

Abstract
Ataxia telangiectasia results from mutations of ATM and is characterized by severe neurodegeneration and defective responses to DNA damage. Inactivation of certain DNA repair genes such as DNA ligase IV results in massive neuronal apoptosis and embryonic lethality in the mouse, indicating the occurrence of endogenously formed DNA double-strand breaks during nervous system development. Here we report that Atm is required for apoptosis in all areas of the DNA ligase IV-deficient developing nervous system. However, Atm deficiency failed to rescue deficits in immune differentiation in DNA ligase IV-null mice. These data indicate that ATM responds to endogenous DNA lesions and functions during development to eliminate neural cells that have incurred genomic damage. Therefore, ATM could be important for preventing accumulation of DNA-damaged cells in the nervous system that might eventually lead to the neurodegeneration observed in ataxia telangiectasia.
Keywords: ATM, DNA ligase IV, apoptosis, neurodegeneration, ataxia telangiectasia
Ataxia telangiectasia (A-T) results from mutations of ATM (ataxia telangiectasia, mutated) and is characterized by progressive neurodegeneration and other defects including immune deficiencies, cancer proneness, chromosomal instability, and ionizing radiation sensitivity (Sedgwick and Boder 1991; Savitsky et al. 1995; Lavin and Shiloh 1997). However, neither the mechanism nor the etiological agent responsible for the neurodegeneration is known. ATM is a serine/threonine protein kinase that is known to function as a DNA-damage sensor that responds to DNA double-strand breaks (Lavin and Shiloh 1997; Canman and Lim 1998; Rotman and Shiloh 1998). In vitro, ATM activation leads to cell cycle arrest via phosphorylation of either p53 (Banin et al. 1998; Canman et al. 1998; Khanna et al. 1998; G1 arrest) NBS1 (Gatei et al. 2000; Lim et al. 2000; Wu et al. 2000; Zhao et al. 2000; S-phase arrest), or Chk2 (Hirao et al. 2000; G2 arrest). In the developing nervous system, Atm is required for ionizing radiation–induced apoptosis (Herzog et al. 1998; Chong et al. 2000). This led to the hypothesis that Atm functions during nervous system development to eliminate neural cells that have incurred genomic damage (Herzog et al. 1998). A number of other human syndromes with defective responses to DNA damage also develop neurological lesions (Rolig and McKinnon 2000). However, until recently, no compelling evidence existed that genotoxic stress was a feature of nervous system development. Remarkably, inactivation of certain repair enzymes such as DNA ligase IV resulted in a substantial perturbation of neurogenesis (Barnes et al. 1998; Gao et al. 1998).
DNA ligase IV (Lig4) is a distinct nuclear ligase that is a critical component of the nonhomologous end-joining machinery involved in V(D)J recombination and DNA repair (Robins and Lindahl 1996; Baumann and West 1998; Jeggo 1998). Yeast mutants lacking Lig4 proliferate normally, although they are strongly deficient in the joining of DNA double-strand breaks. In contrast, inactivation of Lig4 in mice results in widespread apoptosis of the developing nervous system, defective V(D)J recombination and lymphocyte development, and embryonic lethality (Barnes et al. 1998; Gao et al. 1998). Because ATM is a DNA damage sensor whose deficiency leads to neurodegeneration in humans, we asked if Atm signaling mediated the neural apoptosis in Lig4-null mice. Strikingly, we found that all apoptosis in the developing nervous system resulting from ligase IV deficiency required Atm, pinpointing Atm as a critical apoptotic effector after physiological DNA damage.
Results and Discussion
Because Lig4 deficiency most likely leads to accumulation of unrepaired DNA double-strand breaks, we reasoned that these lesions would activate Atm. Therefore, we determined whether the apoptotic signaling pathway activated by Lig4 deficiency was similar to Atm-dependent apoptosis after ionizing radiation. In the Lig4−/− CNS, we found high levels of p53 and active caspase-3 in the regions undergoing apoptosis. This was mostly restricted to the intermediate zone (IZ) of the developing CNS (Fig. 1d–f). Both p53 and caspase-3 are also activated in the developing nervous system after ionizing radiation in an Atm-dependent manner (Chong et al. 2000). In addition, Ser 15 phosphorylation of p53, as determined immunohistochemically, mirrored p53 stabilization in the developing CNS of the Lig4−/− embryos (data not shown). Thus, apoptosis resulting from Lig4 deficiency is similar to that observed after cellular exposure to ionizing radiation, consistent with a genotoxic lesion being present in the Lig4-deficient embryos. Apoptosis in Lig4-deficient embryos is observed very early during neurogenesis from embryonic day 11 (E11) onward and is localized to neural cells that are present in the postmitotic premigratory populations of the developing nervous system. Notably, Atm-dependent apoptosis after ionizing radiation–induced damage also occurs in the IZ that harbors premigratory populations (data not shown). This underscores the newly postmitotic neural populations as a physiologically relevant functional target for Atm. Therefore, we reasoned that, because the lesion associated with Lig4 deficiency activated apoptosis in a similar manner to DNA damage, Atm deficiency might rescue the Lig4-null phenotype.
Figure 1.
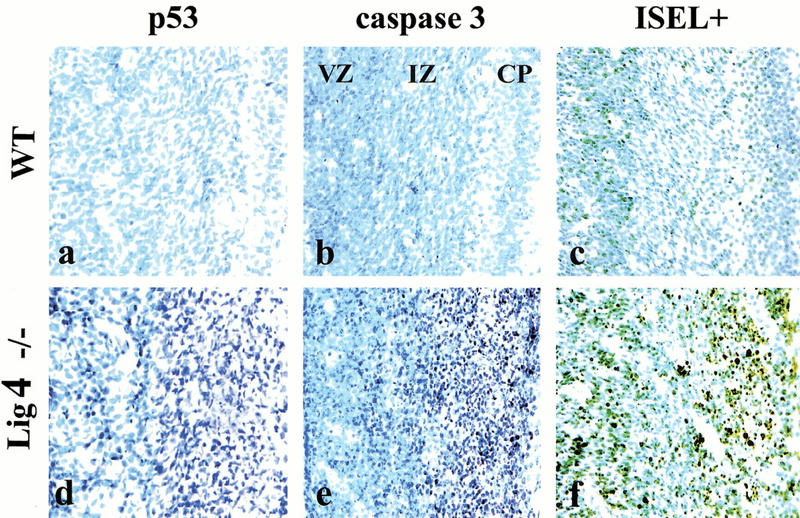
DNA Ligase IV deficiency initiates an apoptotic pathway characteristic of DNA damage. In wild-type (WT) embryos very little p53 (a) or active caspase-3 (b) are present and little apoptosis is seen in either the ventricular zone (VZ), the intermediate zone (IZ), or the cortical plate (CP). In Lig4-deficient embryos, both p53 (d) and activated caspase-3 (e) are abundant, as is apoptosis (f), as indicated by in situ endlabeling (ISEL). Embryos are at E14.5. Magnification is 200×.
To investigate this, we interbred Atm+/− and Lig4+/− mice to generate animals null for both Atm and Lig4. Strikingly, we found a complete attenuation of apoptosis in the embryonic Lig4−/− Atm−/− nervous system, indicating that all apoptosis caused by Lig4 deficiency required Atm (Fig. 2). Extensive apoptosis occurs in Lig4−/− Atm+/+ (Fig. 2b,f) and Lig4−/− Atm+/− (Fig. 2c,g) spinal cord at E11.5 and developing cortex at E14.5, whereas in the Lig4−/− Atm−/− tissue there were no such aberrant events (Fig. 2d,h). Furthermore, we found that apoptosis was rescued in all nervous system structures we examined at each developmental stage between E11.5 and E15.5 in the Lig4−/− Atm−/− mice (data not shown). Thus, the lesion accumulated in Lig4-null neural cells initiates apoptosis in an Atm-dependent manner. This implies that ATM functions to remove cells with endogenously produced DNA double-strand breaks from the developing nervous system.
Figure 2.
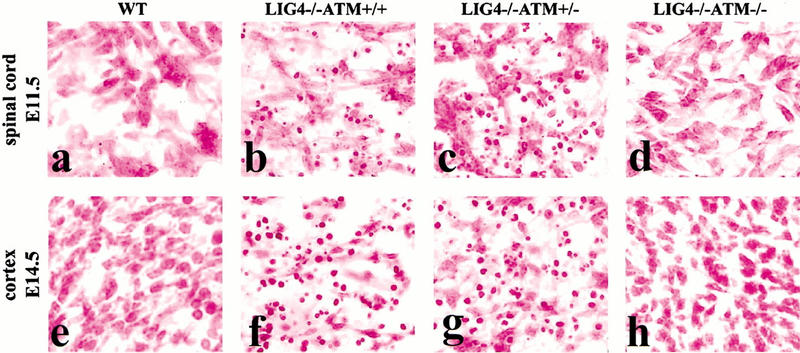
Atm is required for neuronal apoptosis in Lig4−/− embryos. Widespread apoptosis occurring in many CNS regions of the Lig4−/− embryo is dependent on functional Atm. Compared to wild-type (WT) embryos (a,e), Lig4−/− embryos exhibit apoptosis in either an Atm+/+ (b,f) or Atm+/− (c,g) background. However, Lig4−/− embryos also lacking Atm are completely devoid of aberrant apoptosis (d,h) at different developmental times and in different regions of the CNS. Atm−/− embryos are indistinguishable from WT embryos. Magnification is 400×.
The extent of the rescue of the Lig4−/− CNS by Atm deficiency was relatively complete as mice were born alive. However, the Lig4−/− Atm−/− mice were smaller than their wild-type littermates and died around 2 d after birth. Initial histological analysis of newborn brains revealed no significant difference between genotypes. In order to assess developmental variance between the Lig4−/− Atm−/− mice and littermates, we measured overall neural development in the Lig4−/− Atm−/− mice with markers that measure neuronal differentiation. Using Tuj1 to identify immature neurons and NeuN to identify mature neurons, we observed no apparent differences in brain development in the Lig4−/− Atm−/− cerebellum (Fig. 3) or other cortical structures (data not shown). Both the overall histology of the cerebellum (Fig. 3a,d,g) and the abundance and distribution of developing Tuj1+ neurons (Fig. 3b,e,h) or NeuN+ neurons (Fig. 3c,f,i) were not obviously affected in the Lig4−/− Atm−/− mice. Therefore, most of the general processes required for correct neural development in the Lig4−/− Atm−/− mice were functional, such as cellular proliferation, migration, and differentiation. Although these Lig4−/− Atm−/− cell populations are generally intact, they have incurred genomic damage, and this would eventually have serious consequences; a situation that is likely to be paralleled in A-T.
Figure 3.
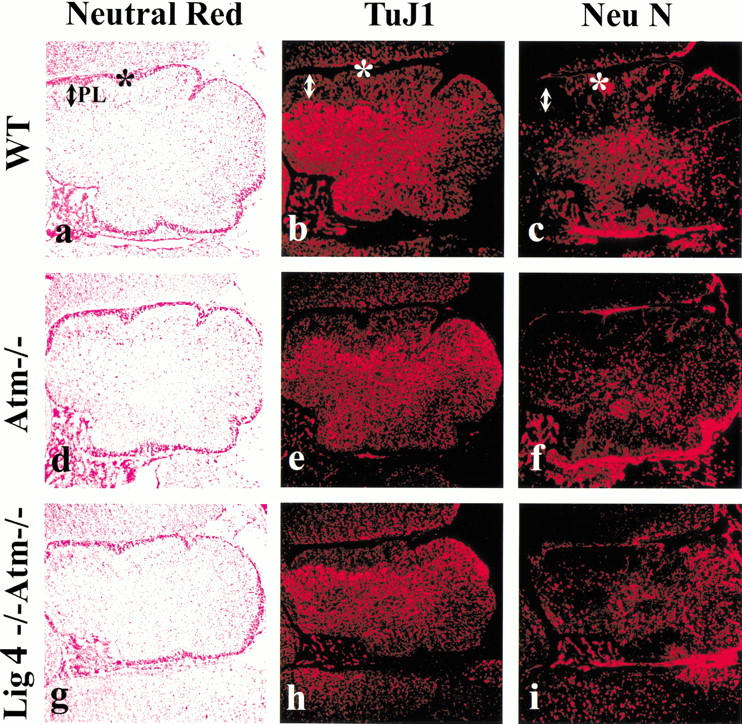
Normal brain development occurs in Lig4−/− Atm−/− mice. Cerebellum from newborn mice was stained with neutral red (a,d,g), Tuj1 (b,e,h), or NeuN (c,f,i). (a–c) Wild type; (d–f) Atm–/–; (g–i) Lig4−/− Atm–/–. The asterisk indicates the external granule layer, and the arrow marks the Purkinje cell layer (PL). Magnification is 100×.
Recently, it was shown that p53 deficiency rescues embryonic lethality in Lig4−/− mice (Frank et al. 2000; Y. Lee, D.E. Barnes, T. Lindahl, and P.J. McKinnon, unpubl.). Therefore, the rescue of Lig4 deficiency by Atm is probably a result of blocking Atm downstream-signal transduction in the nervous system. However, in the case of p53−/− Lig4−/−, rescue was more substantial as these double-null mice survived to around 6 wk of age (Frank et al. 2000). The reason for the difference in rescue of Lig4 deficiency by Atm and p53 is unclear. However, as both an Atm-null and p53-null background rescues neural apoptosis, it is possible that the primary cause for survival differences resides outside the nervous system.
Lig4 deficiency causes substantial defects in the development of the immune system, with both T and B cells showing early developmental arrest (Gao et al. 1998). Although Atm inactivation rescued the neurological defects in Lig4−/− mice, T-cell defects were still present as Lig4−/− Atm−/− animals showed decreased cellularity in the thymus compared to Atm−/− or wild type. Moreover, there was an almost complete lack of CD4+ CD8+ thymocytes in the Lig4−/− Atm−/− animals, consistent with a requirement for Lig4 in order for development to proceed past the CD4− CD8− stage (Fig 4a). In newborn Lig4−/− Atm−/− spleen, there was a decrease in CD45/B220+ B cells, as compared to wild-type controls or Atm null alone, although B-cell differentiation at this stage was similar between genotypes (Fig. 4b). Thus, Atm deficiency does not correct the defective lymphocyte development resulting from Lig4 deficiency, indicating that neuronal rescue is selective and reflects the tissue-specific functionality of Atm. Similarly, loss of p53 restores neural but not lymphocyte development in both Lig4 null and XRCC4-deficient mice (Frank et al. 2000; Gao et al. 2000; Roth and Gellert 2000). This also weakens the parallels between lymphoid development and neuronal development and speculation of somatic DNA recombination as a normal feature of the developing nervous system (Chun and Schatz 1999; Gilmore et al. 2000). However, inactivation of certain other components required for V(D)J recombination, such as Ku70 and Ku80, but not for DNA-PKcs also lead to neuronal apoptosis, with a level of severity that parallels their efficacy in a V(D)J recombination end-joining assay (Gu et al. 2000). Furthermore, while the source of DNA strand breaks during neurogenesis and their existence in the Lig4-null CNS have yet to be identified, they do not appear to depend on the RAG recombinase. Thus, viability of the Lig4 null phenotype is not rescued in a RAG1 or RAG2 null background (data not shown), and RAG1 or RAG2 deficiency did not rescue the embryonic lethality caused by loss of XRCC4 (Sekiguchi et al. 1999). Moreover, embryonic neuronal apoptosis is also observed in DNA polymerase β-deficient mice that are not compromised in double-strand break repair (Sugo et al. 2000). Thus, other types of DNA lesions can trigger apoptosis in developing neurons; a cell type that may in any case be uniquely sensitive to DNA damage (Roth and Gellert 2000).
Figure 4.

Immune differentiation defects are present in Lig4−/− Atm−/− mice. Thymus (a) or spleen (b) were isolated from newborn mice and analyzed for T- or B-cell development. The percentage of CD4+ CD8+ thymocytes for each genotype is indicated (a). IgM- and IgD-positive lymphocytes present in the various genotypes are shown (b). Lig4−/− Atm−/− mice displayed abnormal development of T cells in the thymus (a), while B-cell differentiation in these animals did not differ from wild-type or Atm−/− mice, although a decrease in the number of CD45/B220+ B cells was observed (b).
It is remarkable that Atm deficiency allows neural development to proceed so completely in Lig4-deficient animals. Clearly, a consequence of this is cells that have a genome containing unrepaired or misrepaired DNA double-strand breaks contributing to CNS function. Thus, it is likely that while the form of the CNS is intact, functionality may be compromised, and this may contribute to early postnatal lethality. In view of the severe phenotype, homologous recombination is clearly insufficient in compensating for Lig4 deficiency. In budding yeast, the small amount of residual DNA end-joining observed in cells lacking Lig4 is associated with the introduction of frequent sequence errors and misalignments (Schar et al. 1997). It seems likely that in humans, ATM deficiency allows genomically compromised cells with endogenously produced DNA double-strand breaks to become part of the final form of the nervous system, and that subsequent dysfunction because of this genomic damage leads to neurodegeneration.
Materials and methods
Gene knockout mice
The Lig4−/− and Atm−/− mice used in this study have been described previously (Barnes et al. 1998; Herzog et al. 1998).
Histology and immunohistochemistry
Embryos and postnatal brains were fixed in 4% paraformaldehyde, cryoprotected in 20% sucrose/PBS and cryosectioned (10 μm coronal or sagittal sections). All immunohistochemical analysis was done using antigen retrieval (Midgley et al. 1992). p53 was detected using the CM5 anti-p53 antibody (Vector laboratories) at a dilution of 1 : 500; antiactive caspase-3 was detected with affinity-purified CM1 (Srinivasan et al. 1998) antibody (0.66 mg/mL) at a dilution of 1 : 1500; Tuj1 (BAbCO) and NeuN (Chemicon) were both used at 1 : 500 dilution. ISEL staining was performed on tissue cryosections using the Klenow-FragEL kit (Oncogene Research Products) according to the manufacturer's directions. The Vectastain Elite ABC kit/VIP substrate avidin/biotin immunoperoxidase system (Vector laboratories) were used to visualize the immunopositive signal, and sections were counter stained with 0.1% methyl green (Vector laboratories) and mounted in permount. For Tuj1 and NeuN, immunofluorescence was done with Cy-3 conjugated secondary antibody (Jackson Immunologicals) to visualize primary antibody binding.
Flow cytometry analysis
Cell suspensions of thymus from neonatal pups after birth were stained with anti-CD4 and CD8 coupled with phycoerythrin and fluorescein isothiocyanate, respectively. Single-cell suspensions of spleen from newborn pups for B-cell analysis were labeled with anti-CD45/B220, IgM, and IgD coupled to allophycocyanin, phycoerythrin, and fluorescein isothiocyanate, respectively. B cells were isolated by gating the CD45/B220 positive populations, and IgM/IgD signals were plotted. All labeled cytometry markers were obtained from Pharmingen.
Acknowledgments
We thank Michael Kastan for advice and comments on the manuscript and Janet Gatewood and Mahnaz Paktinat for help with FACS analysis. These studies were supported by the National Institutes of Health (NS-37956 and CA-21765), the A-T Children's Project, the American Lebanese and Syrian Associated Charities (ALSAC) of St. Jude Children's Research Hospital, and the Imperial Cancer Research Fund.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
Comparable results to this study have also been found in other Lig4−/− Atm−/− mice (F.W. Alt, pers. comm.).
Footnotes
E-MAIL peter.mckinnon@stjude.org; FAX (901) 526-2907.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.837100.
References
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol. 1998;8:1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- Baumann P, West SC. DNA end-joining catalyzed by human cell-free extracts. Proc Natl Acad Sci. 1998;95:14066–14070. doi: 10.1073/pnas.95.24.14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman CE, Lim DS. The role of ATM in DNA damage responses and cancer. Oncogene. 1998;17:3301–3308. doi: 10.1038/sj.onc.1202577. [DOI] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Chong MJ, Murray MR, Gosink EC, Russell HR, Srinivasan A, Kapsetaki M, Korsmeyer SJ, McKinnon PJ. Atm and Bax cooperate in ionizing radiation-induced apoptosis in the central nervous system. Proc Natl Acad Sci. 2000;97:889–894. doi: 10.1073/pnas.97.2.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Schatz DG. Alternative ends for a familiar story? Curr Biol. 1999;9:R251–R253. doi: 10.1016/s0960-9822(99)80156-0. [DOI] [PubMed] [Google Scholar]
- Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, Chaudhuri J, Horner J, DePinho RA, Alt FW. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000;404:897–900. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- Gatei M, Young D, Cerosaletti KM, Desai-Mehta A, Spring K, Kozlov S, Lavin MF, Gatti RA, Concannon P, Khanna K. ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat Genet. 2000;25:115–119. doi: 10.1038/75508. [DOI] [PubMed] [Google Scholar]
- Gilmore EC, Nowakowski RS, Caviness VS, Jr, Herrup K. Cell birth, cell death, cell diversity and DNA breaks: How do they all fit together? Trends Neurosci. 2000;23:100–105. doi: 10.1016/s0166-2236(99)01503-9. [DOI] [PubMed] [Google Scholar]
- Gu Y, Sekiguchi J, Gao Y, Dikkes P, Frank K, Ferguson D, Hasty P, Chun J, Alt FW. Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proc Natl Acad Sci. 2000;97:2668–2673. doi: 10.1073/pnas.97.6.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science. 1998;280:1089–1091. doi: 10.1126/science.280.5366.1089. [DOI] [PubMed] [Google Scholar]
- Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- Jeggo PA. Identification of genes involved in repair of DNA double-strand breaks in mammalian cells. Radiat Res. 1998;150:S80–S91. [PubMed] [Google Scholar]
- Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, Taya Y, Gabrielli B, Chan D, Lees-Miller SP, et al. ATM associates with and phosphorylates p53: Mapping the region of interaction. Nat Genet. 1998;20:398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- Lim DS, Kim ST, Xu B, Maser RS, Lin J, Petrini JH, Kastan MB. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- Midgley CA, Fisher CJ, Bartek J, Vojtesek B, Lane D, Barnes DM. Analysis of p53 expression in human tumours: An antibody raised against human p53 expressed in Escherichia coli. J Cell Sci. 1992;101:183–189. doi: 10.1242/jcs.101.1.183. [DOI] [PubMed] [Google Scholar]
- Robins P, Lindahl T. DNA ligase IV from HeLa cell nuclei. J Biol Chem. 1996;271:24257–24261. doi: 10.1074/jbc.271.39.24257. [DOI] [PubMed] [Google Scholar]
- Rolig RL, McKinnon PJ. Linking DNA damage and neurodegeneration. Trends Neurosci. 2000;23:417–424. doi: 10.1016/s0166-2236(00)01625-8. [DOI] [PubMed] [Google Scholar]
- Roth DB, Gellert M. New guardians of the genome. Nature. 2000;404:823–825. doi: 10.1038/35009180. [DOI] [PubMed] [Google Scholar]
- Rotman G, Shiloh Y. ATM: From gene to function. Hum Mol Genet. 1998;7:1555–1563. doi: 10.1093/hmg/7.10.1555. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Schar P, Herrmann G, Daly G, Lindahl T. A newly identified DNA ligase of Saccharomyces cerevisiae involved in RAD52-independent repair of DNA double-strand breaks. Genes & Dev. 1997;11:1912–1924. doi: 10.1101/gad.11.15.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick RP, Boder E. Ataxia-telangiectasia. In: Vinken P, Bruyn G, Klawans H, editors. Handbook of clinical neurology. New York: Elsevier; 1991. pp. 347–423. [Google Scholar]
- Sekiguchi J, Gao Y, Gu Y, Frank KM, Sun Y, Chaudhuri J, Zhu C, Cheng HL, Manis JP, Ferguson DO, et al. Non-homologous end-joining proteins are required for V(D)J recombination, normal growth and neurogenesis. Cold Spring Harbor Symp Quant Biol. 1999;64:169–181. doi: 10.1101/sqb.1999.64.169. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Roth KA, Sayers RO, Shindler KS, Wong AM, Fritz LC, Tomaselli KJ. In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J. 2000;19:1397–1404. doi: 10.1093/emboj/19.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Ranganathan V, Weisman DS, Heine WF, Ciccone DN, T.B. O'Neill TB, Crick KE, Pierce KA, Lane WS, Rathbun G, et al. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature. 2000;405:477–482. doi: 10.1038/35013089. [DOI] [PubMed] [Google Scholar]
- Zhao S, Weng YC, Yuan SS, Lin YT, Hsu HC, Lin SC, Gerbino E, Song MH, Zdzienicka MZ, Gatti RA, et al. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature. 2000;405:473–477. doi: 10.1038/35013083. [DOI] [PubMed] [Google Scholar]