

Abstract
Smad proteins are critical signal transducers downstream of the receptors of the transforming growth factor-β (TGFβ) superfamily. On phosphorylation and activation by the active TGFβ receptor complex, Smad2 and Smad3 form hetero-oligomers with Smad4 and translocate into the nucleus, where they interact with different cellular partners, bind to DNA, regulate transcription of various downstream response genes, and cross-talk with other signaling pathways. Here we show that a nuclear oncoprotein, Ski, can interact directly with Smad2, Smad3, and Smad4 on a TGFβ-responsive promoter element and repress their abilities to activate transcription through recruitment of the nuclear transcriptional corepressor N-CoR and possibly its associated histone deacetylase complex. Overexpression of Ski in a TGFβ-responsive cell line renders it resistant to TGFβ-induced growth inhibition and defective in activation of JunB expression. This ability to overcome TGFβ-induced growth arrest may be responsible for the transforming activity of Ski in human and avian cancer cells. Our studies suggest a new paradigm for inactivation of the Smad proteins by an oncoprotein through transcriptional repression.
Keywords: Ski, TGFβ, Smad proteins, growth inhibition, signal transduction, transcriptional activation
Transforming growth factor-β (TGFβ) is a multipotent cytokine that elicits many biological functions including inhibition of the growth of cells of epithelial, endothelial, and lymphoid origins; production of extracellular matrix components; and regulation of differentiation of many cell types (Roberts and Sporn 1990). Binding of TGFβ1 to the cell-surface type II TGFβ receptor (TβRII) results in the formation of a heteromeric complex containing the type I TGFβ receptor (TβRI) and TβRII, followed by the phosphorylation and activation of TβRI by the TβRII kinase (Heldin et al. 1997; Massague 1998). The activated TβRI then interacts with an adapter molecule SARA (Smad anchor for receptor activation; Tsukazaki et al. 1998), which recruits downstream Smad2 and Smad3 proteins to be phosphorylated by TβRI (Heldin et al. 1997; Massagué 1998).
The Smad family proteins are critical components of the TGF-β signaling pathways (Heldin et al. 1997; Massagué 1998). All Smad proteins share considerable homology in their primary sequences and most of them contain two highly conserved Mad homology domains:—Mad homologous domain 1 (MH1) in the amino-terminal half and MH2 in the carboxy-terminal half—and a diverse linker in between the two. The MH1 domains of Smad3 and Smad4 contain sequence-specific DNA-binding activity (Yingling et al. 1997; Dennler et al. 1998; Jonk et al. 1998; Shi et al. 1998; Song et al. 1998; Zawel et al. 1998; Stroschein et al. 1999), and overexpression of these MH1 and linker domains in Hep3B hepatoma cells results in trancriptional activation of a plasminogen activator inhibitor-1 (PAI-1) promoter construct (Stroschein et al. 1999). The MH2 domain, when fused to the GAL4 DNA-binding domain, can activate transcription, suggesting that it may contain transactivation activity (Liu et al. 1996). This domain also mediates homo- and hetero-oligomerization of the Smad proteins (Heldin et al. 1997; Shi et al. 1997; Massagué 1998). The MH1 and MH2 domains of Smad2 and Smad4 can interact in an intramolecular manner and block the function of each other (Hata et al. 1997). The linker regions of Smad2 and Smad3 contain serine residues that can be phosphorylated by the mitogen-activated protein (MAP) kinase and are involved in cross-talk between different signaling pathways (Kretzschmar et al. 1997, 1999).
On stimulation by TGFβ1, the pathway-restricted Smads, Smad2 and Smad3, interact with the TGFβ receptor complex and become phosphorylated on serine residues located at the carboxyl termini of the molecules (Heldin et al. 1997; Massagué 1998). Phosphorylated Smad2 and Smad3 then form heteromeric complexes with the common mediator Smad4 and translocate into the nucleus where they can bind to the TGFβ-responsive promoter DNA either directly through the Smad-binding elements (SBEs) (Yingling et al. 1997; Dennler et al. 1998; Jonk et al. 1998; Shi et al. 1998; Song et al. 1998; Zawel et al. 1998; Stroschein et al. 1999) or in conjunction with other sequence-specific DNA-binding proteins such as FAST1 and FAST2 (Chen et al. 1996, 1997; Labbe et al. 1998; Zhou et al. 1998b). Smad proteins may form complexes with general transcriptional activators, such as p300/CBP (Feng et al. 1998; Janknecht et al. 1998; Pouponnot et al. 1998; Shen et al. 1998; Topper et al. 1998) and AP-1 (Zhang et al. 1998; Wong et al. 1999), or with a transcriptional corepressor TGIF (Wotton et al. 1999) to regulate TGFβ signaling. Smad3 has also been shown to synergize with transcriptional factors Sp1 (Moustakas and Kardassis 1998), TFE3 (Hua et al. 1998), or NFκB (Kon et al. 1999) to activate transcription from specific promoters. However, many of these interactions are promoter specific and none have been demonstrated to affect TGFβ-induced growth inhibition. Because all three Smad proteins are important tumor suppressors and loss-of-function mutations in Smad2 and Smad4 have been found to associate with many types of human cancers (Massagué 1998), additional cellular proteins may interact with the Smads to modulate their ability to regulate cell growth.
Here we report that Smad2, Smad3, and Smad4 associated with a nuclear oncoprotein, Ski, through their MH2 domains. Ski was first identified as a viral oncogene (v-ski) from the avian Sloan-Kettering retrovirus (SKV) that transforms chicken embryo fibroblasts (Li et al. 1986). The human cellular homolog c-ski was later cloned based on its homology with v-ski and was found to encode a nuclear protein of 728 amino acids (Nomura et al. 1989; Sutrave et al. 1990a). Compared with c-Ski, v-Ski is truncated mostly at the carboxyl terminus (Stavnezer et al. 1989; Sutrave and Hughes 1989). However, this truncation is not responsible for the activation of ski as an oncogene. Overexpression of wild-type c-Ski also results in oncogenic transformation of chicken and quail embryo fibroblasts (Colmenares and Stavnezer 1989; Colmenares et al. 1991). The transforming activity of Ski is likely attributable to overexpression, not truncation, of the c-Ski protein. Consistent with this notion, an elevated level of c-Ski was also detected in several human tumor cell lines derived from neuroblastoma, melanoma, and prostate cancer (Nomura et al. 1989; Fumagalli et al. 1993). c-ski is a unique oncogene in that, in addition to affecting cell growth, it is also involved in regulation of muscle differentiation. Overexpression of Ski resulted in muscle differentiation of quail embryo cells (Colmenares and Stavnezer 1989) and hypertrophy of skeletal muscle in mice (Sutrave et al. 1990b). Furthermore, mice lacking c-ski displayed defective muscle and neuronal differentiation (Berk et al. 1997). Little is known about the pathways that Ski modulates to carry out these diverse functions.
At the molecular level, Ski can function either as a transcriptional activator (Engert et al. 1995; Tarapore et al. 1997) or as a repressor (Nicol and Stavnezer 1998) depending on the specific promoters involved. It has been shown to bind to DNA, but only in conjunction with other yet-to-be-identified cellular proteins (Nagase et al. 1990; Nicol and Stavnezer 1998). Recently, Ski was found to be a component of the histone deacetylase (HDAC1) complex through binding to the nuclear hormone receptor corepressor (N-CoR) and mSin3A, and mediated transcriptional repression of the thyroid hormone receptor, Mad and pRb (Nomura et al. 1999; Tokitou et al. 1999). The interaction between Ski and N-CoR is mediated by the amino-terminal part of Ski (Nomura et al. 1999). This region is also essential for the transforming activity of c-Ski (Zheng et al. 1997) and is conserved among ski family members including v-Ski and c-SnoN (Nomura et al. 1989; Pearson-White 1993). This raises an interesting possibility that the transforming activity of Ski may be linked to its function as a transcriptional corepressor. Despite these observations, the molecular mechanism by which Ski transforms cells and regulates differentiation remains largely unknown. In particular, it is not clear how overexpression of Ski interferes with the growth-regulatory signaling pathways and what the cellular targets of Ski are. Using an affinity purification approach, we have found that Ski interacts with the Smad proteins in vivo and blocks the ability of the Smads to mediate TGFβ-induced growth arrest and transcriptional activation. Furthermore, cells overexpressing Ski became resistant to TGFβ-induced growth inhibition. The identification of Ski as a Smad-associating protein may provide insight into the mechanism for the transforming activity of Ski as well as a mechanism involved in regulation of Smad function.
Results
Smad2, Smad3, and Smad4 interact with Ski through the MH2 domains
To identify Smad4-associated cellular proteins, we stably introduced the Flag-tagged Smad4 amino-terminal domain (Smad4NL, containing the MH1 domain and the linker region) or carboxy-terminal MH2 domain (Fig. 1A) into 293T cells and examined cellular proteins that copurified with Smad4. Cell lysates were prepared from these cell lines and applied to an anti-Flag antibody column. After extensive washes, Smad4 and its associated proteins were eluted with the Flag peptide and visualized by silver staining (Fig. 1A). Two major proteins with apparent molecular masses of 97 and 80 kD were found to associate predominantly with the MH2 domain of Smad4 (lane 2), weakly with the full-length protein (data not shown), but not with the MH1 and linker domains of Smad4 (lane 3). These two proteins were also found to associate with the Smad4 MH2 domain in transfected TGFβ-responsive Hep3B cells (data not shown). Approximately 2 μg of each of the two proteins were affinity purified and sequenced. Amino acid sequences of a total of five peptides derived from the 80-kD protein showed a perfect match to human c-SnoN (Nomura et al. 1989; Pearson-White 1993). Similarly, the 97-kD protein was identified as the human c-Ski protein (Nomura et al. 1989). c-sno and c-ski are two closely related members of the ski family of proto-oncogenes (Nomura et al. 1989; Pearson-White 1993). They share 69% sequence identity in the amino-terminal part of the molecules (Nomura et al. 1989). The relative amounts of Ski and SnoN associated with the Smad4C (Fig. 1A, lane 2) reflected cellular levels of these two proteins (data not shown). In this report, we focused on the interaction between the Smads and Ski. The functional interaction between Smad4 and c-SnoN will be described elsewhere.
Figure 1.
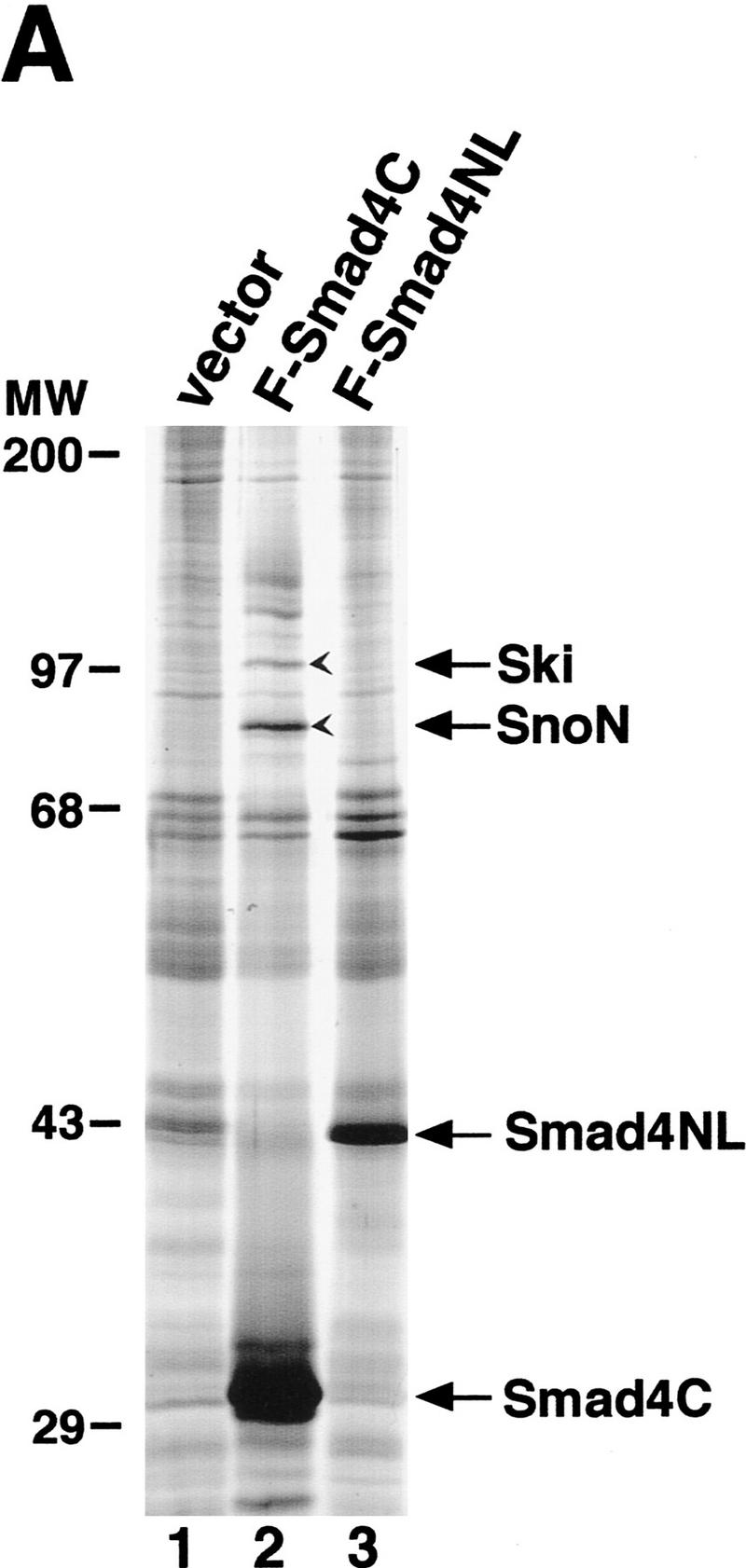

Ski interacts with the MH2 domains of Smad2, Smad3, and Smad4. (A) Cell lysates were prepared from control 293T cells or 293T cells stably expressing Flag–Smad4C or Flag–Smad4N, and Smad4-associated cellular proteins were isolated by affinity purification using anti-Flag agarose followed by elution with the Flag peptide as described in Materials and Methods. The purified Smad4 complexes were visualized by silver staining. The 97-kD protein indicated by an arrow was identified by microsequencing as the human c-Ski protein and the 80-kD protein as the human c-SnoN. (B) Flag-tagged full-length or truncated Smad proteins were cotransfected into 293T cells together with HA-Ski as indicated and isolated by immunoprecipitation with an anti-Flag M2 mAb. The Smad-bound Ski was visualized by Western blotting with an anti-HA mAb as indicated. Cell lysates were blotted directly with an anti-HA mAb as a control for HA–Ski expression (bottom).
To investigate whether Ski also interacts with Smad2 and Smad3 in addition to Smad4, we performed coimmunoprecipitation experiments in lysates of transfected 293T cells. Flag-tagged full-length or truncated Smad2, Smad3, or Smad4 was cotransfected into 293T cells together with HA-tagged Ski and isolated by immunoprecipitation with anti-Flag agarose (Fig. 1B). HA–Ski was found to associate with the full-length and the MH2 domains of Smad2, Smad3, and Smad4, but not with the MH1 domains of these molecules. Interactions of endogenous Ski with Smad2, Smad3, and Smad4 were also detected in cell extracts prepared from human epidermoid carcinoma A431 cells (data not shown), which have been shown to express high levels of Ski (Nomura et al. 1989). The interaction between Ski and Smad2 and Smad3 in TGFβ-responsive cells is dependent on TGFβ treatment (see Fig. 6B, below).
Figure 6.

Overexpression of Ski greatly attenuates TGFβ-induced growth inhibition and activation of JunB expression. (A) Ba/F3 cell lines stably expressing Flag–Ski were generated by retroviral infection. Flag–Ski was isolated by immunoprecipitation with anti-Flag agarose from uninfected Ba/F3 cells or from two stable pools (B/ski-8 and B/ski-12) and analyzed by Western blotting with an anti-Flag mAb. (B) B/ski-8 cells (2 × 108) were stimulated with 200 pm TGFβ1 for 15 or 30 min as indicated. Endogenous Smad proteins associated with Flag–Ski were isolated by immunoprecipitation with anti-Flag agarose and detected by Western blotting with anti-Smad2, anti-Smad3, or anti-Smad4 antibodies. (C) Nuclear extracts were prepared from B/ski-8 cells that had been stimulated with 200 pm TGFβ for 30 min and incubated with 32P-labeled SBE in an EMSA assay. Antibodies used in the supershift assay: (S2/3) anti-Smad2/3; (S4) anti-Smad4; (F) anti-Flag; (NR) nonrelevant control antibody. (*) Non-specific background bands. (D) Growth inhibition assay. Uninfected Ba/F3 cells, B/ski-8 or B/ski-12 cells were incubated for 5 days with various concentrations of TGFβ1 as indicated. The growth of cells was quantified by cell counting and compared to unstimulated cells. The growth rate of B/ski-8 or B/ski-12 cells in the absence of TGFβ1 is similar to that of uninfected Ba/F3 cells. (E) Activation of JunB expression in uninfected Ba/F3 or B/ski-8 cells by Northern blotting. Cells were serum starved for 16 hr and stimulated with 100 pm TGFβ1 for various periods of time as indicated. An analysis of the JunB and human CAC1 RNA is shown. CAC1 was used as a control for equal loading. A quantification of the Northern blot was carried out using the Bio-Rad Molecular Imager FX system and folds of induction of JunB expression are shown in the graph.
Ski binds to SBE in conjunction with Smad3 and Smad4
Ski in nuclear extracts prepared from c-Ski-transformed cells was shown previously to interact with condensed chromatin and recognize a specific DNA element (GTCTAGAC) (Nicol and Stavnezer 1998). However, purified recombinant Ski protein failed to bind DNA directly, suggesting that Ski must bind to this DNA sequence through interaction with yet-to-be-identified DNA-binding partners (Nagase et al. 1990; Nicol and Stavnezer 1998). A close examination of the Ski-binding DNA element revealed a perfect match to SBE (Zawel et al. 1998). Because Smad3 and Smad4 bind directly to SBE (Shi et al. 1998; Zawel et al. 1998), an intriguing possibility is that Smad proteins may be the DNA-binding partners of Ski. To test this hypothesis, a Ski/Smad4 complex was purified from 293T cells transfected with Flag–Ski together with HA–Smad4 and incubated with the 32P-labeled SBE oligonucleotide in an electrophoretic mobility shift assay (EMSA) (Fig. 2). Although Ski alone (GST–Ski or Flag–Ski) did not bind DNA (Fig. 2, lanes 2,3), complexes of Ski with Smad4 resulted in a mobility shift of the SBE oligonucleotide (Fig. 2, lane 4). Antibody supershift experiments demonstrated that the observed DNA–protein complex contained Ski as well as Smad4 (Fig. 2, lanes 6,7). Similarly, Ski/Smad3 complex isolated from the transfected 293T cells also bound the SBE oligo-nucleotide (data not shown). Thus, Ski can interact with Smad proteins on SBE, and Smad proteins may be the long sought-after DNA-binding partners of Ski.
Figure 2.
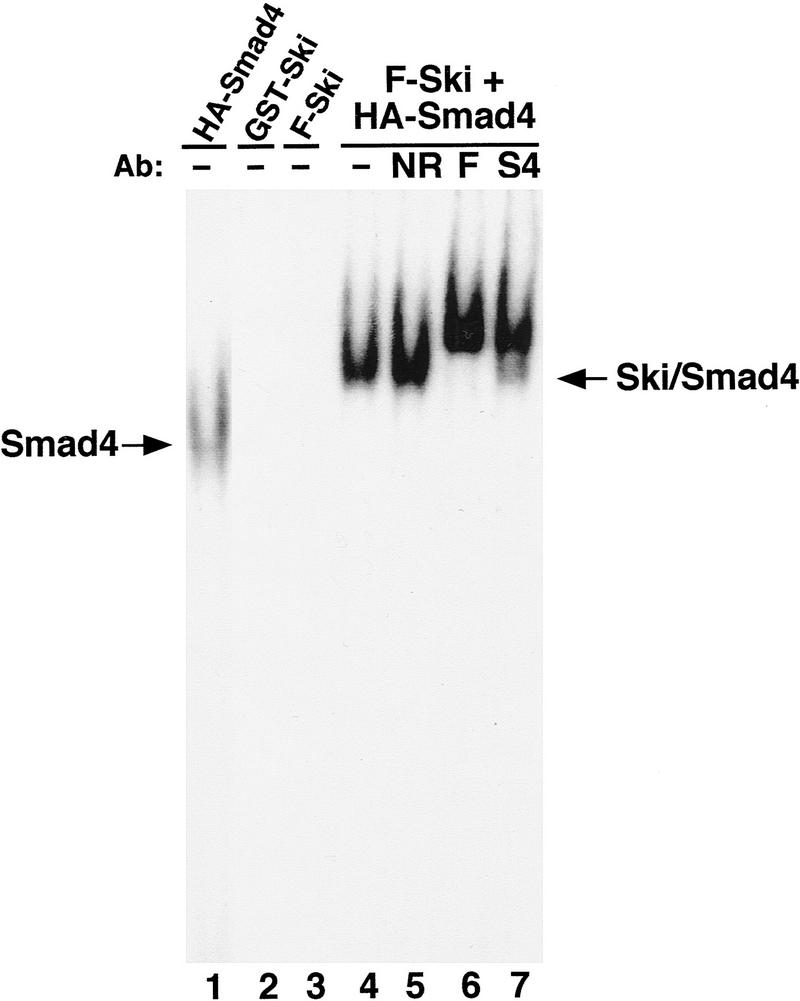
Ski binds to the SBE through interaction with Smad4. 32P-labeled SBE oligonucleotide (2 × 104 cpm) was incubated with HA–Smad4, GST-Ski, Flag–Ski (F-Ski), or purified Ski/Smad4 complex in EMSA reactions. DNA-protein complexes were resolved by nondenaturing PAGE. Proteins used in the EMSA reactions: (Lane 1) 0.5 μg HA–Smad4; (lane 2) 0.8 μg GST–Ski; (lane 3) 0.8 μg Flag-Ski purified from 293T cells transfected with Flag–Ski alone; (lanes 4–7) Flag–Ski/HA–Smad4 complex purified using anti-Flag agarose from cotransfected 293T cells. Antibodies used in supershift reactions: (lane 5) NR (nonrelevant antibody); (lane 6) F (anti-Flag); (lane 7) S4 (anti-Smad4).
Ski represses TGFβ-induced, Smad-dependent transcriptional activation
Smad proteins play an important role in mediating TGFβ-induced transcriptional activation of downstream genes. To examine the effect of the Ski–Smad interaction on TGFβ-induced transcriptional activation, we cotransfected c-ski with the TGFβ-responsive p3TP–lux reporter construct into Hep3B cells. Introduction of increasing amounts of c-ski cDNA resulted in a corresponding decrease in TGFβ-induced transactivation (Fig. 3A), suggesting that Ski functions to repress TGFβ-induced transactivation. Similar results were also obtained using the natural PAI-1 promoter (data not shown). Furthermore, Ski markedly inhibited transcriptional activation of p3TP–lux induced by Smad heteromeric complexes, in particular the Smad3/Smad4 complex (Fig. 3B). Therefore, Ski inhibited TGFβ signaling by blocking the ability of the Smad3/Smad4 complex and, to a lesser extent, the Smad2/Smad4 complex to activate transcription of TGFβ-responsive genes (Fig. 3B).
Figure 3.

Ski represses TGF-β-induced and Smad-dependent transcriptional activation. (A). Ski represses TGF-β-induced transcriptional activation. Hep3B cells were transfected with 0.5 μg p3TP–lux and increasing amounts of ski as indicated. Luciferase activity was measured 16 hr after stimulation with 50 pm TGFβ1. (B) Hep3B cells were cotransfected with the Smad proteins and Ski as indicated. Smad-mediated transcriptional activation of the p3TP–lux in the absence − or presence + of Ski was analyzed.
Ski recruits N-CoR to the Smads
Recently, Ski was found to complex with HDAC through binding to N-CoR and mSin3A and to mediate transcriptional repression by thyroid hormone receptor, Mad (Nomura et al. 1999) and pRb (Tokitou et al. 1999). A similar mechanism may be employed for repression of Smad transactivation by Ski. To examine whether Ski can recruit a nuclear corepressor to the Smads, anti-Flag immunoprecipitates were prepared from cells cotransfected with various Flag–Smads and HA–Ski and examined for the presence of endogenous N-CoR (Fig. 4) by Western blotting. Indeed, Smad2, Smad3, and Smad4 all complexed with N-CoR, but only when Ski was also coexpressed (Fig. 4, lanes 4–6). Expression of Smad proteins alone (lanes 1–3) or coexpression of a mutant Ski protein (Ski 241–323) defective in binding to the Smads (Fig. 5C) and N-CoR (Fig. 4, lane 8) did not result in copurification of N-CoR with the Smads. Consistent with this observation, this mutant Ski(241–323) was defective in repression of TGFβ-induced transactivation of p3TP–lux (Fig. 5D). Because N-CoR has been shown to interact directly with mSin3A and HDAC1, repression of Smad-mediated transcriptional activation by Ski may involve an HDAC complex. Because of technical difficulties, we were not able to detect specific interactions between the Smads and endogenous mSin3A or HDAC (data not shown).
Figure 4.

Smad2, Smad3, and Smad4 complex with N-CoR through Ski. 293T cells were transfected with Flag–Smad2, Flag–Smad3, or Flag–Smad4 either alone (lanes 1–3) or together with HA–Ski (lanes 4–6). Endogenous N-CoR complexed with the Smads was isolated by immunoprecipitation with an anti-Flag mAb and detected by Western blotting with an anti-N-CoR antibody. Cell lysates were also blotted directly with anti-Flag and anti-HA antibodies for control of Smads and Ski expression. As a positive control, N-CoR associated with HA–Ski was isolated by immunoprecipitation with an anti-HA mAb and blotted with an anti-N-CoR antibody (lane 7). (Lane 8) HA-tagged mutant Ski(241–323) isolated from transfected 293T cells by immunoprecipitation with an anti-HA antibody. (Lane 9) Anti-Flag immunoprecipitate prepared from cells cotransfected with Flag–Smad3 and HA–Ski(241–323).
Figure 5.


Smad proteins interact with the amino-terminal region of Ski. (A) Schematic drawings of Ski truncation mutants. The domain in c-Ski that is conserved with v-Ski (residues 24–441) is shaded. This domain contains transforming activity and also mediates binding to N-CoR. (B) Binding of the Ski mutants to Smad3 and Smad4. Flag–Smad3 or Flag–Smad4 were cotransfected with HA-tagged c-Ski and mutants. Association of Smads with various Ski proteins was analyzed by blotting of the Flag immunoprecipitates with an anti-HA antiserum. Cell lysates were blotted directly with an anti-HA antibody as a control for the expression of various Ski mutants. (*) A nonspecific background band. (C) Interaction of Smad3 and Smad4 with Ski(241–323). To detect the smallest Ski truncation mutant, 293T cells transfected with Flag–Smads and HA–Ski(214–323) were labeled with 35S-Express, and Smad-bound Ski(241–323) was isolated by immunoprecipitation with anti-Flag agarose and visualized by autoradiography. Parallel immunoprecipitation with an anti-HA antiserum was carried out to control for the expression of the Ski mutants. Ski(197–441) was used as a positive control for this experiment. (D) Hep3B cells were transfected with p3TP–lux and various Ski mutants. After transfection (30 hr), cells were stimulated with 50 pm TGFβ1 for 16 hr and luciferase activity was measured.
c-ski interacts with the Smads through the Ski homology domain
The amino-terminal portion of c-Ski (residues 24–441) is conserved in v-Ski (Ski homologous region, Fig. 5A). Within this homologous region, a segment containing amino acid residues 76–304 in c-Ski has been shown to be sufficient for the transforming activity (Zheng et al. 1997), and a segment containing residues 99–274 is responsible for binding to the corepressor N-CoR (Nomura et al. 1999). To determine whether this region can mediate Smad binding and transcriptional repression, Ski deletion mutants were tested in both binding and transcription assays. As shown in Figure 5B, a Ski fragment between residues 197 and 441 within the highly conserved ski homologous region was sufficient for interaction with Smad4 (Fig. 5B, right panels), Smad3 (left panels), and Smad2 (data not shown) and for transcriptional repression (Fig. 5D). Within this fragment, residues 203–239 appeared to be important for binding to Smad4, but not to Smad3 (Fig. 5B) or Smad2 (data not shown), as an internal deletion of Ski lacking residues 203–239 failed to interact with Smad4, but still associated with Smad3 (Fig. 5B) and Smad2 (data not shown). Binding to Smad3 and Smad2 required the region between residues 241 and 441. Because Smad3 (Smad2) and Smad4 bind to adjacent, but distinct, regions in Ski, Ski could contact two Smad molecules in a Smad hetero-oligomer simultaneously. A short fragment containing residues 241–323 did not interact with the Smads (Fig. 5C), was unable to recruit N-CoR complex to the Smads (Fig. 4), and failed to repress TGFβ-induced transactivation of p3TP–lux (Fig. 5D). Therefore, the domain in Ski that confers transforming activity also contains Smad-interacting regions. Interaction with Smads could have an important role in the transforming activity of Ski.
Overexpression of Ski attenuates TGFβ-induced growth inhibition and JunB expression
A critical function of TGFβ is the inhibition of growth of many cell types including epithelial, endothelial, and lymphoid cells. Because Ski repressed TGFβ-induced transcriptional activation, we speculated that overexpression of Ski may abolish the ability of these cells to undergo growth arrest in response to TGFβ, and this could be the molecular basis for the transforming activity of Ski. To test this hypothesis, stable Ba/F3 pro-B cell lines expressing different levels of Flag-tagged Ski were generated (Fig. 6A) and tested first for the ability of Flag–Ski to interact with endogenous Smad proteins on SBE DNA (Fig. 6B,C). Association of Flag–Ski with Smad2 or Smad3 was induced by TGFβ1, whereas its interaction with Smad4 occurred in both the presence and the absence of TGFβ1 (Fig. 6B). Nuclear extracts prepared from the B/ski-8 cells that had been stimulated with TGFβ1 contained a specific DNA-binding activity that can be supershifted by anti-Flag, anti-Smad2/3, and anti-Smad4 antibodies (Fig. 6C), confirming that Ski interacted with the endogenous Smad proteins on the SBE.
We next examined the abilities of B/ski-8 and B/ski-12 cells to undergo growth arrest in response to TGFβ. Overexpression of Ski rendered these cells resistant to TGFβ-induced growth arrest, and the degree of resistance correlated with the level of Ski expression (Fig. 6D). Furthermore, TGFβ-induced activation of JunB expression was also attenuated in these cells (Fig. 6E). Unlike the control Ba/F3 cells in which JunB expression was activated markedly 1 hr after TGFβ1 stimulation (Fig. 6E, lane 2), the level of JunB mRNA in Ski-overexpressing B/ski-8 cells did not change significantly in response to TGFβ1 (Fig. 6E, lanes 6–8). The lack of response to TGFβ in the B/ski-8 cells was not caused by the decreased expression of cell surface TGFβ receptors, as an affinity labeling experiment using 125I-labeled TGFβ1 showed that B/ski-8 cells expressed the same amounts of TβRI and TβRII on the cell surface as the uninfected Ba/F3 cells (data not shown). Thus, overexpression of Ski can directly block the ability of TGFβ to inhibit cell growth, and this could be an important mechanism by which high levels of Ski result in transformation of mammalian cells (Colmenares and Stavnezer 1989; Colmenares et al. 1991; Nomura et al. 1989; Fumagalli et al. 1993).
Discussion
Ski induces morphogenic transformation and anchorage-independent growth when overexpressed in chicken and quail embryo fibroblasts (Colmenares and Stavnezer 1989; Colmenares et al. 1991). Overexpression of Ski protein has also been detected in human tumor cells derived from neuroblastoma, melanoma, and prostate cancer (Nomura et al. 1989; Fumagalli et al. 1993). However, the mechanism by which high levels of Ski proteins regulate cell proliferation and oncogenic transformation is not fully understood. We have shown here that Ski can interact with Smad2, Smad3, and Smad4 and repress TGFβ-induced transactivation by recruitment of N-CoR and possibly its associated HDAC complex to the TGFβ-responsive promoter DNA. The domain in Ski that mediates interaction with the Smads overlaps with the domain responsible for the transforming activity of Ski (Zheng et al. 1997) as well as that mediating transcriptional repression (Fig. 5D; Nomura et al. 1999). Furthermore, we showed that overexpression of Ski in the Ba/F3 pro-B cell line rendered these cells resistant to TGFβ-induced growth arrest. Thus, Ski can modulate the TGFβ signaling pathway by directly blocking the transactivation activity of Smad proteins. Because Smad proteins are essential tumor suppressors, repression of their activity may be an important mechanism in oncogenic transformation by Ski.
Inactivation of Smad function may play an important role in epithelial carcinogenesis, as evidenced by a large number of loss-of-function Smad mutations in human cancers (Massagué 1998). However, in many cancer cells, despite the fact that the growth inhibitory pathway of TGFβ is intact and the Smads are fully functional when assayed in vitro, these tumor cells do not undergo growth arrest in response to TGFβ in vivo. Alternative mechanisms to disable TGFβ signaling must be employed by these transformed cells. Various oncoproteins have been shown to directly block key steps in Smad signaling. For example, phosphorylation of Smad3 in the linker region by MAP kinase in Ras-transformed cells prevents its nuclear translocation (Kretzschmar et al. 1999), whereas the Evi-1 oncoprotein inhibits the DNA-binding activity of Smad3 (Kurokawa et al. 1998). Here we showed that the Ski oncoprotein represses TGFβ signaling by recruitment of a transcriptional repressor complex to the TGFβ-responsive promoters through interaction with the Smads, and thus revealed a novel mechanism for inactivation of Smad signaling during mammalian carcinogenesis.
It is not clear what causes elevated expression of Ski in human tumor cells. We have not detected any regulation of the Ski protein expression by either TGFβ or other activated type I receptors of the TGFβ superfamily (Alk2–Alk4) (data not shown). It is also important to note that in normal mammalian cells, the endogenous level of Ski proteins is low and the expression level is tightly controlled during development or cell differentiation. The cellular level of Ski was reported to be induced during early stages of differentiation of myoblasts (Leferovich et al. 1995) and hematopoietic cells (Namciu et al. 1994). Up-regulation of Ski by these differentiation signaling pathways may modulate the activity of TGFβ signaling pathway. Indeed, injection of Ski in Xenopus embryos results in the cell-autonomous induction of neural axis formation and neural-specific gene expression, a phenotype similar to that induced by antagonizing bone morphogenetic protein (BMP) and activin signaling (Amaravadi et al. 1997). Thus, Ski may play a role in mediating cross-talk between the differentiation signaling pathways and the TGFβ pathway. Because TGFβ also regulates these processes, antagonizing interactions between Ski and the Smads could result in a precise control of the differentiation program. Because Smad4 is a common mediator that also functions downstream of BMP and activin receptors, Ski may also be involved in regulation of BMP or activin signaling. Future experiments will determine whether Ski interacts with other pathway-restricted Smads, such as Smad1 or Smad5, and represses the function of these proteins.
Smad3 and Smad4 as DNA-binding partners of Ski
Ski has been shown to bind DNA, but only in conjunction with other cellular proteins (Nagase et al. 1990; Nicol and Stavnezer 1998). Using a nuclear extract from c-ski-transformed cells, a specific DNA-binding site for Ski and its associated proteins was identified (GTCTAGAC) by cyclic amplification and selection of targets (CASTing) and was found to mediate transcriptional repression by Ski (Nicol and Stavnezer 1998). This sequence is identical to SBE (Zawel et al. 1998), suggesting that Ski may bind to DNA through interaction with the Smads. We showed here that Ski/Smad3 and Ski/Smad4 complexes can bind to SBE and repress Smad-mediated transcriptional activation. Thus, Smad3 and Smad4 are the DNA-binding partners of Ski in these c-ski-transformed cells. In addition to SBE, Ski was also found to interact with the nuclear factor I (NFI) binding site through interaction with the NFI protein (Tarapore et al. 1997). However, in this context, Ski functions to potentiate, not repress, NFI-stimulated transcriptional activation. Thus, Ski may interact with different DNA-binding factors and regulate transcription both positively and negatively depending on the proper cellular context or interacting partners.
Ski as trancriptional corepressor of the Smads
Because Ski interacts with the MH2 domains of the Smads, it may repress Smad-mediated transcriptional activation either by blocking the ability of the Smads to homo- and hetero-oligomerize or by recruitment of a transcriptional repressor complex to the Smads. We found that the ability of Smad3 to homo- and hetero-oligomerize with Smad4 was not affected by coexpression of Ski (data not shown). On the other hand, we showed that Ski recruited the nuclear repressor N-CoR to the Smads. Thus, Ski functions as a corepressor of the Smad proteins by recruitment of a transcriptional repressor complex. In addition to the Smad proteins, Ski was recently reported to bind directly to Rb (Tokitou et al. 1999) and retinoic acid receptor (Dahl et al. 1998) and to repress transactivation induced by these proteins, probably through similar mechanisms. N-CoR was originally identified as a corepressor that mediates transcriptional repression by the thyroid hormone receptor and Mad (Horlein et al. 1995). It is a protein of 270 kD and contains three repressor domains in its amino-terminal region (Horlein et al. 1995). It shows a striking homology to another corepressor, SMRT, and represses transcription by forming complexes with mSin3 and HDAC (Alland et al. 1997; Heinzel et al. 1997). Although we were not able to detect specific interactions between the Smads and endogenous mSin3A or HDAC because of technical difficulties, the recruitment of a N-CoR complex to the Smads suggests that repression of Smad-mediated transcription by Ski may involve deacetylation of nucleosomal histones. Recently, Smad2 has been shown to interact with TGIF, another transcriptional corepressor that recruits HDAC to the Smads (Wotton et al. 1999). Thus, repression of Smad-mediated transactivation may involve multiple corepressors. Future studies will allow us to determine whether Ski, Smads, N-CoR, and TGIF are in the same complex or whether Smads interact with different corepressors depending on the expression level of these corepressors in different cell types or at different developmental stages.
Materials and methods
Cells, antisera, and constructs
293T and Bing cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Hep3B, a human hepatoma cell line (ATCC), was maintained in minimum essential medium (MEM) supplemented with 10% FBS. Ba/F3, a pro-B cell line, was grown in RPMI supplemented with 10% FBS and 10% WEHI cell-conditioned medium as a source of interleukin-3 (Luo and Lodish 1996). TGFβ1 was purchased from R&D Systems. Antisera against Smad4 (C-20), Smad2/3 (N-19), and N-CoR were purchased from Santa Cruz Biotechnology. An anti-Smad3-specific antibody was raised against a peptide (amino acid residues 193–212) located in the linker region of the human Smad3 protein. Anti-Ski antibody was raised against a glutathione S-transferase (GST)-fusion protein containing amino acid residues 1–605 of human c-Ski.
Full-length and truncated Smad2, Smad3, and Smad4 were generated as described previously (Stroschein et al. 1999). Flag- or HA-tagged full-length or truncated ski were generated by PCR, cloned into pCMV5B, and used for transient transfection experiments.
Transfection and retroviral infection
293T, Bing, and Hep3B cells were transiently transfected using the Lipofectamin-Plus protocol (GIBCO BRL). To generate a stable Ba/F3 cell line overexpressing ski, Flag-ski was cloned into the retroviral vector, pBabe-puro, that also expresses a puromycin-resistance gene. The construct was used to transfect Bing cells to generate retroviruses expressing Flag–Ski. Forty-eight hr after transfection, 1 × 106 Ba/F3 cells were cocultivated with the transfected Bing cells for 24 hr, and the infected cells were selected in medium containing puromycin.
Purification of Smad4-associated proteins
Stable 293T cell lines expressing Flag–Smad4NL (amino acid residues 1–318) or Flag–Smad4C (319–551) were generated by transfection. For large-scale purification of Smad4-associated proteins, cells from 96 tissue culture dishes (150 mm) were lysed in buffer containing 50 mm HEPES at pH 7.8, 500 mm NaCl, 5 mm EDTA, 1% NP-40, 3 mm dithiothreitol (DTT) and 0.5 mm phenylmethylsulfonyl fluoride. Clarified cell lysates were then incubated with anti-Flag M2 agarose (Sigma), and bound proteins were eluted with 0.4 mg/ml Flag peptide (Sigma) (Zhou et al. 1998a; Stroschein et al. 1999). Approximately 2 μg of the 97-kD protein was resolved on an SDS–polyacrylamide gel, transferred to nitrocellulose, and microsequenced. Amino acid sequence derived from two of the peptides showed a perfect match to human c-Ski: Peptide 1 (RLSAFRPWSPAV) was mapped to amino acid residues 375–386 and peptide 2 (KELQEQLWP) was mapped to residues 703–711.
Immunoprecipitation and Western blotting
Flag- and/or HA-tagged Smads and Ski were isolated from transfected 293T cells by immunoprecipitation with anti-Flag agarose, followed by elution with the Flag peptide, and analyzed by Western blotting as described previously (Zhou et al. 1998a; Stroschein et al. 1999).
Growth inhibition and transcriptional reporter assays
For growth inhibition assay, 3 × 104 Ba/F3 cells were incubated with various concentrations of TGFβ1 for 5 days. The growth of cells was determined by cell counting and compared with unstimulated cells (Luo and Lodish 1996).
For transcriptional reporter assay, 2.5 μg DNA (0.5 μg p3TP–lux, 1 μg of Smad, and 0.5–1 μg of ski) was used to transfect Hep3B cells in a six-well cluster plate. Twenty-four hr after transfection, Hep3B cells were starved in serum-free media for 8 hr and stimulated with 50 pm TGFβ1 for 16 hr as described (Stroschein et al. 1999).
EMSA
The Ski/Smad4 complex isolated by affinity purification or nuclear extracts prepared from TGFβ-stimulated Ba/F3 cells (Lee et al. 1987) was incubated with the 32P-labeled SBE probe (5′-CTCTATCAATTGGTCTAGACTTAACCGGA-3′) in binding buffer (25 mm Tris-Cl at pH 7.5, 80 mm NaCl, 35 mm KCl, 5 mm MgCl2, 10% glycerol, 1 mm DTT, 15 μg/ml poly[d(I-C)], 300 μg/ml bovine serum albumin, 2% NP-40), and the protein–DNA complexes were resolved on a 4% nondenaturing polyacrylamide gel (Stroschein et al. 1999). For antibody supershift assays, the Ski/Smad4 complex was preincubated with 4 μg of specific antibodies for 1 hr at 4°C.
Northern blotting
Ba/F3 cells were serum starved for 16 hr and then stimulated with 200 pm TGFβ1 for the indicated period of time. Total RNA was prepared from these cells using RNeasy kit (Qiagen), and 20 μg RNA was resolved on a 1% formaldehyde gel, transferred to a Nylon membrane, and analyzed by Northern blotting. The probes (JunB and CAC1) were labeled by random priming (Stratagene) and hybridized with the RNA samples in QuikHyb (Stratagene). Human chromatin assembly factor-1 (CAC1) was used as a loading control.
Acknowledgments
We thank Drs. N. Nomura for the c-ski cDNA, P. Kaufman for the human CAC1 probe, G.S. Martin for the A431 cell line, J. Wrana and L. Attisano for the human Smad2 and Smad4 cDNA, and R. Derynck for human Smad3 cDNA. This work was supported by Department of Energy (DOE)/LBNL grant DE-AC03-76SF00098, Department of Energy grant DE-AC03-76SF00099, Wendy Will Case Cancer Fund 009476-001, and California breast cancer research program award 4KB-0151 to K.L. S.S. was supported by a predoctoral fellowship from the National Science Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL k_luo@ux5.lbl.gov; FAX (510) 643-9290.
References
- Alland L, Muhle R, Hou H, Jr, Potes J, Chin L, Schreiber-Agus N, DePinho RA. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387:49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- Amaravadi LS, Neff AW, Sleeman JP, Smith RC. Autonomous neural axis formation by ecotopic expression of the protooncogene c-ski. Dev Biol. 1997;192:392–404. doi: 10.1006/dbio.1997.8780. [DOI] [PubMed] [Google Scholar]
- Berk M, Desai SY, Heyman HC, Colmenares C. Mice lacking the ski proto-oncogene have defects in neurulation, craniofacial, patterning, and skeletal muscle development. Genes & Dev. 1997;11:2029–2039. doi: 10.1101/gad.11.16.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Rubock MJ, Whitman M. A transcriptional partner for MAD proteins in TGF-beta signalling. Nature. 1996;383:691–696. doi: 10.1038/383691a0. [DOI] [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- Colmenares C, Stavnezer E. The ski oncogene induces muscle differentiation in quail embryo cells. Cell. 1989;59:293–303. doi: 10.1016/0092-8674(89)90291-2. [DOI] [PubMed] [Google Scholar]
- Colmenares C, Sutrave P, Hughes SH, Stavnezer E. Activation of the c-ski oncogene by overexpression. J Virol. 1991;65:4929–4935. doi: 10.1128/jvi.65.9.4929-4935.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl R, Kieslinger M, Beug H, Hayman MJ. Transformation of hematopoietic cells by the Ski oncoprotein involves repression of retinoic acid receptor signaling. Proc Natl Acad Sci. 1998;95:11187–11192. doi: 10.1073/pnas.95.19.11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert JC, Servaes S, Sutrave P, Hughes SH, Rosenthal N. Activation of a muscle-specific enhancer by the Ski proto-oncogene. Nucleic Acids Res. 1995;23:2988–2994. doi: 10.1093/nar/23.15.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X-H, Zhang Y, Wu R-Y, Derynck R. The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for Smad3 in TGF-β-induced transcriptional activation. Genes & Dev. 1998;12:2153–2163. doi: 10.1101/gad.12.14.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli S, Doneda L, Nomura N, Larizza L. Expression of the c-ski proto-oncogene in human melanoma cell lines. Melanoma Res. 1993;3:23–27. doi: 10.1097/00008390-199304000-00004. [DOI] [PubMed] [Google Scholar]
- Hata A, Lo RS, Wotton D, Lagna G, Massagué J. Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature. 1997;388:82–87. doi: 10.1038/40424. [DOI] [PubMed] [Google Scholar]
- Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- Heldin C-H, Miyazono K, ten Dijke P. TGF-β signaling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- Hua X, Liu X, Ansari DO, Lodish HF. Synergistic cooperation of TFE3 and Smad proteins in TGFβ-induced transcription of the plasminogen activator inhibitor-1 gene. Genes & Dev. 1998;12:3084–3095. doi: 10.1101/gad.12.19.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janknecht R, Wells NJ, Hunter T. TGF-β-stimulated cooperation of Smad proteins with the coactivators CBP/p300. Genes & Dev. 1998;12:2114–2119. doi: 10.1101/gad.12.14.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonk LJ, Itoh S, Heldin CH, ten Dijke P, Kruijer W. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J Biol Chem. 1998;273:21145–21152. doi: 10.1074/jbc.273.33.21145. [DOI] [PubMed] [Google Scholar]
- Kon A, Vindevoghel L, Kouba DJ, Fujimura Y, Uitto J, Mauviel A. Cooperation between SMAD and NF-kappaB in growth factor regulated type VII collagen gene expression. Oncogene. 1999;18:1837–1844. doi: 10.1038/sj.onc.1202495. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Massagué J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature. 1997;389:618–622. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Timokhina I, Massagué J. A mechanism of repression of TGFβ/Smad signaling by oncogenic Ras. Genes & Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa M, Mitani K, Irie KM, Takahashi T, Chiba S, Yazaki Y, Matsumoto K, Hirai H. The oncoprotein Evi-1 represses TGFβ signaling by inhibiting Smad3. Nature. 1998;394:92–96. doi: 10.1038/27945. [DOI] [PubMed] [Google Scholar]
- Labbe E, Silvestri C, Hoodless PA, Wrana JL, Attisano L. Smad2 and Smad3 positively and negatively regulate TGF beta-dependent transcription through the forkhead DNA-binding protein FAST2. Mol Cell. 1998;2:109–120. doi: 10.1016/s1097-2765(00)80119-7. [DOI] [PubMed] [Google Scholar]
- Lee KAW, Bindereif A, Green MR. A small-scale procedure for preparation of nuclear extracts that support efficient transcription and pre-mRNA splicing. Genet Anal Technol. 1987;5:22–31. doi: 10.1016/0735-0651(88)90023-4. [DOI] [PubMed] [Google Scholar]
- Leferovich JM, Lana DP, Sutrave P, Hughes SH, Kelly AM. Regulation of c-ski transgene expression in developing and mature mice. J Neurosci. 1995;15:596–603. doi: 10.1523/JNEUROSCI.15-01-00596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Turck CM, Teumer JK, Stavnezer E. Unique sequence, ski, in Sloan-Kettering avian retroviruses with properties of a new cell-derived oncogene. J Virol. 1986;57:1065–1072. doi: 10.1128/jvi.57.3.1065-1072.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Hata A, Baker JC, Doody J, Carcamo J, Harland RM, Massagué J. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature. 1996;381:620–623. doi: 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- Luo K, Lodish HF. Signaling by chimeric erythropoietin-TGF-beta receptors: Homodimerization of the cytoplasmic domain of the type I TGF-beta receptor and heterodimerization with the type II receptor are both required for intracellular signal transduction. EMBO J. 1996;15:4485–4496. [PMC free article] [PubMed] [Google Scholar]
- Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Kardassis D. Regulation of the human p21/WAF1/Cip1 promoter in hepatic cells by functional interactions between Sp1 and Smad family members. Proc Natl Acad Sci. 1998;95:6733–6738. doi: 10.1073/pnas.95.12.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase T, Mizuguchi G, Nomura N, Ishizaki R, Ueno Y, Ishii S. Requirement of protein co-factor for the DNA-binding function of the human ski proto-oncogene product. Nucleic Acids Res. 1990;18:337–343. doi: 10.1093/nar/18.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namciu S, Lieberman MA, Stavnezer E. Induction of the c-ski proto-oncogene by phorbol ester correlates with induction of megakaryocyte differentiation. Oncogene. 1994;9:1407–1416. [PubMed] [Google Scholar]
- Nicol R, Stavnezer E. Transcriptional repression by v-Ski and c-Ski mediated by a specific DNA binding site. J Biol Chem. 1998;273:3588–3597. doi: 10.1074/jbc.273.6.3588. [DOI] [PubMed] [Google Scholar]
- Nomura N, Sasamoto S, Ishii S, Date T, Matsui M, Ishizaki R. Isolation of human cDNA clones of ski and the ski-related gene, sno. Nucleic Acids Res. 1989;17:5489–5500. doi: 10.1093/nar/17.14.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Khan MM, Kaul SC, Dona H-D, Wadhwa R, Colmenares C, Kohno I, Ishii S. Ski is a component of the histone deacetylase complex required for transcriptional repression by Mad and thyroid horomone receptor. Genes & Dev. 1999;13:412–423. doi: 10.1101/gad.13.4.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson-White S. SnoI, a novel alternatively spliced isoform of the ski protooncogene homolog, sno. Nucleic Acids Res. 1993;21:4632–4638. doi: 10.1093/nar/21.19.4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouponnot C, Jayaraman L, Massagué J. Physical and functional interaction of SMADs and p300/CBP. J Biol Chem. 1998;273:22865–22868. doi: 10.1074/jbc.273.36.22865. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB. The transforming growth factor-βs. In: Roberts AB, Sporn MB, editors. Peptide growth factors and their receptors. Heidelberg, Germany: Springer-Verlag; 1990. pp. 421–472. [Google Scholar]
- Shen X, Hu PP, Liberati NT, Datto MB, Frederick JP, Wang XF. TGF-beta-induced phosphorylation of Smad3 regulates its interaction with coactivator p300/CREB-binding protein. Mol Biol Cell. 1998;9:3309–3319. doi: 10.1091/mbc.9.12.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Hata A, Lo RS, Massagué J, Pavletich NP. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature. 1997;388:87–93. doi: 10.1038/40431. [DOI] [PubMed] [Google Scholar]
- Shi Y, Wang Y-F, Jayaraman L, Yang H, Massagué J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: Insights on DNA binding in TGF-β signaling. Cell. 1998;94:585–594. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
- Song C-Z, Siok TE, Gelehrter TD. Smad4/DPC4 and Smad3 mediate transforming growth factor-β (TGF-β) signaling through direct binding to a novel TGF-responsive element in the human plasminogen activator inhibitor-1 promoter. J Biol Chem. 1998;273:29287–29290. doi: 10.1074/jbc.273.45.29287. [DOI] [PubMed] [Google Scholar]
- Stavnezer E, Brodeur D, Brennan LA. The v-ski oncogene encodes a truncated set of c-ski coding exons with limited sequence and structural relatedness to v-myc. Mol Cell Biol. 1989;9:4038–4045. doi: 10.1128/mcb.9.9.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroschein SL, Wang W, Luo K. Cooperative binding of Smad proteins to two adjacent DNA elements in the plasminogen activator inhibitor-1 promoter mediates transforming growth factor β-induced Smad-dependent transcriptional activation. J Biol Chem. 1999;274:9431–9441. doi: 10.1074/jbc.274.14.9431. [DOI] [PubMed] [Google Scholar]
- Sutrave P, Hughes SH. Isolation and characterization of three distinct cDNAs for the chicken c-ski gene. Mol Cell Biol. 1989;9:4046–4051. doi: 10.1128/mcb.9.9.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutrave P, Copeland TD, Showalter SD, Hughes SH. Characterization of chicken c-ski oncogene products expressed by retrovirus vectors. Mol Cell Biol. 1990a;10:3137–3144. doi: 10.1128/mcb.10.6.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutrave P, Kelly AM, Hughes SH. ski can cause selective growth of skeletal muscle in transgenic mice. Genes & Dev. 1990b;4:1462–1472. doi: 10.1101/gad.4.9.1462. [DOI] [PubMed] [Google Scholar]
- Tarapore P, Richmond C, Zheng G, Cohen SB, Kelder B, Kopchick J, Kruse U, Sippel AE, Colmenares C, Stavnezer E. DNA binding and transcriptional activation by the Ski oncoprotein mediated by interaction with NFI. Nucleic Acids Res. 1997;25:3895–3903. doi: 10.1093/nar/25.19.3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokitou F, Nomura T, Khan MM, Kaul SC, Wadhwa R, Yasukawa T, Kohno I, Ishii S. Viral ski inhibits retinoblastoma protein (Rb)-mediated transcriptional repression in a dominant negative fashion. J Biol Chem. 1999;274:4485–4488. doi: 10.1074/jbc.274.8.4485. [DOI] [PubMed] [Google Scholar]
- Topper JN, DiChiara MR, Brown JD, Williams AJ, Falb D, Collins T, Gimbrone MAJ. CREB binding protein is a required coactivator for smad-dependent, transforming growth factor beta transcriptional responses in endothelial cells. Proc Natl Acad Sci. 1998;95:9506–9511. doi: 10.1073/pnas.95.16.9506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95:779–791. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- Wong C, Rougier-Chapman EM, Frederick JP, Datto MB, Liberati NT, Li JM, Wang XF. Smad3-Smad4 and AP-1 complexes synergize in transcriptional activation of the c-Jun promoter by transforming growth factor beta. Mol Cell Biol. 1999;19:1821–1830. doi: 10.1128/mcb.19.3.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton D, Lo RS, Lee S, Massagué J. A Smad transcriptional corepressor. Cell. 1999;97:29–39. doi: 10.1016/s0092-8674(00)80712-6. [DOI] [PubMed] [Google Scholar]
- Yingling JM, Datto MB, Wong C, Frederick JP, Liberati NT, Wang XF. Tumor suppressor Smad4 is a transforming growth factor beta-inducible DNA binding protein. Mol Cell Biol. 1997;17:7019–7028. doi: 10.1128/mcb.17.12.7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Feng X-H, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature. 1998;394:909–913. doi: 10.1038/29814. [DOI] [PubMed] [Google Scholar]
- Zheng G, Teumer J, Colmenares C, Richmond C, Stavnezer E. Identification of a core functional and structural domain of the v-Ski oncoprotein responsible for both transformation and myogenesis. Oncogene. 1997;15:459–471. doi: 10.1038/sj.onc.1201205. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Chen D, Pierstorff E, Luo K. Transcription elongation factor P-TEFb mediates Tat activation of HIV-1 transcription at multiple stages. EMBO J. 1998a;17:3681–3691. doi: 10.1093/emboj/17.13.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Zawel L, Lengauer C, Kinzler KW, Vogelstein B. Characterization of human FAST-1, a TGF beta and activin signal transducer. Mol Cell. 1998b;2:121–127. doi: 10.1016/s1097-2765(00)80120-3. [DOI] [PubMed] [Google Scholar]