

Abstract
Far1p is a bifunctional protein that is required to arrest the cell cycle and to establish cell polarity during yeast mating. Far1p is localized predominantly in the nucleus but accumulates in the cytoplasm in cells exposed to pheromones. Here we show that Far1p functions in both subcellular compartments: nuclear Far1p is required to arrest the cell cycle, whereas cytoplasmic Far1p is involved in the establishment of cell polarity. The subcellular localization of Far1p is regulated by two mechanisms: (1) Far1p contains a functional bipartite nuclear localization signal (NLS), and (2) Far1p is exported from the nucleus by Msn5p/Ste21p, a member of the exportin family. Cells deleted for Msn5p/Ste21p failed to export Far1p in response to pheromones, whereas overexpression of Msn5p/Ste21p was sufficient to accumulate Far1p in the cytoplasm in the absence of pheromones. Msn5p/Ste21p was localized in the nucleus and interacted with Far1p in a manner dependent on GTP-bound Gsp1p. Two-hybrid analysis identified a small fragment within Far1p that is necessary and sufficient for binding to Msn5p/Ste21p, and is also required to export Far1p in vivo. Finally, similar to Δmsn5/ste21 strains, cells expressing a mutant Far1p, which can no longer be exported, exhibit a mating defect, but are able to arrest their cell cycle in response to pheromones. Taken together, our results suggest that nuclear export of Far1p by Msn5p/Ste21p coordinates the two separable functions of Far1p during mating.
Keywords: Export, cell cycle, mating, Msn5p/Ste21p, NLS
In eukaryotic cells, a double membrane system known as the nuclear envelope separates the nucleus from the cytoplasm, thus forming two distinct subcellular compartments. The localization of proteins to either the nucleus or the cytoplasm can confer regulation of their function (Görlich and Mattaj 1996; Nigg 1997). In many instances this regulation is imposed by the cell cycle stage or by activation of a signal transduction pathway in response to extracellular signals. For example, the transcription factor Swi5p is nuclear only during the G1 phase of the cell cycle (Moll et al. 1991), whereas multiple mitogen-activated protein kinases (MAPKs) translocate into the nucleus in response to growth factors (Feldherr and Akin 1994).
Yeast-mating pheromones regulate the subcellular localization of Far1p; Far1p is nuclear in the absence of pheromones but is found predominantly in the cytoplasm in cells treated with pheromones (Butty et al. 1998). Pheromones trigger a MAPK signal transduction pathway, which results in transcriptional activation of many genes, cell cycle arrest, and changes in cell polarity and morphology (Sprague and Thorner 1992; Leberer et al. 1997). These responses are initiated by binding of pheromones to a seven-transmembrane receptor, which is coupled to a heterotrimeric G protein. Gβγ then transduces the signal through its effectors Ste5p and Ste20p to a MAPK cascade composed of Ste11p, Ste7p, and Fus3p (Herskowitz 1995; Leeuw et al. 1998). Fus3p is thought to phosphorylate the transcriptional repressors Dig1p and Dig2p resulting in activation of the transcription factor Ste12p (Cook et al. 1996; Pi et al. 1997; Tedford et al. 1997) and regulates the activity of Far1p, which is required to arrest the cell cycle presumably by inhibiting cyclin-dependent kinases (Peter and Herskowitz 1994; Gartner et al. 1998). Far1p also functions as an adaptor that targets cytoplasmic polarity establishment proteins to the heterotrimeric G protein (Butty et al. 1998; Nern and Arkowitz 1999).
We are interested in understanding how the subcellular localization of Far1p is regulated in response to pheromones. Subcellular localization of proteins can be controlled by regulating import into or export from the nucleus. Two types of targeting signals mediate nuclear transport of proteins: nuclear localization signal (NLS) sequences promote nuclear import and generally consist of a cluster of basic amino acids (Kalderon et al. 1984; Dingwall and Laskey 1991). Second, nuclear export signal (NES) sequences promote export of proteins from the nucleus to the cytoplasm. A small leucine-rich sequence was the first NES identified, and was shown to be necessary and sufficient to export the human immunodeficient virus (HIV) Rev protein and protein kinase inhibitor (PKI), an inhibitor of cAMP-dependent protein kinase A (Nakielny and Dreyfuss 1997). Targeting signals are recognized by a family of soluble receptors, which are heterodimers consisting of importin α and importin β. The complex translocates with the cargo into or out of the nucleus and disassembles in the new compartment (Nigg 1997). In several cases, binding of import or export receptors to the targeting signal of the cargo is regulated by phosphorylation (Moll et al. 1991; Sidorova et al. 1995; Beals et al. 1997; DeVit et al. 1997; Kaffmann et al. 1998a).
Both genetic and biochemical experiments demonstrate a crucial role of the small GTPase Ran (or Gsp1p in yeast) in both nuclear import and export (Koepp and Silver 1996). Ran is found in the nucleus and cytoplasm, but because the regulators of Ran are localized differentially, the nucleus is thought to contain Ran predominantly in its GTP form, whereas Ran–GDP is predominantly cytoplasmic. This asymmetric distribution of GDP– and GTP–Ran controls assembly and disassembly of transport complexes. Binding of Ran–GTP to importin β family members involved in export promotes interaction with the NES-containing cargo in the nucleoplasm, whereas in the cytoplasm binding of importin β members involved in import allows translocation of NLS-containing proteins into the nucleus. In the nucleus, exchange of Ran–GDP to Ran–GTP by the exchange factor Rcc1 (or Rna1p in yeast) facilitates release of the cargo from importin β. Thus, the nucleotide state of Ran serves as a marker for nuclear and cytoplasmic compartments and imparts directionality to transport processes (Görlich et al. 1996; Izauralde et al. 1997).
Searches of the yeast genome database revealed at least 13 proteins with significant homology to importin β, and several family members have now been shown to function as import or export receptors. The uncharacterized receptors are thought to define additional import and export pathways. A major challenge is to identify targets of the multiple import and export receptors and to understand their role in controlling the subcellular localization of the target proteins in response to extracellular signals.
Here we show that the subcellular localization of Far1p is regulated by two pathways: (1) A bipartite NLS in the amino terminus of Far1p is necessary for efficient import into the nucleus in a cell cycle- and pheromone-independent manner; and (2) we have identified Msn5p/Ste21p as a nuclear export receptor for Far1p. Msn5p/Ste21p was localized in the nucleus and was required to export Far1p in response to pheromones. Msn5p/Ste21p bound Far1p through a novel NES sequence in a manner dependent on the Ran homolog Gsp1p. Accumulation of Far1p in the cytoplasm required activation of the pheromone response pathway but not transcriptional activation of Msn5p/Ste21p, suggesting that post-translational mechanisms regulate relocalization of Far1p in response to pheromones. Finally, our results suggest distinct roles for nuclear and cytoplasmic Far1p during yeast mating and may serve as a paradigm for how cell cycle arrest and polarity establishment are coordinated.
Results
Far1p contains a functional bipartite NLS
To identify sequences within Far1p required for nuclear localization, we determined the subcellular localization of fusions between portions of Far1p and the green fluorescent protein (GFP). We found that the amino-terminal domain of Far1p was required for nuclear localization; deletion of 50 amino-terminal amino acids resulted in a truncated Far1 protein, which was found predominantly in the cytoplasm (Fig. 1A). Importantly, a fusion protein between this amino-terminal domain of Far1p and GFP was found in the nucleus (Fig. 1C), demonstrating that these 50 amino acids of Far1p are not only required but also sufficient for nuclear localization. Closer examination of the sequence revealed two potential bipartite NLSs located between amino acids 11 and 30 (nls1) and 38 and 48 (nls2) of Far1p (Fig. 1B). To address the functional importance of these putative NLS sequences for the localization of Far1p, we mutated the lysine residues 29 and 30 (Far1p–K29A/K30A; nls1) and 41 and 42 (Far1p-R41A/K42A; nls2) to alanine residues. Whereas Far1p–nls2 was still localized predominantly in the nucleus, Far1p–nls1 was found largely in the cytoplasm, even in the absence of pheromones, demonstrating that NLS1 comprises a functional NLS (Fig. 1A). However, some remaining nuclear staining of Far1p–nls1 was still visible, suggesting that NLS2 may contribute to efficient nuclear localization of Far1p. Consistent with this notion, a Far1p mutant protein that has both putative NLS sequences inactivated (Far1p–nls1/nls2) was almost exclusively cytoplasmic (Fig. 1A). NLS1 and NLS2 may function as two separate bipartite NLS or they may be part of the same NLS sequence. Taken together, these results demonstrate that Far1p contains a functional bipartite NLS sequence in the amino-terminal 50 amino acids; NLS1 plays a major role, whereas NLS2 contributes to nuclear localization of Far1p to a minor extent (see also below).
Figure 1.



Far1p contains a functional NLS in the amino terminus. (A) Wild-type or various Far1p mutant proteins were expressed as fusions to GFP from the inducible GAL promoter and visualized by fluorescence microscopy. The introduced mutations are indicated schematically of left. Photographs show GFP fluorescence superimposed with phase contrast. (B) Schematic representation of Far1p and the 50 amino-terminal amino acids that are required for nuclear localization of Far1p. The mutated basic amino acids that are part of a bipartite nuclear localization signal are highlighted in bold. (C) The 50 amino-terminal amino acids of Far1p are sufficient to localize GFP in the nucleus. GFP fused to the 50 amino-terminal amino acids of Far1p (far11–50; left) or GFP alone (right) were expressed in cells and visualized as described above.
Msn5p/Ste21p functions as an exportin for Far1p
Next, we examined whether Far1p might be exported from the nucleus in response to pheromones. Nuclear export is mediated by exportins, which bind to target proteins and export them in an ATP- and Ran-dependent manner (Göhrlich and Mattaj 1996). Because cells lacking MSN5/STE21 exhibit reduced mating efficiency (Akada et al. 1996) and Msn5p/Ste21p displays significant sequence homology with exportins (Weis 1998) and interacts with Ran–GTP (Göhrlich et al. 1997), we tested whether Msn5p/Ste21p may be involved in localization of Far1p. Interestingly, we observed that Far1p remained exclusively nuclear in Δmsn5/ste21 cells treated with pheromones (Fig. 2A), suggesting that Msn5p/Ste21p is involved in exporting Far1p. Strikingly, although Far1p–nls1 was predominantly cytoplasmic in wild-type cells, it accumulated in the nucleus of cells deleted for STE21/MSN5 (Fig. 2B). Both defects were fully corrected by a plasmid expressing endogenous levels of Msn5p/Ste21p (Fig. 2B, right; data not shown), confirming that the defects are caused by lack of Msn5p/Ste21p. We conclude that Msn5p/Ste21p is required for cytoplasmic localization of Far1p in response to pheromones. These results further indicate that Far1p is very dynamic and shuttles between the nucleus and the cytoplasm even in the absence of pheromones.
Figure 2.

The exportin Msn5p/Ste21p is required to export Far1p. (A) Far1p–GFP was expressed in wild-type cells (right) or cells deleted for MSN5/STE21 (left) and treated (right row) or not treated (left row) with α-factor for 6 hr. Photographs show GFP fluorescence superimposed with phase contrast. Note that Far1p–GFP remains nuclear in Δmsn5/ste21 cells treated with α-factor. (B) Δmsn5/ste21 cells expressing Far1p–nls1–GFP were transformed with either an empty control plasmid (vector; left row) or a low copy number plasmid encoding MSN5/STE21 (pCEN-STE21; right row). Cells were analyzed as above. Note that Far1p–nls1–GFP is predominantly nuclear in Δmsn5/ste21.
To test whether expression of Msn5p/Ste21p is sufficient to accumulate Far1p in the cytoplasm, we overexpressed Msn5p/Ste21p from the inducible GAL promoter. Strikingly, Far1p relocalized efficiently under these conditions and was found predominantly in the cytoplasm (Fig. 3A). Addition of α-factor further increased the cytoplasmic pool of Far1p, suggesting that pheromones may activate export of Far1p by Msn5p/Ste21p or may inhibit its nuclear import. Moreover, no remaining nuclear staining of Far1p–nls1 was observed in cells overexpressing Msn5p/Ste21p (data not shown). In contrast, overexpression of Msn5p/Ste21p did not alter nuclear localization of Rap1p–Δ303–416–GFP (Fig. 3B), demonstrating that Msn5p/Ste21p is specific and does not perturb indiscriminately nuclear transport. Importantly, overexpression of Msn5p/Ste21p did not activate the pheromone response pathway as measured by the induction of the reporter FUS1–lacZ (Fig. 3D). Moreover, expression of Msn5p/Ste21p was able to trigger relocalization of Far1p in strains deleted for STE7 or STE20, demonstrating that activation of the mating pathway was not required to export Far1p under these conditions (Fig. 3C). Taken together, these results demonstrate that overexpression of Msn5p/Ste21p is sufficient to relocalize Far1p to the cytoplasm in a mating pheromone pathway-independent manner, and strongly suggest that Msn5p/Ste21p functions as an exportin for Far1p in vivo.
Figure 3.

Overexpression of Msn5p/Ste21p is sufficient to export Far1p. (A,B) Cells expressing Far1p–GFP (A) or for control Rap1p–Δ303–416-GFP (B) were transformed with a control plasmid (vector; left rows) or a plasmid allowing overexpression of Msn5p/Ste21p from the inducible GAL promoter (pRS–STE21; right rows). Cells were grown in the presence of galactose and analyzed by fluorescence microscopy. Where indicated α-factor was added for 3 hr. Note that complete cytoplasmic localization of Far1p requires overexpression of Msn5p/Ste21p and addition of α-factor. (C) Redistribution of Far1p to the cytoplasm does not require an intact mating pathway. Δste20 (top) or Δste7 cells (bottom) expressing Far1p–GFP were transformed with a control plasmid (vector; left row) or a plasmid allowing overexpression of Msn5p/Ste21p from the inducible GAL promoter (pRS–STE21; right row) and analyzed as described above. (D) The ability of cells carrying a control plasmid (pRS) or plasmids allowing overexpression of Msn5p/Ste21p (pRS–STE21) or Ste4p (pRS–STE4) to induce the reporter FUS1–lacZ was determined either in the absence (− αF) or presence of α-factor (+ αF). Bars show mean β-galactosidase activity ±s.d. for four independent transformants. Note that overexpression of Msn5p/Ste21p does not activate the pheromone response pathway.
Msn5p/Ste21p binds to Far1p in a manner dependent on Gsp1p–GTP
Because exportins have been shown to bind directly to their target proteins in a Ran–GTP-dependent manner, we tested whether Msn5p/Ste21p and Far1p are able to interact with each other by coimmunoprecipitation (Fig. 4) and two-hybrid assays (Table 1; Fig. 5). Myc-tagged Msn5p/Ste21p was immunoprecipitated with 9E10 antibodies from extracts prepared from wild-type cells (Fig. 4A,B, lanes 1–6,8,9) or cells harboring a temperature-sensitive GSP1 allele (lanes 10–12), which express Far1p from the inducible GAL promoter. The immunoprecipitates were then examined for the presence of Far1p by immunoblotting. Far1p readily coimmunoprecipitated with myc-tagged Msn5p/Ste21p (lanes 4,6), whereas no interaction was detected in cells expressing untagged Msn5p/Ste21p (lanes 2,8). Likewise, no interaction between Msn5p/Ste21p and Far1p could be detected when extracts were prepared from gsp1 cells shifted to 35°C for 3 hr (lanes 10,11), indicating that Gsp1p is required for binding of Far1p to Msn5p/Ste21p. Expression of a GTP-locked mutant form of Gsp1p (Gsp1p–G21V) restored binding of Far1p and Msn5p/Ste21p in gsp1-1 cells (Fig. 4B, lane 12), although the strain was still unable to grow at the restrictive temperature (data not shown). To further corraborate these results, we performed in vitro binding assays (Fig. 4C): Gsp1p–Myc expressed in Escherichia coli was immunoprecipitated, loaded with either GTPγS or GDP, and incubated with yeast extracts containing Msn5p/Ste21p, Far1p, or both proteins as indicated. Interestingly, Far1p bound Gsp1p–Myc only in the presence of Msn5p/Ste21p (cf. Lanes 14 and 18), suggesting that all three proteins together form a complex. Furthermore, Far1p interacted preferentially with Gsp1p–Myc in its GTP-bound form (cf. lanes 17 and 18). Taken together, these results suggest that Far1p and Msn5p/Ste21p interact with each other in a Gsp1p–GTP-dependent manner.
Figure 4.




Far1p interacts with Msn5p/Ste21p in a Gsp1p-GTP-dependent manner. (A,B) Msn5/Ste21p–myc was immunoprecipitated with 9E10 antibodies from extracts prepared from wild-type (K699; lanes 1–6, 8,9) or temperature-sensitive gsp1 cells (YSH80; lanes 10–12) expressing Far1p and either untagged Msn5p/Ste21p (lanes 1,2,8), Msn5p/Ste21p–mycn (lanes 3,4,9–12) or Msn5p/Ste21p–myc3 (lanes 5,6). The immunoprecipitates (IP; lanes 2, 4, 6, 8-12) and an aliquot of the supernatant before immunoprecipitation (SN; lanes 1,3,5,7) were analyzed for the presence of Far1p (top) or Msn5p/Ste21p–myc (bottom) by immunoblotting. Expression of Gsp1–GTP (YBM100; gsp1–G21V, lane 12) restored the ability of Msn5p/Ste21p to interact with Far1p. Cells lacking Far1p (YMP1054, lane 7) were included to control for the specificity of the antibodies. The arrowhead marks the position of Far1p (top) or Msn5p/Ste21p–myc (bottom); the asterisk points to the position of proteins that cross-reacts with the antibodies. Note that Far1p and Msn5p/Ste21p interact in a Gsp1p-dependent manner. (C) Sepharose beads containing immunoprecipitated Gsp1p–Myc expressed in E. coli and loaded with either GTPγS (lanes 14,16,18) or GDP (lanes 13,15,17) were incubated with yeast extracts containing as indicated Msn5p/Ste21p and Far1p expressed from the GAL promoter. Bound proteins were eluted and analyzed by immunoblotting for the presence of Far1p (top) and Gsp1p–Myc (bottom). Note that Far1p preferentially bound Gsp1p–GTP, but only in the presence of Msn5p/Ste21p. (D) Two-hybrid analysis of Msn5p/Ste21p and either wild-type (wt) or cytoplasmic Far1p mutant proteins in response to pheromones (times in minutes after addition of α-factor). The interaction was quantified as described and shown as percentage of Miller units relative to wild-type controls without pheromones. Note that the interaction between Msn5p/Ste21p and wild-type Far1p but not cytoplasmic mutant forms of Far1p decreases in an α-factor-dependent manner.
Table 1.
Two-hybrid analysis of the interaction between Msn5p/Ste21p and Far1p
| Activation domain fusion
|
Miller units ± s.d.
|
|---|---|
| WT(EGY48) | |
| Far1p(1–830) | 415 ± 117 |
| Far1p(353–830) | 0 |
| Far1p(1–389) | 877 ± 194 |
| Far1p(174–285) | 0 |
| Far1p(89–389) | 327 ± 110 |
| Far1p(189–389) | 255 ± 71 |
| Far1p(289–389) | 253 ± 88 |
| Far1pΔ(285–390) | 0 |
| far1Δ | |
| Far1p(1–389) | 958 ± 73 |
| Far1p(1–392) A26 A87 A114 A324 V341 A346 | 1044 ± 78 |
| Far1p(1–392) A26 A87 A114 A306 V341 A346 | 951 ± 126 |
| far1Δ fus3Δ | |
| Far1p(1–830) | 492 ± 18 |
| Far1p(1–389) | 801 ± 30 |
Full-length or various mutants of Far1p fused to an activation domain (AD) were tested for their ability to interact with full-length Msn5p/Ste21p fused to a DNA-binding domain by two-hybrid analysis. The numbers in parenthesis indicate the amino acids of Far1p fused to the AD, except in Far1pΔ(285–390), where they indicate the deleted amino acids. The mutations in the Fus3p phosphorylation sites are indicated in small letters (Gartner et al. 1998). Expression of the β-Gal reporter was quantified as described and shown as Miller units with standard deviations. The interaction was tested in either a wild-type strain (EGY48), a strain deleted for FAR1 (YMP290), or a strain deleted for both FAR1 and FUS3 (YMP291). Note that a small domain within Far1p (amino acids 289–389) is necessary and sufficient to bind Msn5p/Ste21p.
Figure 5.

Binding of Far1p and Msn5p/Ste21p is required for efficient mating but not cell cycle arrest. Cells expressing wild-type Far1p–GFP (Far1wt; top row) or mutant Far1p–GFP lacking various parts of the Msn5p/Ste21p binding domain (bottom rows) were transformed with a control plasmid (pRS; left row) or a plasmid expressing Msn5p/Ste21p from the inducible GAL promoter (pRS–STE21; right rows). Where indicated cells were treated with α-factor for 3 hr. Full-length or the various deletion mutants of Far1p are schematically represented on the left; deleted amino acids are indicated in parentheseis. The Far1p mutants were also tested for their ability to interact with Msn5p/Ste21p by two-hybrid assay; Miller units with standard deviations are shown. Cells deleted for FAR1 (YMP1054) transformed with low copy number plasmids expressing wild-type Far1p (top row) or the indicated Far1p mutant proteins (bottom rows) from the endogenous promoter were tested for their ability to arrest the cell cycle by halo assay, or for their ability to mate against the mating tester IH2625. (++) Wild-type mating; (+/−) strongly reduced mating; (−) sterile; (ND) not determined. Note that the Msn5p/Ste21p-binding domain of Far1p is required for nuclear export and efficient mating in vivo.
Far1p also interacted with Msn5p/Ste21p by two-hybrid assay (Table 1; Fig. 4D and 5). Deletion analysis of Far1p revealed that the domain of Far1p, which binds Msn5p/Ste21p, was located between amino acids 285 and 390 (Table 1). Interestingly, this domain overlaps with the binding site for Cdc28p–Cln2p (Peter et al. 1993; Gartner et al. 1998), suggesting that Msn5p/Ste21p and Cdc28p–Cln2p kinase might compete for binding to Far1p. Although this segment of Far1p does not contain a classic leucine-rich hydrophobic (NES) sequence, we found a motif that is conserved in Far1p from Candida albicans and is also present in Ste5p, suggesting that Far1p may use a novel type of NES. Consistent with this notion, we observed that Ste5p–GFP was exported after overexpression of Msn5p/Ste21p, and conversely Ste5p–GFP remained nuclear in Δmsn5/Δste21 cells treated with α-factor, suggesting that Ste5p is also a target of Msn5p/Ste21p (data not shown). To test whether this conserved motif is required to export Far1p in vivo we deleted the Msn5p/Ste21p-binding site on Far1p (Far1p–Δ285–390). As shown in Table 1, Far1p–Δ285–390 was unable to interact with Msn5p/Ste21p and importantly, both α-factor treatment and overexpression of Msn5p/Ste21p were unable to export Far1p–Δ285–390 from the nucleus (data not shown, see below). Thus, the ability of Far1p to bind to Msn5p/Ste21p correlates with the ability of Far1p to relocalize to the cytoplasm, suggesting that binding of Far1p to Msn5p/Ste21p is required to export Far1p in response to pheromones. However, this putative NES fused to GFP containing the NLS of Pho4p (Kaffmann et al. 1998a) was only able to induce nuclear export weakly even when Msn5p/Ste21p was overexpressed (data not shown), suggesting that this domain may not be sufficient to function as an export signal in vivo.
Binding of Far1p and Msn5p/Ste21p may not be regulated by pheromones
To test whether the interaction between Far1p and Msn5p/Ste21p is regulated by pheromones we performed coimmunoprecipitation and two-hybrid analysis in cells treated or not treated with α-factor. As shown in Figure 4D, the interaction between wild-type Far1p and Msn5p/Ste21p decreased in a time-dependent manner in cells exposed to pheromones with kinetics that mirror the cytoplasmic accumulation of Far1p. We interpret this result to indicate that pheromones do not increase the interaction between Far1p and Msn5p/Ste21p, and that cytoplasmic accumulation of Far1p reduces the transcriptional readout of the two-hybrid assay that occurs in the nucleus. Consistent with this explanation, the interaction between Msn5p/Ste21p and several cytoplasmic Far1p mutants lacking their nuclear localization signal was decreased to levels comparable to wild-type Far1p in pheromone-treated cells, and importantly, no further decrease was observed after pheromone treatment (Fig. 4D). Likewise, Far1p and Msn5p/Ste21p were able to coimmunoprecipitate with similar efficiency in cells treated or not treated with α-factor (data not shown), suggesting that phosphorylation of Far1p does not increase their interaction. In addition, although redistribution of Far1p in response to pheromones was dependent on Fus3p in vivo (M. Blondel and M. Peter, unpubl.), the interaction between Far1p and Msn5p/Ste21p as assayed by two-hybrid analysis was neither dependent on FUS3 (Table 1) nor on the sites on Far1p, which are phosphorylated by Fus3p in response to pheromones (Table 1; Gartner et al. 1998). Taken together, we conclude that the binding of Far1p and Msn5p/Ste21p may not be regulated by pheromones, although it remains possible that a weak effect could have been masked because the proteins were overexpressed. Therefore, nuclear export of Far1p may be constitutive or regulated by nuclear retention.
The requirements for binding of Far1p to Cdc28p–Clnp and the exportin Msn5p/Ste21p can be mutationally separated
Far1p–Δ285–390 remained in the nucleus in α-factor-treated cells (data not shown), but no longer interacts with Cdc28p–Clnp and therefore is unable to arrest the cell cycle in response to pheromones (Peter et al. 1993). To separate the cell cycle arrest and export functions we constructed several short deletion mutants within this domain (Fig. 5). Any Far1p deletion mutant that removed threonine 306 (T306) was unable to arrest the cell cycle as determined by halo assay (Fig. 5; data not shown), consistent with the result that phosphorylation of T306 by Fus3p regulates its binding to Cdc28p–Clnp (Peter et al. 1993; Gartner et al. 1998). In contrast, Far1p–Δ338–382, which lacks the carboxy-terminal half of this domain, was able to arrest efficiently the cell cycle, but failed to interact with Msn5p/Ste21p by two-hybrid assay and as a consequence was unable to exit from the nucleus in vivo (Fig. 5). These experiments demonstrate that the requirements on Far1p for interacting with Cdc28p–Clnp or Msn5p/Ste21p are mutationally separable. Importantly, cells expressing Far1p–Δ338–382 exhibited a bilateral mating defect, demonstrating that nuclear export of Far1p is needed for efficient mating, most likely for oriented cell polarity.
Msn5p/Ste21p is a nuclear protein that is not induced in response to α-factor
Because overexpression of Msn5p/Ste21p was sufficient to export Far1p from the nucleus, we tested whether pheromone may relocalize Far1p by increasing the levels of Msn5p/Ste21p. However, we found that the levels of Msn5p/Ste21p were not altered in response to pheromones (Fig. 6A), suggesting that post-translational modifications of either Far1p or Msn5p/Ste21p regulate export of Far1p in response to pheromones. To examine the subcellular localization of Msn5p/Ste21p, we epitope tagged Msn5p/Ste21p at its amino terminus with GFP and visualized the functional fusion protein by fluorescence microscopy. Consistent with Msn5p/Ste21p functioning as an exportin, the protein was found predominantly in the nucleus (Fig. 6B). We did not observe any differences in localization of Msn5p/Ste21p through the cell cycle or in cells exposed to α-factor (bottom). Similar results were also obtained if Msn5p/Ste21p was localized by indirect immunofluorescence microscopy using myc-tagged Msn5p/Ste21p (data not shown).
Figure 6.
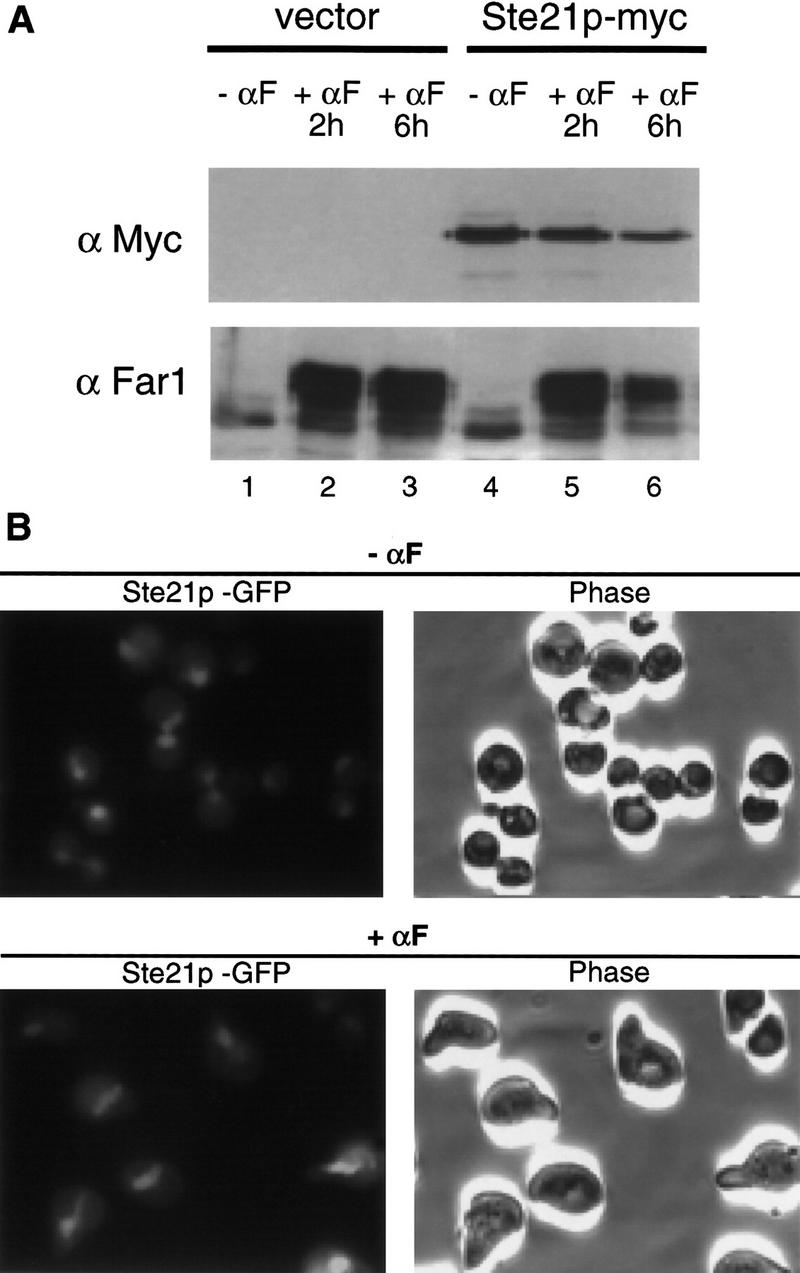
Msn5p/Ste21p is a nuclear protein that is not induced in response to pheromones. (A) Cells expressing epitope-tagged Msn5p/Ste21p (Ste21p–myc; lanes 4–6) or untagged Msn5p/Ste21p (vector; lanes 1–3) from the endogenous promoter were treated (lanes 2,3,5,6) or not treated with α-factor for the times indicated (lanes 1,4). Extracts were immunoblotted with 9E10 antibodies (top) or antibodies specific for Far1p (bottom). Note that in contrast to Far1p the levels of Msn5p/Ste21p do not increase in α-factor-treated cells. (B) Cells expressing Msn5p/Ste21p fused to GFP (Ste21p–GFP) from the endogenous promoter were treated (top) or not treated (bottom) with α-factor and analyzed by fluorescence microscopy (left). The corresponding phase contrast pictures are shown on the right. Note that Msn5p/Ste21p is nuclear in α-factor arrested cells and during all phases of the cell cycle.
Msn5p/Ste21p plays multiple roles during yeast mating
Next, we investigated the mating phenotype of cells deleted for STE21/MSN5. As observed previously (Akada et al. 1996), Δste21/msn5 cells mate with reduced efficiency (Fig. 7A, top); this mating defect is bilateral as the mating efficiency was decreased dramatically if both mating partners were deleted for STE21/MSN5 (Fig. 7A, bottom). Interestingly, Δmsn5/ste21 cells were able to induce the transcripts of FUS1 and FAR1 efficiently in response to pheromones (Fig. 7C), demonstrating that Msn5p/Ste21p is not required for signal transduction. Likewise, cells lacking Msn5p/Ste21p were able to arrest the cell cycle in response to pheromones in a FAR1-dependent manner (Fig. 7B), suggesting that cytoplasmic Far1p is not required for cell cycle arrest (see also below). However, Δmsn5/ste21 cells displayed a reduced ability to form mating projections (shmoos), and even after several hours in pheromones >80% of the cells remained unpolarized (Fig. 7D), suggesting that Msn5p/Ste21p is involved in exporting a protein involved in shmoo formation. This protein is unlikely to be Far1p, because Far1p is not needed to form shmoos (Valtz et al. 1995; M. Peter, unpubl.). Consistent with these observations, we found that overexpression of cytoplasmic Far1p was not sufficient to suppress the mating defect of Δmsn5/ste21 cells (data not shown). In addition, MSN5/STE21 and FAR1 were synthetic sterile (data not shown), supporting an additional role of Msn5p/Ste21p during mating. Thus, besides Far1p, Msn5p/Ste21p must export yet unknown targets involved in shmoo formation and perhaps other steps of mating.
Figure 7.
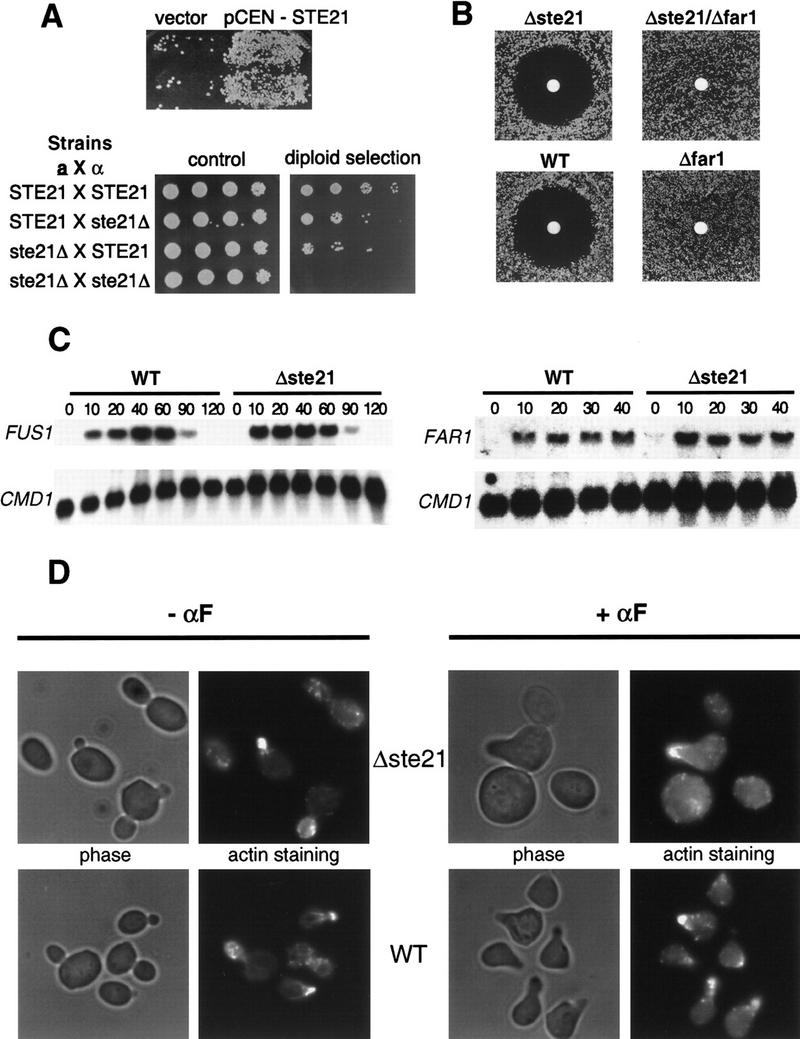
Cells lacking Msn5p/Ste21p exhibit a bilateral mating defect but are able to arrest their cell cycle. (A; top) Mating assay with the mating tester IH2625 and Δmsn5/ste21 cells (PAY20) transformed with a control plasmid (vector) or a plasmid carrying MSN5/STE21 (pCEN–STE21). (Bottom) Mating assays with either MATa or MATα wild-type or Δmsn5/ste21 cells as indicated. Serial dilutions of the mating reactions were spotted on control plates (left) or selective plates where only diploid cells are able to grow (right). Note that Δmsn5/ste21 cells exhibit a bilateral mating defect. (B) The indicated strains were analyzed by halo assay for their ability to arrest the cell cycle in response to α-factor. Note that cells lacking Msn5p/Ste21p efficiently arrest their cell cycle in a manner dependent on Far1p. The following strains were analyzed: Δste21 (PAY20); Δste21 Δfar1 (YMP1067); wt (K699) and Δfar1 (YMP1054). (C) Δmsn5/ste21 cells (PAY20) were able to efficiently induce mating-specific transcription of FAR1 (right) and FUS1 (left). Northern analysis of total RNA isolated from wild-type (K699; left lanes) or Δmsn5/ste21 cells (PAY20, right lanes) treated with α-factor for the times indicated (in minutes). A probe against CMD1 RNA was used as total RNA loading control (bottom). (D) The morphology of cells deleted for Msn5p/Ste21p (PAY20; top row) or wild-type cells (K699; bottom row) in the absence (left rows) or presence of α-factor (right rows) was analyzed by phase contrast microscopy (left). Actin distribution is directed toward the growing bud or shmoo tip as visualized after staining with rhodamine–phalloidin (right).
Nuclear and cytoplasmic Far1p play distinct roles during mating
Next, we examined the functional importance of Far1p localization for its cell cycle arrest and mating function. Far1p is required for oriented cell polarization during mating (Dorer et al. 1995; Valtz et al. 1995) and is thought to function as a cytoplasmic adaptor that recruits polarity establishment proteins to the site of extracellular signaling marked by the heterotrimeric G protein Gβγ (Bähler and Peter 1999). Several lines of evidence suggest that cytoplasmic Far1p is required for this polarization function. First, like Δmsn5/ste21 cells, Δfar1 cells expressing Far1p–Δ338–382 mutant protein, which can no longer be exported in response to pheromones, exhibited a mating defect (see Fig. 5). Second, wild-type cells expressing cytoplasmic Far1p–nls1 or cells overexpressing Msn5p/Ste21p mated with comparable or slightly increased efficiency (Fig. 8E, right column), suggesting that additional Far1p in the cytoplasm may improve the mating function of Far1p. Finally, ∼60% of haploid cells overexpressing cytoplasmic Far1p–nls1, but not wild-type Far1p, budded in a bipolar or random instead of axial pattern (Table 2; Chant 1996), suggesting that compartmentalization in the nucleus prevents Far1p from interfering with polarity establishment proteins in the absence of pheromones. Taken together, these results indicate that the polarization function of Far1p during mating requires cytoplasmic Far1p.
Figure 8.

Nuclear Far1p is required to arrest the cell cycle in response to pheromones, whereas cytoplasmic Far1p is necessary for efficient mating. (A) Wild-type cells (K699) were transformed with plasmids allowing overexpression of wild type or various mutant Far1p from the inducible GAL promoter as indicated, and grown on plates containing glucose (GLU; Far1p not expressed) or galactose (GAL; Far1p expressed). Extracts were prepared from cells and immunoblotted with specific antibodies against Far1p (top) or actin (bottom). Note that Far1p-22 is only toxic in the presence of a functional NLS. (B) Wild-type strains (K699, right) or strains deleted for MSN5/STE21 (Δste21; left) were transformed with the plasmids as indicated above and grown on media containing galactose. Note that in contrast to wild-type cells Far1–nls1/22 is toxic in Δmsn5/ste21 cells. (C) Cell deleted for FAR1 but harboring an empty plasmid (vector, left) or low copy number plasmids expressing Far1p-22 (middle) or Far1p–nls1/22 (right) from the endogenous promoter were analyzed by halo assay for their ability to arrest the cell cycle in response to pheromones. The circles contained 2, 10, and 20 μg of α-factor. (D) The minimal concentration of α-factor required to arrest cells expressing wild type or the indicated mutant Far1p from the endogenous promoter was determined by serial dilutions as described. Note that efficient cell cycle arrest in response to pheromones requires Far1p with a functional NLS. (E) Cells deleted for FAR1 but harboring an empty plasmid (vector) or low copy number plasmids expressing wild type or the indicated Far1p mutants from the endogenous promoter were analyzed for their ability to mate with wild-type (IH1793; left) or mating-reduced FAR1-C mating testers (IH2625, right). Note that cytoplasmic Far1p does not reduce the mating efficiency. Wild-type cells (K699) harboring an empty plasmid (vector; left patch) or a plasmid allowing overexpression of Msn5p/Ste21p (pRS–STE21; right patch) were mated to wild-type mating testers (IH1793). Note that overexpression of Msn5p/Ste21p slightly increases the mating efficiency.
Table 2.
Overexpression of cytoplasmic Far1p interferes with the budding pattern
| Plasmid
|
Budding pattern (%)
|
No. of cells counted
|
||
|---|---|---|---|---|
| axial
|
bipolar
|
random
|
||
| pRD53 | 76 | 23.5 | 0.5 | 200 |
| pRD53–Far1wt | 72 | 26.5 | 1.5 | 400 |
| pRD53–far1–nls1 | 34.5 | 54 | 11.5 | 400 |
| pRD53–far1–nls1,2 | 42 | 44 | 14 | 200 |
Cells deleted for FAR1 (YACB169) were tranformed with an empty control plasmid (pRD53) or plasmids allowing overexpression of wild-type Far1p, Far1p–nls1 or Far1p–nls1/2 from the inducible GAL promoter. Cells were grown in galactose at 30°C, the budding pattern was determined as described and presented as percent of total cells. Only cells with three or more bud scars were included in the analysis.
In contrast, nuclear Far1p appears to be required for cell cycle arrest in response to pheromones. We have shown above that Δste21/msn5 cells failed to export Far1p but were able to arrest efficiently their cell cycle in response to pheromones, suggesting that nuclear Far1p is sufficient for cell cycle arrest. Likewise, Far1p–Δ338–382 can no longer be exported in response to pheromones but is able to fully complement the cell cycle arrest defect of Δfar1 cells. Previously, we have found that overexpression of a nuclear, stable mutant form of Far1p (Far1p-22) arrests cells in the G1 phase of the cell cycle by inhibiting the Cdc28p–Clnp kinase (Henchoz et al. 1997). In contrast, cells overexpressing a cytoplasmic double mutant between Far1p-22 and NLS1 (Far1p–nls1/22) were viable and able to divide, although the Far1p–nls1/22 protein was expressed at similar or even higher levels than Far1p-22 (Fig. 8A). Interestingly, cytoplasmic Far1p was stable and no longer subjected to ubiquitin-mediated degradation (M. Blondel and M. Peter, unpubl.), explaining the increased steady-state levels of Far1p–nls1 and Far1p–nls1/22. However, overexpression of Far1p–nls1/22 is toxic in cells deleted for MSN5/STE21 (Fig. 8B), which accumulated Far1p–nls1/22 in the nucleus (data not shown) because of a defect in the export system, demonstrating that Far1p–nls1/22 is functional and able to arrest the cell cycle when localized in the nucleus. Consistent with these findings, Δfar1 cells expressing Far1p–nls1/22 from the endogeneous promoter exhibited an eightfold reduced ability to arrest the cell cycle in response to pheromones compared to Δfar1 cells expressing Far1p-22 (Fig. 8C,D). Taken together, we conclude that nuclear Far1p is required to arrest the cell cycle in response to pheromones.
Discussion
The subcellular localization of Far1p is controlled by activation of the pheromone response pathway during mating; in the absence of pheromones, Far1p is predominantly nuclear, whereas in the presence of pheromones Far1p accumulates in the cytoplasm. We show here that the localization of Far1p is under the control of both nuclear import and export pathways and have identified Msn5p/Ste21p as a specific exportin for Far1p. Export of Far1p in response to pheromones requires Msn5p/Ste21p, which interacts with Far1p in a manner dependent on Gsp1p–GTP. Our results further suggest that nuclear Far1p is required for its cell cycle arrest function, whereas cytoplasmic Far1p interacts with Gβγ and the polarity establishment proteins to orient cell polarity during mating.
The subcellular localization of Far1p is mediated by nuclear import and export
In the absence of pheromones Far1p is localized predominantly in the nucleus (Henchoz et al. 1997). As shown here nuclear localization depends on at least one bipartite NLS, which is located in the amino-terminal domain of Far1p. Far1p lacking this region or harboring point mutations in the basic residues within the bipartite NLS accumulates predominantly in the cytoplasm. NLSs are recognized by import receptors that target specific proteins to the nucleus. For example, Kap123p was shown to be involved in the import of the ribosomal protein L25 (Rout et al. 1997; Schlenstedt et al. 1997), and Pse1p is required to import Pho4p in response to phosphate starvation (Kaffmann et al. 1998a). At present, we do not know which import receptor is required for nuclear localization of Far1p.
In contrast, several lines of evidence strongly suggest that Msn5p/Ste21p functions as an exportin for Far1p, although we cannot rigorously exclude the possibility that Msn5p/Ste21p may indirectly inhibit nuclear import of Far1p. First, cytoplasmic accumulation of Far1p in response to pheromones was abolished in cells deleted for Msn5p/Ste21p. Second, Far1p accumulated in the cytoplasm of cells overexpressing Msn5p/Ste21p even in the absence of pheromones, and third, a cytoplasmic mutant of Far1p that harbors mutations in the major NLS becomes predominantly nuclear when expressed in Δmsn5/ste21 cells. Fourth, Msn5p/Ste21p is localized in the nucleus, consistent with its proposed function as an exportin for Far1p. Fifth, Far1p coimmunoprecipitated with Msn5p/Ste21p in a manner dependent on GTP-bound Gsp1p and also interacted with Msn5p/Ste21p by two-hybrid analysis. Finally, a small segment of Far1p, which is necessary and sufficient to interact with Msn5p/Ste21p, is required to export Far1p in vivo in response to pheromones. Signals have been defined that target proteins from the nucleus to the cytoplasm (Nakielny and Dreyfuss 1997; Weis 1998). The NES contained in the HIV Rev protein and PKI, an inhibitor of cAMP-dependent protein kinase A, is a small sequence rich in leucine residues, which is bound by the export receptor Crm1p/Xpo1p and Ran–GTP in the nucleus. The interaction domain between Far1p and Msn5p/Ste21p does not contain an obvious leucine-rich NES, suggesting that Far1p uses a novel type of NES. However, the Msn5p/Ste21p-binding domain of Far1p was not able to export efficiently a nuclear GFP fusion protein, suggesting that this motif may not be sufficient to mediate Msn5p/Ste21p-dependent export in vivo. Nevertheless, together with the recently identified Msn5p/Ste21p targets Msn2p, Msn4p, and Pho4p it may be possible to deduce a consensus sequence, which may facilitate the identification of additional proteins exported by Msn5p/Ste21p.
Regulation of the subcellular localization of Far1p by pheromones
The subcellular localization of Far1p is surprisingly dynamic and is regulated by both nuclear import and export. In the absence of α-factor, Far1p is predominantly nuclear and import appears to overcome export. Addition of pheromones shifts the equilibrium and Far1p accumulates in the cytoplasm. It is not understood how pheromones alter this balance, but available results suggest a role for the MAPK Fus3p (M. Blondel and M. Peter, unpubl.). It is possible that Fus3p inhibits nuclear import, increases nuclear export, or both. Fus3p may also regulate the activity of nuclear or cytoplasmic docking sites that may retain Far1p in the nucleus in the absence of pheromones, or in the cytoplasm in the presence of pheromones (Hood and Silver 1999; Kaffmann and O’Shea 1999).
Nuclear import is regulated in many cases by phosphorylation of sites within or close to the NLS (Moll et al. 1991; Kaffmann et al. 1998a). Two consensus phosphorylation sites for Cdc28p kinase or MAPK are present within the major NLS of Far1p and it has been shown that this region of Far1p is heavily phosphorylated by Cdc28p kinases in vivo and in vitro (McKinney and Cross 1995). Thus, phosphorylation of these sites by Fus3p may prevent nuclear import of Far1p in response to pheromones. Alternatively, because Fus3p is required to inhibit Cdc28p–Clnp activity in response to pheromones (Elion et al. 1990; Peter et al. 1993), it is possible that phosphorylation of Far1p by Cdc28p–Clnp may be required for nuclear import. However, we found that Far1p is localized in the nucleus of G1 cells arrested by depletion of the G1 cyclins, suggesting that nuclear import was not dependent on Cdc28p kinase activity (data not shown). In addition, nuclear localization of Far1p is independent of the cell cycle position (Henchoz et al. 1997) and did not require Fus3p or any other component of the mating pathway (data not shown). Finally, a fusion protein between the 50 amino-terminal amino acids of Far1p (containing the NLS and the putative phosphorylation sites) with GFP was localized efficiently in the nucleus of cells treated (data not shown) or not treated with α-factor (Fig. 1C); however, because we do not know whether this amino-terminal fragment of Far1p is phosphorylated efficiently in vivo, we cannot exclude the possibility that phosphorylation of these sites may inhibit nuclear import of full-length Far1p in response to pheromones.
It is clear that nuclear export of Far1p must occur even in the absence of pheromones, because Far1p–nls1 is nuclear in cells lacking Msn5p/Ste21p, whereas it is predominantly cytoplasmic in wild-type cells. Thus, a decrease in the rate of nuclear import can be compensated by decreasing the rate of nuclear export. Overexpression of Msn5p/Ste21p was sufficient to export Far1p in the absence of pheromones or a functional pheromone response pathway, suggesting that increased levels of Msn5p/Ste21p are able to shift the equilibrium. However, endogenous Msn5p/Ste21p levels were not altered in response to pheromones, indicating that post-translational mechanisms may regulate nuclear export of Far1p. Because Msn5p/Ste21p is required to export multiple proteins, not only during mating, but also in response to environmental conditions such as high phosphate levels, we favor a model where regulation of nuclear export occurs at the level of the substrate rather than at the level of the exportin or the export machinery. In support of this notion, recently it has been shown that Msn5p/Ste21p specifically interacts with phosphorylated Pho4p and that phosphorylation of Pho4p is required for its nuclear export in vivo (Kaffmann et al. 1998b). However, although Far1p is a substrate of Fus3p (Peter et al. 1993; Tyers and Futcher 1993; Kranz et al. 1994; Gartner et al. 1998), the interaction between Far1p and Msn5p/Ste21p as assayed by two-hybrid analysis was not increased in response to pheromones and was neither dependent on the presence of Fus3p nor on the pheromone-dependent phosphorylation of Far1p (Table 1). Thus, these results suggest that Fus3p may not regulate directly binding of Far1p with its exportin Msn5p/Ste21p. Further work is required to elucidate the mechanism of regulation of cytoplasmic accumulation of Far1p by pheromones.
Multiple roles of Msn5p/Ste21p during mating and response to various extracellular signals
Several proteins have now been shown to be targets of the exportin Msn5p/Ste21p: the transcription factor Pho4p is exported by Msn5p/Ste21p under high phosphate conditions (Kaffmann et al. 1998b), and Msn5p/Ste21p keeps Msn2p and Msn4p in the cytoplasm in the absence of stress conditions (Gorner et al. 1998; Alepuz et al. 1999). As shown here Msn5p/Ste21p exports Far1p in response to pheromones and cells lacking Msn5p/Ste21p exhibit a bilateral mating defect. The signal transduction and cell cycle arrest functions of Δmsn5/ste21 cells in response to pheromones are intact, but the cells exhibit a defect in projection formation and in orienting growth toward the mating partner. The latter defect is thought to result from a failure to export Far1p, which is necessary to target the polarity establishment proteins Bem1p, Cdc24p, and Cdc42p to the site of the incoming pheromone signal marked by Gβγ (Butty et al. 1998; Nern and Arkowitz 1999). However, cells lacking Far1p are able to form mating projections and thus Far1p cannot be the only Msn5p/Ste21p target that needs to be exported during mating, an observation that is supported by the synthetic sterility of cells lacking both Msn5p/Ste21p and Far1p. In addition, we found that overexpression of cytoplasmic Far1p was not sufficient to bypass the need for Msn5p/Ste21p during mating, although cytoplasmic Far1p increased the mating efficiency of wild-type cells. Interestingly expression of a membrane-bound version of Ste5p (Ste5p–CTM; Pryciak and Huntress 1998) partially restored shmoo formation in Δmsn5/ste21 cells (data not shown), suggesting that export of Ste5p may be necessary to form mating projections efficiently.
The function of Far1p is required in two subcellular compartments
Far1p is known to play two separable roles during yeast mating (Valtz et al. 1995): Far1p is required to arrest the cell cycle presumably through the inhibition of the Cdc28p–Clnp kinase (Peter and Herskowitz 1994; Gartner et al. 1998) and Far1p is necessary for oriented cell polarity by linking the polarity establishment proteins to Gβγ (Butty et al. 1998; Nern and Arkowitz 1999). Several lines of evidence suggest that the cell cycle arrest function of Far1p requires nuclear localization of Far1p. First, cells expressing Far1p–nls1 with reduced ability to enter into the nucleus exhibit a modest cell cycle arrest defect in response to pheromones. Second, a dominant Far1p, which when overexpressed arrests the cell cycle by inhibiting the Cdc28p–Clnp kinase (Henchoz et al. 1997), is no longer toxic if the dominant mutation is combined with a mutation that inactivates the NLS. Finally, cells unable to export Far1p either because they lack the exportin Msn5p/Ste21p or because they express a mutant Far1p deleted for its NES (Far1p–Δ338–382), are able to arrest efficiently the cell cycle in response to pheromones. In contrast, Δfar1 cells expressing Far1p–Δ338–382 exhibit a mating defect, although these cells are able to arrest and form normal mating projections. Similarly, Δmsn5/ste21 cells mate with reduced efficiency; we presume that this mating defect is partly due to the requirement of cytoplasmic Far1p to interact with Ste4p to perform its function as an adaptor for polarity establishment (Butty et al. 1998; Nern and Arkowitz 1999). Consistent with that notion, we found that cytoplasmic Far1p was sufficient for the mating function and even slightly increased the mating efficiency. In addition, overexpression of cytoplasmic Far1p interferes with the budding pattern of wild-type cells (Table 2), presumably through its interaction with polarity establishment proteins. It has been found previously that overexpression of a truncated Far1 protein lacking 50 amino acids of the amino terminus interfered with bud formation after release from α-factor arrest (McKinney and Cross 1995). Our results now show that this truncated Far1p protein lacks its major NLS and therefore, may interfere with the polarity establishment machinery during bud formation. Thus, these results suggest that sequestration in the nucleus might prevent Far1p from interacting with polarity establishment proteins in the absence of the physiological stimulus.
On the basis of these results we propose a model for how the cell cycle arrest and polarity establishment functions of Far1p may be coordinated (Fig. 9). In the absence of pheromones, low levels of Far1p are present because the expression of FAR1 is controlled at the transcriptional level by the pheromone pathway (Chang and Herskowitz 1990; Oehlen et al. 1996). Expressed Far1p is sequestered in an inactive form in the nucleus, where it is unable to interfere with the function of cytoplasmic polarity establishment proteins. Activation of the pheromone response pathway increases expression of Far1p. In addition, Far1p is phosphorylated by Fus3p, which enables Far1p to bind to the Cdc28p–Clnp kinase in the cell nucleus leading to cell cycle arrest (Peter et al. 1993; Gartner et al. 1998). Pheromones also trigger export of Far1p into the cytoplasm by a mechanism that requires the exportin Msn5p/Ste21p. Cytoplasmic Far1p interacts with Bem1p, Cdc24p, and Cdc42p and targets them to the activated heterotrimeric G protein to organize the actin cytoskeleton toward the incoming signal (Arkowitz 1999; Bähler and Peter 1999). Because Cdc28p–Clnp and Msn5p/Ste21p have at least partially overlapping binding sites it is possible that Msn5p/Ste21p and Cdc28p–Clnp compete for binding to Far1p. Such a mechanism may be important to coordinate the cell cycle arrest and polarity functions of Far1p (Fig. 9).
Figure 9.

A schematic representation of the Far1p functions. In the absence of pheromones Far1p is exclusively nuclear because of the presence of functional NLS sequences. Nuclear Far1p is inactive as a cyclin-dependent kinase inhibitor (CKI) because it needs to be phosphorylated by the MAPK Fus3p to be able to bind to the Cdc28p–Clnp kinase (Peter et al. 1993; Gartner et al. 1998). Nuclear Far1p interacts with Msn5p/Ste21p in a Gsp1p-dependent manner, which then transports Far1p into the cytoplasm. In the presence of pheromones the balance between import and export is shifted toward export presumably because of activation of Fus3p. Nuclear Far1p is required for cell cycle arrest, whereas cytoplasmic Far1p is needed for the establishment of oriented cell polarity.
Materials and methods
Yeast strains, genetic manipulations, and database searches
Yeast strains are described in Table 3. The genotypes of the yeast strains are W303, ade2-1, trp1-1, can1-100, leu2-3,112, his3-11,15, ura3, GAL+, psi+, ssd1-d2; A364a, trp1-289, leu2-3,112, his3-11,15, ura3-52, GAL+; and EG123: trp1-Δ99, leu2-Δ1, ura3-52, ade2-101, unless noted otherwise. Standard yeast growth conditions and genetic manipulations were used as described (Guthrie and Fink 1991). Yeast transformations were performed by lithium acetate procedure (Ito et al. 1983). Strains deleted for FAR1 marked with LEU2 or URA3 were constructed using plasmids pMT870 digested with PvuII, or pFC13 digested with NotI. Strains deleted for MSN5/STE21 were constructed using plasmids pLH64 digested with NotI and XhoI, and strains deleted for STE7 were constructed using the plasmid pSL2270 digested with PstI and XhoI (Pinten and Sprague 1994). STE20 was deleted using the kanR cassette as described in Longtine et al. (1998). Strain YBM100 was constructed by integration of GAL–gsp1(G21V) at the URA3 locus of YSH80 by digesting the plasmid pSH125-1 with StuI. Transformants were selected on SD–URA plates at 25°C and tested for their inability to grow on medium containing galactose and thermosensitivity at 37°C. Database searches were performed using the SGD (Stanford University) and the NCBI BLAST programs (National Institutes of Health).
Table 3.
Strains list
| Strain name
|
Relevant genotype
|
Background
|
Source
|
|---|---|---|---|
| K699 | MATa | W303 | Kim Nasmyth (IMP, Vienna, Austria) |
| YMP1054 | MATa far1::LEU2 | W303 | Henchoz et al. (1997) |
| PAY20 | MATa ste21::HIS3 | W303 | this study |
| YMP1067 | MATa ste21::HIS3 far1::LEU2 | W303 | this study |
| MJ 234 | MATa fus1::lacZ::URA3 | A364a | this study |
| GA682 | MATa/α RAP1Δ303–416-GFP–LEU2::rap1 | SK1 | Monika Tsai (ISREC) |
| YFD162 | MATa ste7::ADE2 bar1-1 far1Δ | W303 | Frank van Drogen (ISREC) |
| YFD163 | MATa ste20::kanR bar1-1 far1Δ | W303 | Frank van Drogen |
| YSH80 | MATa gsp1::hisG his3-1,15::gsp1ts(HIS3) | W303 | Shai Shaham |
| IH1793 | MATα lys1 | collection | |
| IH2625 | MATα far1c lys1 | collection | |
| YACB169 | MATa far1::LEU2 | EG123 | this study |
| LH90 | MATa ste21::TRP1 | JC-2B | this study |
| YMP290 | MATa far1Δ bar1-1 | W303 | Gustav Ammerer |
| YMP291 | MATa far1Δ fus3Δ bar1-1 | W303 | this study |
| YBM100 | MATa gsp1::hisG his3-1,15::gsp1ts(HIS3) GAL–gsp1(G21V)–URA3::ura3 | W303 | this study |
Pheromone response and mating assays
Pheromone response and mating assays were carried out as described (Valtz and Peter 1997). Mating assays were performed with both wild-type (IH1793) and orientation-defective far1-c mating testers (IH2625). Quantitative cell cycle arrest assays were performed in microtiter plates as described (Grishin et al. 1998). Each series uses twofold dilutions from well to well starting with 100 μg/ml α-factor; the last well contains no α-factor. To analyze the expression of Msn5p/Ste21p in response to pheromones, cells (LH90) harboring a plasmid-allowing expression of Msn5p/Ste21p from the endogenous promoter (pLH287) were grown in selective media to early log phase, at which time α-factor was added to 25 μg/ml final concentration (time 0). Aliquots were removed after the times indicated and the expression of Msn5p/Ste21p and Far1p was analyzed by immunoblotting as described. Induction of FAR1 or FUS1 mRNA in Δmsn5/ste21 (PAY20) or wild-type cells (K699) was determined by Northern analysis as described previously (Martinez-Pastor et al. 1996).
DNA manipulations
Plasmids are described in Table 4. Standard procedures were used for recombinant DNA manipulations (Sambrook et al. 1989; Ausubel et al. 1991). PCR reactions were performed with the Expand polymerase kit as recommended by the manufacturer (Boehringer Mannheim). Oligonucleotides were synthesized by Genset (France) and the sequences are available upon request. Mutations were introduced by PCR and the correct sequence confirmed by sequencing. Internal deletion mutants of FAR1 were constructed by PCR by introducing in frame BglII restriction sites. The XhoI–SphI fragment of FAR1 containing the various mutations was ligated into pTP68 (Henchoz et al. 1997) to express fusions to the GFP (Heim et al. 1995), into pBM18 for expression from the endogenous FAR1 promoter, and pTP62 (Henchoz et al. 1997) for expression from the inducible GAL promoter. For two-hybrid analysis FAR1 fragments were amplified by PCR, digested with NcoI and XhoI and subcloned into the two-hybrid vectors pEG203 or pJG4-6 as described previously (Butty et al. 1998). pEG203 and pJG4-6 are derivatives of pEG202 and pJG4-5, respectively (Gyuris et al. 1993), with an altered polylinker. The fragment encoding the Msn5p/Ste21p-binding domain of Far1p (amino acids 289–389) was amplified with specific primers introducing HindIII and XhoI restriction sites and ligated to a fragment encoding GFP in frame with the nuclear localization signal of Pho4p (Kaffmann et al. 1998a). The resulting fragment was then ligated into pRS416(ADH), resulting into plasmid pNP124. This plasmid allows expression of a PHO4(NLS)–GFP–NES(289–389) fusion protein from the constitutive ADH promoter. STE21 was amplified by PCR, digested with XhoI and ApaI or SalI and ligated into pEG203 (pBM41) for two-hybrid analysis, or pRS414(G) for expression from the inducible GAL promoter (pBM43). To introduce multiple copies of the 9E10 (myc) epitope at the amino terminus of Msn5p/Ste21p, the BamHI site in MSN5/STE21 was eliminated with a silent mutation, replaced with a new BamHI site at the ATG start codon and ligated with a myc3 cassette flanked by BamHI sites. pLH132 contains one copy, whereas pLH133 contains several copies of the myc cassette. The NotI–XhoI fragment from pLH132 or pLH133 was also cloned into pRS316 (Sikorski and Hieter 1989) yielding plasmid pLH287 and pRS424 yielding plasmids pBM55 and pBM56. The plasmid allowing expression of Msn5p/Ste21p–GFP was constructed as follows: the NotI–XhoI fragment from pLH133 was ligated into pRS316 to yield pLH287; pLH287 was then digested with BamHI to remove the myc3 cassette and replaced with a GFP fragment isolated from pPP356 (Pryciak and Huntress 1998), to yield plasmid pLH266. Both pLH133 and pLH266 are fully functional and able to complement the mating defect of cells deleted for MSN5/STE21 (data not shown).
Table 4.
Plasmids list
| Plasmids
|
Relevant characteristics
|
Source
|
|---|---|---|
| pTP68 | GAL FAR1wt–GFP URA3 CEN | Henchoz et al. (1997) |
| pTP487 | GAL Far1Δ50–GFP URA3 CEN | this study |
| pBM9 | GAL Far1–NLS1–GFP URA3 CEN | this study |
| pBM10 | GAL Far1–NLS2-GFP URA3 CEN | this study |
| pBM11 | GAL Far1–NLS1/2-GFP URA3 CEN | this study |
| pTP88 | GAL Far11–50–GFP URA3 CEN | this study |
| pTP419 | GAL GFP URA3 CEN | this study |
| pmsn5-D3 | Ste21::HIS3 | this study |
| pBM45 | STE21wt LEU2 CEN | this study |
| pBM43 | GAL STE21wt TRP1 CEN | this study |
| pLH287 | STE21-mycn URA3 CEN | this study |
| pSL2270 | Ste7::ADE2 | Printen and Sprague (1994) |
| pBM41 | pLG203 STE21wt | this study |
| ACB412 | pJG4-6 FAR1wt | Butty et al. (1998) |
| ACB418 | pJG4-6 Far1353–830 | Butty et al. (1998) |
| ACB413 | pJG4-6 Far11–389 | Butty et al. (1998) |
| ACB415 | pJG4-6 Far1174–285 | Butty et al. (1998) |
| pBC100 | pJG4-6 Far189–389 | this study |
| pBC101 | pJG4-6 Far1189–389 | this study |
| pBC102 | pJG4-6 Far1289–389 | this study |
| pTP577 | pJG4-6 Far1 Δ285–390 | this study |
| GA2162 | pJG4-6 Far1 A26 A87 A114 A324 V341 A346 | this study |
| GA2163 | pJG4-6 Far1 A26 A87 A114 A306 V341 A346 | this study |
| pLH132 | STE21–Myc3 TRP1 2μ | this study |
| pLH133 | STE21–Mycn TRP1 2μ | this study |
| pLH58 | STE21wt URA3 CEN | this study |
| pLH64 | STE21::TRP1 LEU2 2μ | this study |
| pBM56 | STE21–Mycn TRP1 2μ | this study |
| pBM55 | STE21–Myc3 TRP1 2μ | this study |
| ACB435 | GAL FAR1wt LEU2 CEN | Butty et al. (1998) |
| pSH125 | GAL Gsp1 (G21V) URA3 integrative | this study |
| pLH266 | STE21wt–GFP URA3 CEN | this study |
| pTP62 | GAL FAR1wt URA3 CEN | Henchoz et al. (1997) |
| pBM5 | GAL Far1 NLS1 URA3 CEN | this study |
| pBM8 | GAL Far1 NLS1/2 URA3 CEN | this study |
| pTP63 | GAL Far1-22 URA3 CEN | Henchoz et al. (1997) |
| pBM14 | GAL Far1 NLS1/22 URA3 CEN | this study |
| pBM18 | FAR1wt URA3 CEN | this study |
| pBM19 | Far1–22 URA3 CEN | this study |
| pBM20 | Far1–NLS1 URA3 CEN | this study |
| pBM29 | Far1–NLS1/22 URA3 CEN | this study |
| pLH59 | STE21wt URA3 2μ | this study |
| pBM58 | GAL Far1Δ285–308 GFP URA3 CEN | this study |
| pBM76 | GAL Far1Δ312–382 GFP URA3 CEN | this study |
| pBM77 | GAL Far1Δ338–382 GFP URA3 CEN | this study |
| pBM78 | GAL Far1Δ312–338 GFP URA3 CEN | this study |
| pBM79 | Far1Δ312–382 URA3 CEN | this study |
| pBM80 | Far1Δ338–382 URA3 CEN | this study |
| pBM81 | Far1Δ312–338 URA3 CEN | this study |
| pTP595 | pJG4-6 Far1 Δ312–382 | this study |
| pTP596 | pJG4-6 Far1 Δ338–382 | this study |
| pTP597 | pJG4-6 Far1 Δ312–338 | this study |
| pTP598 | pJG4-6 Far1 Δ304–308 | this study |
| pNP124 | ADH Pho4(NLS)–GFP–Far1(NES) URA3 CEN | this study |
| pTP594 | ADH Pho4(NLS)–GFP URA3 CEN | this study |
| EB0806 | pT7-Myc–GSP1 | Kaffmann et al. (1998b) |
Antibodies and Western blots
Standard procedures were used for yeast cell extract preparation and immuno blotting (Brown et al. 1997; Harlow and Lane 1988). Polyclonal anti-Far1p antibodies have been described previously (Henchoz et al. 1997) and 9E10 antibodies were obtained from the ISREC antibody facility. Antibodies specific for actin were purchased from Boehringer Mannheim and used as recommended by the manufacturer.
Coimmunoprecipitation and in vitro binding experiments
Wild-type (K699) or gsp1-1 cells (YSH80) were transformed with a multicopy plasmid expressing myc-tagged Msn5p/Ste21p from the endogenous promoter (pBM56 or pBM55) or for control an empty vector (pRS424) and a plasmid expressing Far1p from the inducible GAL promoter (ACB435; Butty et al. 1998). Cells were grown in selective media containing raffinose (2% final concentration) to early log phase, at which time galactose (2% final concentration) was added for 6 hr at 30°C (25°C for experiments with gsp1-1 cells followed by a shift at 35°C for 3 hr) to induce expression of Far1p. Cells were pelleted, resuspended in RIPA buffer [50 mm Tris (pH 7.5), 50 mm NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS] containing the protease inhibitors PMSF, aprotinin, leupeptin, and pepstatin (Complete, Boehringer Mannheim), and lysed with a one-shot cell disruptor (Constant Systems Ltd.) set to the maximum pressure (2.8 kBar). The soluble extract was then incubated for 2 hr at 4°C with 9E10 monoclonal antibodies and Sepharose beads coupled to protein G (Pharmacia). The beads were washed four times with RIPA buffer, bound proteins eluted with gel-sample buffer, and subjected to immunoblot analysis with polyclonal antibodies against Far1p and 9E10 antibodies to control for the presence of myc-tagged Msn5p/Ste21p.
In vitro binding assays were carried out as follows: Gsp1p–Myc was immunoprecipitated with 9E10 antibodies from extracts prepared from DH5α cells containing the pT7–MycGsp1 expression plasmid (EB0806). The immunoprecipitate was divided; One-half was incubated for 2 hr at 4°C with 2 mm GTPγS; the other half with 2 mm GDP in 10 mm Tris-HCl (pH 7.5) containing 20 mm EDTA and 2 mm DTT. The reaction was stopped by adding MgCl2 to a final concentration of 50 mm. Yeast extracts were prepared from ste21Δ far1Δ cells (YMP1067) transformed with control vectors, or plasmids expressing Far1p (CMP62; Henchoz et al. 1997) and Msn5p/Ste21p (pBM43) from the inducible GAL promoter. Cells were grown at 30°C to early log phase in selective media containing raffinose (2% final concentration), at which time galactose (2% final concentration) was added for 6 hr. Cells were lysed in phosphate-buffered saline [PBS: 137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4-7H2O, 1.4 mm KH2PO4 (pH 7.3)] containing the protease inhibitors PMSF, aprotinin, leupeptin, and pepstatin (Complete, Boehringer Mannheim) as described above and the extracts incubated for 2 hr at 4°C with Gsp1p–Myc-containing beads. The beads were washed four times with PBS, bound proteins eluted with gel-sample buffer, and analyzed by immunoblotting as described.
Two-hybrid assays
Two-hybrid assays were performed as described (Brown et al. 1997) in EGY48- or W303-based yeast strains using pEG202-based plasmids expressing LexA DNA-binding domain fusions (DBD), and pJG4-5-based plasmids containing fusions to the B42 transcriptional activation domain (AD) (Gyuris et al. 1993). Miller units are averages of at least six independent experiments (with independent colonies) with standard deviations. For experiments with pheromones, cells were grown in selective media containing galactose (2% final concentration) to early log phase, at which time the culture was divided and pheromone (25 μg/ml final concentration) was added to one half. After the times indicated, an aliquot was analyzed as described above. The values were expressed as percent Miller units compared to wild-type controls grown in the absence of pheromones; each time point represents the average of at least four independent experiments (with independent colonies).
Microscopy and budding assays
Yeast actin was visualized with rhodamine–phalloidin (Molecular Probes, Inc., Leiden, The Nctherlands). Briefly, cells were fixed with formaldehyde (3.7% final concentration) for 60 min, washed and stained for 20 min on ice with rhodamine–phalloidin (diluted 1:5 in methanol), washed three times with PBS, and viewed on a Zeiss Axiophot fluorescence microscope. At least 200 cells were counted for the morphological analysis. Proteins tagged with GFP were visualized using a Chroma GFPII filter (excitation 440–470 nm), photographed with a Photometrics CCD camera, and analyzed with Photoshop 4.0 software (Adobe). Photographs shown are overlays of phase contrast and fluorescence images. Cells expressing Far1p–GFP from the inducible GAL promoter were grown to early log phase at 25°C in selective media containing raffinose (2% final concentration), at which time galactose was added (2% final concentration) for 6 hr. Where indicated α-factor (25 μg/ml final concentration) was added during the last 3 hr. LH90 cells expressing Msn5p/Ste21p–GFP from the endogenous promoter (pLH58) were grown in selective media at 30°C to early log phase. Where indicated α-factor (25 μg/ml final concentration) was added for 2 hr.
The budding pattern of cells overexpressing wild-type or cytoplasmic mutant forms of Far1p was determined after staining bud scars with calcofluor as described (Guthrie and Fink 1991). Briefly, cells were grown in selective media containing raffinose (2% final concentration) to early log phase, at which time galactose (2% final concentration) was added; cells were kept in log phase for 36 hr by successive dilutions. Cells were stained with calcofluor and counted on a Zeiss Axiophot fluorescence microscope using a UV filter. Only cells with more than three bud scars were included in the analysis.
Acknowledgments
We thank members of the laboratory for helpful discussions, M. Funk, P. Pryciak, E. Elion, E. O’Shea, B. Catarin, E. Schwob, A.-C. Butty, F. van Drogen, and M. Tsai for plasmids and strains. K. Hoffmann kindly provided a detailed alignment of Msn5p/Ste21p with importins/exportins. S. Henchoz is acknowledged for help during early aspects of this work, T. Laroche and A. de Bruyn Kops for advice with the microscope, J.-M. Galan and A.-C. Butty for help with coimmunoprecipitation and budding pattern experiments, N. Perrinjaquet for excellent technical assistance, and B. Amati, V. Simanis, and R. Iggo for critical reading of the manuscript. M.B. is supported by Association pour La Recherche sur le Cancer (ARC) and an European Molecular Biology Organization (EMBO) long-term fellowship, and L.S.H. by an NIH and a Herbert W. Boyer postdoctoral fellowship. M.P. is supported by the Swiss National Science Foundation, the Swiss Cancer League, and a Helmut Horten Incentive award.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Matthias.Peter@esrec.unel.ch; FAX (41) 21-652-6933
References
- Akada R, Kallal L, Johnson DI, Kurjan J. Genetic relationships between the G-protein beta gamma complex, Ste5p, Ste20p and Cdc42p: Investigation of effector roles in the yeast pheromone response pathway. Genetics. 1996;143:103–117. doi: 10.1093/genetics/143.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alepuz, P.M., D. Matheos, K.W. Cunningham, and F. Estruch. 1999. The Saccharomyces cerevisiae RanGTP binding protein Msn5p is involved in different signal transduction pathways. Genetics (in press). [DOI] [PMC free article] [PubMed]
- Arkowitz RA. Responding to attraction: Chemotaxis and chemotropism in Dictyostelium and yeast. Trends Cell Biol. 1999;9:20–27. doi: 10.1016/s0962-8924(98)01412-3. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York, NY: Greene Publishing Associates and Wiley-Interscience; 1991. [Google Scholar]
- Bähler J, Peter M. Cell polarity in yeast. In: Drubin DG, editor. Frontiers in molecular biology: cell polarity. Oxford University Press; 1999. (In press). [Google Scholar]
- Beals CR, Clipstone NA, Ho SN, Crabtree GR. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes & Dev. 1997;11:824–834. doi: 10.1101/gad.11.7.824. [DOI] [PubMed] [Google Scholar]
- Brown JL, Jaquenoud M, Gulli MP, Chant J, Peter M. Novel Cdc42-binding proteins Gic1 and Gic2 control cell polarity in yeast. Genes & Dev. 1997;11:2972–2982. doi: 10.1101/gad.11.22.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butty A-C, Pryciak P, Huang L, Herskowitz I, Peter M. The role of Far1p in linking the heterotrimeric G-protein to polarity establishment proteins during yeast mating. Science. 1998;282:1511–1516. doi: 10.1126/science.282.5393.1511. [DOI] [PubMed] [Google Scholar]
- Chang F, Herskowitz I. Identification of a gene necessary for cell cycle arrest by a negative growth factor of yeast: FAR1 is an inhibitor of a G1 cyclin, CLN2. Cell. 1990;63:999–1011. doi: 10.1016/0092-8674(90)90503-7. [DOI] [PubMed] [Google Scholar]
- Chant J. Generation of cell polarity in yeast. Curr Opin Cell Biol. 1996;8:557–565. doi: 10.1016/s0955-0674(96)80035-4. [DOI] [PubMed] [Google Scholar]
- Cook JG, Bardwell L, Kron SJ, Thorner J. Two novel targets of the MAP kinase Kss1 are negative regulators of invasive growth in the yeast Saccharomyces cerevisiae. Genes & Dev. 1996;10:2831–2848. doi: 10.1101/gad.10.22.2831. [DOI] [PubMed] [Google Scholar]
- De Vit MJ, Waddle JA, Johnston M. Regulated nuclear translocation of the Mig1 glucose repressor. Mol Biol Cell. 1997;8:1603–1618. doi: 10.1091/mbc.8.8.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingwall C, Laskey RA. Nuclear targeting sequences—A consensus? Trends Biochem Sci. 1991;16:478–481. doi: 10.1016/0968-0004(91)90184-w. [DOI] [PubMed] [Google Scholar]
- Dorer R, Pryciak PM, Harwell LH. Saccharomyces cerevisiae cells execute a default pathway to select a mate in the absence of pheromone gradients. J Cell Biol. 1995;131:845–861. doi: 10.1083/jcb.131.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldherr CM, Akin D. Role of nuclear trafficking in regulating cellular activity. Int Rev Cytol. 1994;151:183–228. doi: 10.1016/s0074-7696(08)62633-9. [DOI] [PubMed] [Google Scholar]
- Elion EA, Grisafi PL, Fink GR. FUS3 encodes a cdc2+/CDC28-related kinase required for the transition from mitosis into conjugation. Cell. 1990;60:649–664. doi: 10.1016/0092-8674(90)90668-5. [DOI] [PubMed] [Google Scholar]
- Gartner A, Jovanovic A, Jeoung DI, Bourlat S, Cross FR, Ammerer G. Pheromone-dependent G1 cell cycle arrest requires Far1 phosphorylation, but may not involve inhibition of Cdc28-Cln kinase, in vivo. Mol Cell Biol. 1998;18:3681–3691. doi: 10.1128/mcb.18.7.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göhrlich D, Mattaj I. Nucleocytoplasmic transport. Science. 1996;271:1513–1518. doi: 10.1126/science.271.5255.1513. [DOI] [PubMed] [Google Scholar]
- Göhrlich D, Pante N, Kutay U, Aebi U, Bischoff FR. Identification of different roles for RanGDP and RanGTP in nuclear protein import. EMBO J. 1996;15:5584–5594. [PMC free article] [PubMed] [Google Scholar]
- Göhrlich D, Dabrowski M, Bischoff FR, Kutay U, Bork P, Hartmann E, Prehn S, Izaurralde E. A novel class of RanGTP binding proteins. J Cell Biol. 1997;14:65–80. doi: 10.1083/jcb.138.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorner W, Durchschlag E, Martinez-Pastor MT, Estruch F, Ammerer G, Hamilton B, Ruis H, Schuller C. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes & Dev. 1998;12:586–597. doi: 10.1101/gad.12.4.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin AV, Rothenberg M, Downs MA, Blumer KJ. Mot3, a Zn finger transcription factor that modulates gene expression and attenuates mating pheromone signaling in Saccharomyces cerevisiae. Genetics. 1998;149:879–892. doi: 10.1093/genetics/149.2.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to yeast genetics and molecular biology. San Diego, CA: Academic Press; 1991. [Google Scholar]
- Gyuris J, Golemis E, Chertkov H, Brent R. Cdi, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Henchoz S, Chi Y, Catarin B, Herskowitz I, Deshaies RJ, Peter M. Phosphorylation- and ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor Far1p in budding yeast. Genes & Dev. 1997;11:3046–3060. doi: 10.1101/gad.11.22.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz I. MAP kinase pathways in yeast: For mating and more. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- Hood JK, Silver PA. In or out? Regulating nuclear transposrt. Curr Opin Cell Biol. 1999;11:241–247. doi: 10.1016/s0955-0674(99)80032-5. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izauralde E, Kutay U, von Kobbe C, Mattaj IW, Göhrlich D. The asymmetric distribution of the constituents of the Ran system is essential for transport in and out of the nucleus. EMBO J. 1997;16:6535–6547. doi: 10.1093/emboj/16.21.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffmann, A. and E.K. O’Shea. 1999. Regulation of nuclear localization: A key to a door. Annu. Rev. Cell Dev. Biol. (in press). [DOI] [PubMed]
- Kaffmann A, Miller Rank N, O’Shea EK. Phosphorylation regulates association of the transcription factor Pho4p with its import receptor Pse1/Kap121. Genes & Dev. 1998a;12:2673–2683. doi: 10.1101/gad.12.17.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffmann A, Miller Rank N, O’Neill EM, Huang LS, O’Shea EK. Msn5 is a nuclear export receptor for the phosphorylated transcription factor Pho4. Nature. 1998b;396:482–486. doi: 10.1038/24898. [DOI] [PubMed] [Google Scholar]
- Kalderon D, Richardson WD, Markham AF, Smith AE. Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature. 1984;311:33–38. doi: 10.1038/311033a0. [DOI] [PubMed] [Google Scholar]
- Koepp D, Silver PA. A GTPase controlling nuclear trafficking: Running the right way or walking RANdomly? Cell. 1996;87:1–4. doi: 10.1016/s0092-8674(00)81315-x. [DOI] [PubMed] [Google Scholar]
- Kranz JE, Satterberg B, Elion EA. The MAP kinase Fus3 associates with and phosphorylates the upstream signaling component Ste5. Genes & Dev. 1994;8:313–327. doi: 10.1101/gad.8.3.313. [DOI] [PubMed] [Google Scholar]
- Leeuw T, Wu C, Schrag JD, Whiteway M, Thomas DY, Leberer E. Interaction of a G-protein beta subunit with a conserved sequence in Ste20/PAK family protein kinases. Nature. 1998;391:191–195. doi: 10.1038/34448. [DOI] [PubMed] [Google Scholar]
- Leberer E, Thomas DY, Whiteway M. Pheromone signalling and polarized morphogenesis in yeast. Curr Opin Genet Dev. 1997;7:59–66. doi: 10.1016/s0959-437x(97)80110-4. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;10:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Martinez-Pastor M T, Marchler G, Schuler C, Marchler-Bauer A, Ruis H, Estruch F. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress-response element (STRE) EMBO J. 1996;15:2227–2235. [PMC free article] [PubMed] [Google Scholar]
- McKinney JD, Cross FR. FAR1 and the G1 phase specificity of cell cycle arrest by mating factor in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:2509–2516. doi: 10.1128/mcb.15.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll T, Tebb G, Surano U, Robitsch H, Nasmyth K. The role of phosphorylation and the CDC28 protein kinase in cell cycle-regulated nuclear import of the S. cerevisiae transcription factor SWI5. Cell. 1991;66:743–758. doi: 10.1016/0092-8674(91)90118-i. [DOI] [PubMed] [Google Scholar]
- Nakielny S, Dreyfuss G. Nuclear export of proteins and RNAs. Curr Opin Cell Biol. 1997;9:420–429. doi: 10.1016/s0955-0674(97)80016-6. [DOI] [PubMed] [Google Scholar]
- Nern A, Arkowitz RA. A Cdc24p–Far1p–Gbetagamma protein complex required for yeast orientation during mating. J Cell Biol. 1999;144:1187–1202. doi: 10.1083/jcb.144.6.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA. Nucleocytoplasmic transport: Signals, mechanisms and regulation. Nature. 1997;386:779–787. doi: 10.1038/386779a0. [DOI] [PubMed] [Google Scholar]
- Oehlen LJ, McKinney JD, Cross FR. Ste12 and Mcm1 regulate cell cycle-dependent transcription of FAR1. Mol Cell Biol. 1996;16:2830–2837. doi: 10.1128/mcb.16.6.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M, Herskowitz I. Direct inhibition of the yeast cyclin-dependent kinase Cdc28-Cln by Far1. Science. 1994;265:1228–1231. doi: 10.1126/science.8066461. [DOI] [PubMed] [Google Scholar]
- Peter M, Gartner A, Horecka J, Ammerer G, Herskowitz I. FAR1 links the signal transduction pathway to the cell cycle machinery in yeast. Cell. 1993;73:747–760. doi: 10.1016/0092-8674(93)90254-n. [DOI] [PubMed] [Google Scholar]
- Pi H, Chien CT, Fields S. Transcriptional activation upon pheromone stimulation mediated by a small domain of Saccharomyces cerevisiae Ste12p. Mol Cell Biol. 1997;17:6410–6418. doi: 10.1128/mcb.17.11.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinten JA, Sprague GF. Protein–protein interactions in the yeast pheromone response pathway: Ste5p interacts with all members of the MAP kinase cascade. Genetics. 1994;138:609–619. doi: 10.1093/genetics/138.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryciak PM, Huntress FA. Membrane recruitment of the kinase cascade scaffold protein Ste5 by the Gβγ complex underlies activation of the yeast pheromone response pathway. Genes & Dev. 1998;12:2684–2697. doi: 10.1101/gad.12.17.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, Blobel G, Aitchison JD. A distinct nuclear import pathway used by ribosomal proteins. Cell. 1997;89:715–725. doi: 10.1016/s0092-8674(00)80254-8. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schlenstedt G, Smirnova E, Deane R, Solsbacher J, Kutay U, Görlich D, Ponstingl H, Bischoff FR. Yrb4, a yeast ran-GTP-binding protein involved in import of ribosomal protein L25 into the nucleus. EMBO J. 1997;16:6237–6249. doi: 10.1093/emboj/16.20.6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova JM, Miskesell GE, Breeden LL. Cell cycle-regulated phosphorylation of Swi6 controls its nuclear localization. Mol Biol Cell. 1995;6:1641–1658. doi: 10.1091/mbc.6.12.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague GF, Thorner JW. Pheromone response and signal transduction during the mating process of Saccharomyces cerevisiae. In: Jones EW, Pringle JR, Broach JR, editors. The molecular and cellular biology of the yeast Saccharomyces. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. pp. 657–744. [Google Scholar]
- Tedford K, Kim S, Sa D, Stevens K, Tyers M. Regulation of mating pheromone and invasive growth responses in yeast by two MAP kinase substrates. Curr Biol. 1997;7:228–238. doi: 10.1016/s0960-9822(06)00118-7. [DOI] [PubMed] [Google Scholar]
- Tyers M, Futcher B. Far1 and Fus3 link the mating pheromone signal transduction pathway to three G1-phase Cdc28 kinase complexes. Mol Cell Biol. 1993;13:5659–5669. doi: 10.1128/mcb.13.9.5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtz N, Peter M. Functional analysis of FAR1 in yeast. Methods Enzymol. 1997;283:350–365. doi: 10.1016/s0076-6879(97)83029-7. [DOI] [PubMed] [Google Scholar]
- Valtz N, Peter M, Herskowitz I. FAR1 is required for oriented polarization of yeast cells in response to mating pheromones. J Cell Biol. 1995;131:863–873. doi: 10.1083/jcb.131.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis K. Importins and exportins: How to get in and out of the nucleus. Trends Biochem Sci. 1998;23:185–189. doi: 10.1016/s0968-0004(98)01204-3. [DOI] [PubMed] [Google Scholar]