hmg/ddh254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.