

Abstract
The relationship between cerebellar dysfunction, motor symptoms, and neuronal loss in the inherited ataxias, including the polyglutamine disease spinocerebellar ataxia type 3 (SCA3), remains poorly understood. We demonstrate that before neurodegeneration, Purkinje neurons in a mouse model of SCA3 exhibit increased intrinsic excitability resulting in depolarization block and the loss of the ability to sustain spontaneous repetitive firing. These alterations in intrinsic firing are associated with increased inactivation of voltage-activated potassium currents. Administration of an activator of calcium-activated potassium channels, SKA-31, partially corrects abnormal Purkinje cell firing and improves motor function in SCA3 mice. Finally, expression of the disease protein, ataxin-3, in transfected cells increases the inactivation of Kv3.1 channels and shifts the activation of Kv1.2 channels to more depolarized potentials. Our results suggest that in SCA3, early Purkinje neuron dysfunction is associated with altered physiology of voltage-activated potassium channels. We further suggest that the observed changes in Purkinje neuron physiology contribute to disease pathogenesis, underlie at least some motor symptoms, and represent a promising therapeutic target in SCA3.
Introduction
Spinocerebellar ataxia type 3 (SCA3) is the most common dominantly inherited ataxia and one of nine neurodegenerative disorders and six hereditary ataxias caused by abnormal polyglutamine (polyQ) expansions. As in other polyQ disorders, the disease protein in SCA3 causes selective neuronal degeneration despite being widely expressed in the nervous system. Symptoms in SCA3 primarily reflect neuropathological changes in the brainstem and cerebellum. Intriguingly, many affected neuronal populations in SCA3, including cerebellar Purkinje neurons (Munoz et al., 2002) and neurons of the deep cerebellar nuclei (Riess et al., 2008), exhibit autonomous pacemaker firing (Aizenman and Linden, 1999; Raman and Bean, 1999). This special physiological property may make these neurons particularly vulnerable to degeneration in SCA3 and other polyglutamine ataxias.
In neurodegenerative ataxias, a prolonged period of neuronal dysfunction likely precedes neuronal death. Both the nature of this neuronal dysfunction and its causal relationship to motor symptoms, however, remain poorly understood. Defining early physiologic changes in SCA3 and other SCAs is important because they may represent proximal events in disease pathogenesis. For example, expression of the SCA3 disease protein, polyglutamine-expanded ataxin-3, in neural cells has been shown to reduce the resting membrane potential and shift the activation curve of delayed rectifier potassium currents well before the onset of ultrastructural morphological changes (Jeub et al., 2006). Changes in physiology also offer attractive therapeutic targets for symptomatic and preventive treatment of ataxia. In the ducky and tottering mouse models of ataxia, mutations in the P/Q calcium channel perturb pacemaker firing of Purkinje neurons, which can be corrected pharmacologically with an activator of calcium-activated potassium channels, thereby restoring normal motor function (Walter et al., 2006). Likewise, pharmacologic reduction of intracellular calcium with dantrolene improves motor coordination and reduces neuronal loss in SCA2 and SCA3 transgenic mice (Chen et al., 2008; Liu et al., 2009). Despite these promising leads, no proven symptomatic or preventive therapies currently exist for any degenerative ataxia.
Here, using a transgenic mouse model of SCA3 that expresses the full human ATXN3 disease gene, we investigate whether expanded polyglutamine-mediated changes in neuronal function that precede cell death might explain motor impairment in SCA3. Our results in SCA3 mice establish that altered Purkinje cell firing is a prominent early manifestation of disease that occurs before degeneration and show that correcting this aberrant physiology through activation of calcium-activated potassium channels is a potential route to therapy. Our findings underscore the point that early electrophysiological defects in neuronal function may be a target for therapeutic intervention in degenerative polyglutamine disorders.
Materials and Methods
Mice
All animal procedures were approved by the University of Michigan Committee on the Use and Care of Animals. Genotyping was performed on DNA isolated from tail biopsy at the time of weaning as described previously (Cemal et al., 2002). These mice were generated and maintained in a C57BL/6 background strain and have been maintained by us on the same background since 2006. Mice of either sex were used in all the experiments. In SCA3 transgenic mice, hemizygosity/ homozygosity was determined by quantitative real-time PCR by amplification of a fragment of the ATXN3 transgene using the pair of primers 5′GAATATTTTAGCCCTGTGGAATT and 5′AGTTTAAAATCAGTACCTGTAAAAACG and normalizing for a genomic fragment of the mouse Hprt1 gene amplified with primers 5′GTGCAACCATTGCCCTAAGT and 5′CAGCCAGCATCTCAGGTGTA.
Western blotting
Membrane-enriched fractions of cerebellum were prepared following rapid dissection of cerebella in ice-cold PBS. Cerebella were homogenized in a detergent-free buffer containing 50 mm Tris-HCl, 10 mm EGTA, and protease inhibitors (Roche). Following low speed centrifugation at 1500 × g, samples were centrifuged at 38,000 rpm in a Beckman SW41 rotor (Beckman Coulter). The supernatant was discarded, and the membrane pellet was resuspended in fresh buffer. For immunoblotting, samples were resuspended in Laemmli buffer, electrophoresed, blotted, and probed with the appropriate Kv channel antibody, followed by HRP-labeled goat anti-mouse IgG (Jackson ImmunoResearch Laboratories) and chemiluminescent detection (Pierce Chemical). Kv channel monoclonal antibodies were obtained from the UC Davis/NIH NeuroMab facility.
Immunohistochemistry
Mice were anesthetized with ketamine/xylazine and perfused transcardially with 0.9% normal saline followed by 4% paraformaldehyde. Brains were removed and postfixed in 4% paraformaldehyde overnight, immersed in 30% sucrose in PBS, and sectioned on a sledge microtome. Free-floating 40 μm parasagittal sections (6- to 8-week-old mice) or 30 μm coronal sections (79- to 82-week-old mice) were processed for immunohistochemistry. Purkinje cells were labeled with 1:1000 anti-calbindin antibody (C9848; Sigma) and a secondary goat anti-mouse Alexa Fluor 568 antibody (Invitrogen). Sections were imaged using a FV500 Olympus confocal microscope and single-plane images were obtained from three midline cerebellar sections from each mouse. Measurements of molecular layer thickness were made for each section, 100 μm from the depth of the primary fissure, as described previously (Duvick et al., 2010). Purkinje neuron cell counts were obtained as the total number of neurons in the region of the folium within 100 μm of the depth of the primary fissure in three sections from each mouse.
Phenotype analysis
Motor coordination and balance were evaluated by assessing the ability of mice to traverse a square 5-mm-wide, 41-cm-long Plexiglas balance beam to reach an enclosed black platform (Heng et al., 2007). Time to traverse the beam was recorded for each trial with a 20 s maximum cutoff, and falls were scored as 20 s. Mice were trained to cross the beam for two trials on each of 3 consecutive days. Data were reported as the mean of two trials on the fourth day. Gait abnormalities were quantified from the footprint pattern of the mice. Forepaws and hindpaws of the mice were painted with different colors, and the gait of the animals was obtained through their walk across an 83-cm-long corridor (7 cm wide by 11 cm high) lined with paper into the escape area. Stride length, base length, front base width between left and right paws, and forepaw/hindpaw overlap were determined. Mice were weighed on a digital balance periodically. Littermate mice with the different genotypes were evaluated at 6, 12, and 24 weeks of age (±1 week).
Trial of SKA-31
Ten SCA3 tg/tg and 10 wild-type mice between 6 and 9 months of age were used for the trial. Mice were randomly assigned to drug and placebo groups. Mice were trained on the balance beam before the initiation of the trial to saturate effects of learning. Mice were coaxed gently to proceed walking if they paused on the beam. The trial of SKA-31 was performed over 4 consecutive days. On the first 2 d, a baseline performance was established to confirm that the mice had attained their peak performance and that there was no significant variability from one day to the next. On the subsequent 2 d, mice were administered either SKA-31 or the vehicle by intraperitoneal injection. SKA-31 was administered at a dose of 10 mg/kg dissolved in peanut oil as a vehicle, reconstituted on the morning of the trial. The injections were staggered so that all mice were tested exactly 1.5 h after administration of SKA-31 or vehicle. Mice were tested in triplicate with a 15 s interval between trials. The investigator scoring the performance of mice was blinded to the phenotype as well as the treatment groups. Following a washout period of 3–5 d, the groups were crossed over, so that animals that had received SKA-31 now received vehicle, and vice versa. The performance of animals in each group between the trials was pooled, and data were analyzed with a repeated-measures ANOVA.
Electrophysiology
Preparation of brain slices for electrophysiological recordings.
Mice (6–8 weeks old) were anesthetized by isofluorane inhalation and decapitated, and the brains were chilled in an ice-cold solution containing the following (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 1 NaH2PO4, 0.5 CaCl2, 7 MgCl2, 75 sucrose, and 10 glucose, bubbled with 5% CO2/95% O2. Parasagittal cerebellar slices (300 μm thick) were cut using a vibratome. Slices were incubated at 33°C in artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 3.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 10 glucose, bubbled with 5% CO2 + 95% O2 (carbogen) for 45 min. Slices were then placed in an unheated recording chamber and perfused continuously with carbogen-bubbled ACSF at 2–3 ml/min.
Whole-cell recordings.
Purkinje neurons were identified for whole-cell recordings in parasagittal cerebellar slices prepared from 6- to 8-week-old mice. Borosilicate glass patch pipettes (with resistances of 4–5 MΩ) were filled with internal recording solution containing the following (in mm): 119 K gluconate, 2 Na gluconate, 6 NaCl, 2 MgCl2, 10 EGTA, 10 HEPES, 14 Tris-phosphocreatine, 4 MgATP, and 0.3 Tris-GTP. The calcium buffering in the pipette solution was chosen to closely approximate the physiological milieu of Purkinje neurons. Others have noted the high endogenous calcium buffering in Purkinje cells (Fierro and Llano, 1996), an order of magnitude higher than other neurons. It has been reported previously that Purkinje cell firing frequency and spike morphology do not differ at EGTA concentrations between 0.9 and 9 mm (Khaliq et al., 2003). We also performed recordings with 0.9 mm EGTA in the internal solution, and the amplitude of the afterhyperpolarization potential (AHP) and the amount of current required for depolarization block were similar with 0.9 and 10 mm EGTA (four wild-type and five SCA3 tg/− neurons). Whole-cell recordings were made in ACSF 1–5 h after slice preparation using an Axopatch 200B amplifier, Digidata 1440A interface, and pClamp-10 software (MDS Analytical Technologies). Voltage data were acquired in the fast current-clamp mode of the amplifier and filtered at 2 kHz. The fast current-clamp mode is necessary to reduce distortion of action potentials observed when patch-clamp amplifiers are used in current-clamp mode (Magistretti et al., 1996). We could obtain stable recordings without oscillations in fast current-clamp mode with electrode resistances above 3 MΩ, as has been reported previously (Swensen and Bean, 2003). Series resistance was monitored but not compensated; cells were rejected if the series resistance exceeded 20 MΩ. Total cell capacitance was calculated from measurement of the area under current transients evoked from a 10 mV depolarizing step from −80 mV. Input resistance was calculated from the change in the leak current from an applied 10 mV voltage step from −80 mV. Data were digitized at 100 kHz. Voltage traces were corrected for a 10 mV liquid junction potential.
Somatic patches.
Patch pipettes were wrapped with Parafilm (Pechiney Plastic Packaging) to reduce pipette capacitance. Somatic outside-out patches were excised from Purkinje neurons following the establishment of the whole-cell patch-clamp configuration. The pipette solution and external solution were identical to those used for whole-cell voltage recordings. Patches were rejected if they did not reseal immediately to >1 GΩ. Series resistance was compensated at >85%. Leak was compensated by linear extrapolation of the current evoked by a step depolarization from −80 to −70 mV (in some cases, a P/−4 leak correction protocol was used). Under our recording conditions, patches lasted for a few minutes, enabling 15–20 sweeps with a 10 s intersweep interval.
Nucleated patches.
Excised outside-out nucleated patches to record voltage-activated sodium currents were generated using a pipette solution containing the following (in mm): 120 CsF, 8 NaCl, 10 HEPES, 4.5 tetraethyl ammonium (TEA)-Cl, 10 EGTA, and 2 MgCl2. Following recovery of the slice in ACSF as described above, slices were placed in a recording chamber that was perfused continuously with an external solution bubbled with carbogen and containing the following (in mm): 100 NaCl, 26 NaHCO3, 19.5 TEA-Cl, 3 KCl, 2 MgCl2, 2 CaCl2, 2 BaCl2, 0.1 CdCl2, 4 4-aminopyridine, and 11 glucose. Divalent ions were added from stock solutions following the establishment of a stable pH in the presence of carbogen to prevent precipitation. Series resistance was compensated by 50–70%, leak was compensated using a P/−4 subtraction protocol, and five sweeps were averaged for each applied voltage. Traces were corrected for a 5 mV liquid junction potential.
Dissociated Purkinje neurons.
Dissociated Purkinje neurons were generated using a modification of a previously described protocol (Raman and Bean, 1999). Briefly, the cerebellums were removed from 14- to 18-d-old mice. Brains were chilled in ice-cold dissociation solution, minced, and placed in 10 ml of Proteinase XIII (Sigma-Aldrich) at 37°C for 7 min, with oxygen blown over the surface. The tissue was then placed in dissociation solution containing 1 mg/ml BSA and trypsin inhibitor (Sigma-Aldrich). The tissue was washed in a modified Tyrode's solution containing 50 NaCl, 156 sucrose, 4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose, pH 7.4, and plated on laminin/polylysine-coated coverslips (Becton Dickinson). Recordings were performed in the same low-sodium modified Tyrode's solution to improve the quality of voltage clamp. The internal solution contained 119 K gluconate, 2 Na gluconate, 6 NaCl, 2 MgCl2, 0.9 EGTA, 10 HEPES, 14 Tris-phosphocreatine, 4 MgATP, and 0.3 tris-GTP. Whole-cell recordings were made between 1 and 3 h after dissociation using an Axopatch 200B amplifier, Digidata 1440A interface, and pClamp-10 software (MDS Analytical Technologies). Series resistance was <10 MΩ and compensated by 50–70%.
Kv channel cell lines.
Fibroblast cell lines stably expressing Kv3.1b or Kv1.2 were maintained as described previously (Grissmer et al., 1994). Cells were transfected with previously described GFP-tagged ataxin-3 constructs (Chai et al., 2002) with Lipofectamine LTX (Invitrogen) using the manufacturer's recommended protocol.
Channel biophysics.
To analyze voltage dependence of ion channel activation, patches were subjected to depolarizing voltage steps from a negative holding potential (−80 mV for Kv currents and −100 mV for voltage-activated sodium currents) to test voltages (−80 +10N mV, N = 1–13 for 100 ms for Kv currents; −100 +5N mV, N = 1–21 for 30 ms for voltage-activated sodium currents). The voltage dependence of activation was determined by calculating normalized peak conductance values from the peak current at each membrane voltage and fitting a Boltzmann function to the data using the formula f(Vm) = 1/(1+e[V1/2(act) − Vm]/k), where f(Vm) is the fraction of open channels, V1/2(act) is the voltage of half-maximal activation, and k is the slope factor.
Kv channel inactivation was measured as the fraction of available peak current remaining at the end of the 100 ms depolarizing step at each voltage. The fractional inactivation was fit to a single exponential.
Deactivation was determined by first opening the channels with a 15 ms conditioning step to 40 mV and then repolarizing the membrane to different potentials. In both wild-type and SCA3 neurons, deactivation at −50 mV, a voltage where no current activates, could be fit to a single exponential with a decay time constant of ∼2 ms. With a two-exponential fit, the fast time constant was 1.9 ms, and the slow time constant was 18 ms. The current amplitudes corresponding to each of the time constants was determined from the fit, and the ratio of the amplitude coefficients for the slow to the fast current decay was 0.04, indicating that 96% of the Kv current in our somatic patches was rapidly deactivating. The R2 was ≥0.97 for both the single and double exponential fit.
The voltage dependence of sodium channel inactivation was measured in nucleated patches using a protocol in which membrane voltage was stepped from −100 mV to preconditioning test voltages for 125 ms, followed by further depolarization to 0 mV. The peak sodium current at 0 mV measures the fraction of channels still available. The plot of the percentage of channels available versus membrane voltage was fit to the Boltzmann equation with the formula f(Vm) = 1/(1+e[V1/2(inact) − Vm]/k), where V1/2(inact) is the voltage of half inactivation, and k is the slope factor.
The slowly inactivating component of the sodium current was measured as the average residual sodium current measured 25–30 ms following depolarization from −45 to −30 mV.
Statistics
Statistical significance was assessed by either an unpaired Student's t test or a Fisher's exact test. A paired Student's t test was used to determine the effect of pharmacologic agents on firing properties. Ion channel biophysical properties were analyzed by a two-way repeated-measures ANOVA. Values of p < 0.05 were considered significant. Data are expressed as mean ± SEM unless specified otherwise. Data were analyzed using SigmaPlot (Systat Software), GraphPad Prism, and Excel (Microsoft).
Results
Motor dysfunction before neurodgeneration in SCA3 mice is associated with loss of repetitive firing of Purkinje neurons
For our studies we used the yeast artificial chromosome (YAC) SCA3 Q84 transgenic mouse (Cemal et al., 2002) (hereafter referred to as SCA3 mice). SCA3 mice express the full human mutant ATXN3 gene in its normal tissue distribution, including throughout the CNS. Mice hemizygous for the transgene (SCA3 tg/− mice) are smaller than littermate controls and display mild gait abnormalities at 6–8 weeks (Fig. 1A–C), as initially reported (Cemal et al., 2002). Calbindin immunostaining revealed no significant reduction in Purkinje neuron number or change in molecular layer thickness in 8-week-old SCA3 tg/− mice, indicating that the motor deficits in SCA3 tg/− mice precede overt neuropathologic cerebellar changes (Fig. 1D,E,H). The neurodegeneration in the cerebellum was not fully quantified in the original description of this mouse model (Cemal et al., 2002). SCA3 tg/− mice develop only mild Purkinje neuron loss at 80 weeks of age (Fig. 1G). Whereas SCA3 tg/− mice do not develop a motor deficit on the balance beam by 6 months of age (Fig. 1C), homozygous SCA3 mice (SCA3 tg/tg) expressing double the dose of mutant ataxin-3 have a prominent early motor deficit on the balance beam (Fig. 1C) and show significant reduction in the thickness of the molecular layer by 8 weeks (Fig. 1D,E), before any loss of Purkinje neurons (Purkinje neuron count was 20.7 ± 2.6). This reduction in the thickness of the cerebellar molecular layer may represent either a defect in development or an early degenerative change. Consistent with the more severe phenotype associated with the double dose of mutant ataxin-3, SCA3 tg/tg mice die prematurely, with a mean survival of 42.7 ± 2.8 weeks (n = 28 mice).
Figure 1.

Motor dysfunction precedes Purkinje neuron degeneration in SCA3 mice. A, Six- to 8-week-old SCA3 tg/− mice have a broader limb base than wild-type littermates (numbers of animals of each genotype analyzed are shown within the bars). *p < 0.05. B, SCA3 mice gain weight more slowly than wild-type littermates (n > 10 animals for each time point). C, SCA3 tg/tg mice display significant impairment of motor performance on the balance beam (p < 0.0001; n >12 animals for each group; legend is the same as in B). D, Calbindin immunostaining of cerebellar sections from 6- to 8-week-old wild-type (left), SCA3 tg/− (middle), and SCA3 tg/tg mice (right) shows significant thinning of the molecular layer in SCA3 tg/tg mice but not SCA3 tg/− mice compared to wild-type littermate controls. Scale bars, 100 μm. These data are summarized in E (n = 3 mice per group; p = 0.08; NS, not significant). F, Loss of Purkinje neurons, particularly in the depth of the folia, is seen in 80-week-old SCA3 tg/− mice compared to litter mate controls. Scale bars, 100 μm. G, The age-dependent loss of Purkinje neurons in SCA3 tg/− mice is not accompanied by a change in molecular layer thickness (H).
Because SCA3 tg/− mice most closely model the genetics of human SCA3 and manifest early gait abnormalities before morphological changes, we determined whether functional changes in cerebellar Purkinje neurons might explain the mild gait changes in SCA3 tg/− mice. Purkinje neurons normally exhibit spontaneous repetitive firing even in the absence of synaptic input (Raman and Bean, 1999). In recordings from cerebellar slices, Purkinje neurons from 6- to 8-week-old wild-type littermate controls uniformly exhibited normal repetitive firing (Fig. 2A,B). In contrast, less than half of Purkinje neurons in SCA3 tg/− mice exhibited tonic repetitive firing (Fig. 2A,C). Most of the remaining SCA3 tg/− neurons were silent, with a depolarized membrane potential (Fig. 2A,D), though a few exhibited repetitive bursts of action potentials (n = 4; Fig. 2A,E). SCA3 tg/tg mice also displayed this abnormal Purkinje neuron physiology. Only 38% of SCA3 tg/tg neurons exhibited normal tonic repetitive firing, compared to 47% of SCA3 tg/− neurons and 100% of wild-type littermate controls (p < 0.001 compared to wild-type controls; Fig. 2A).
Figure 2.
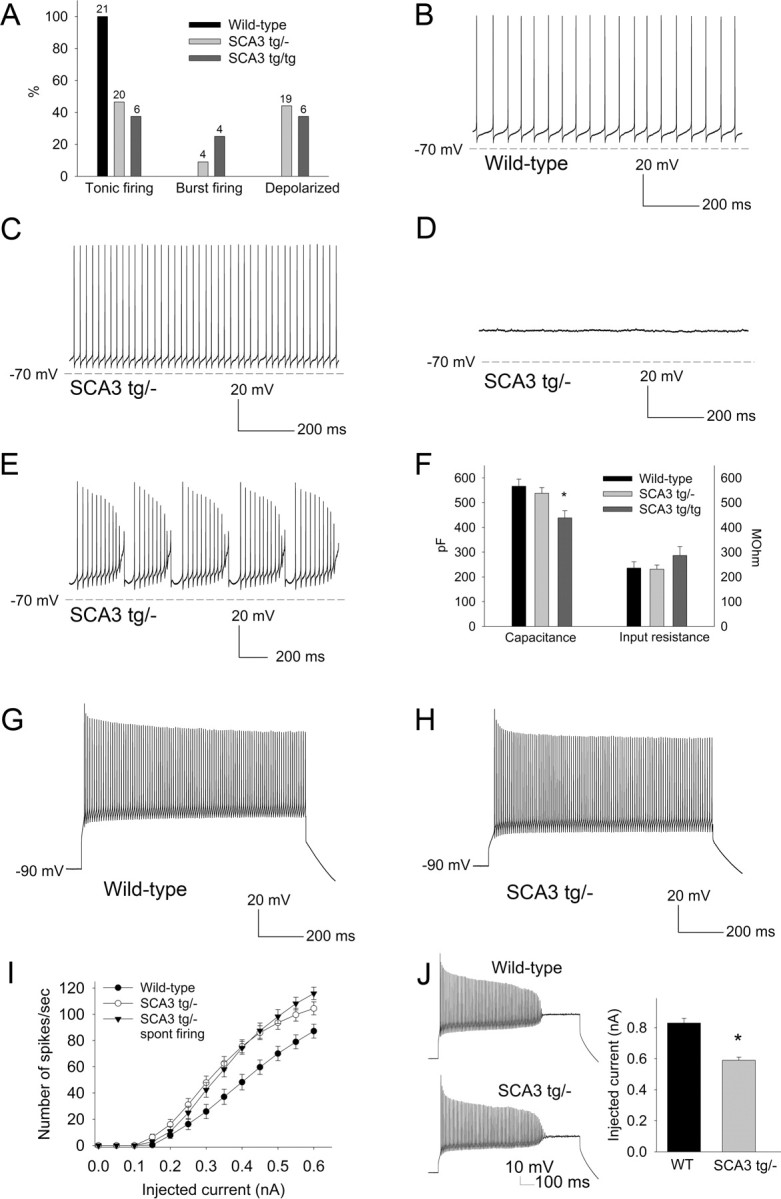
SCA3 Purkinje neurons fail to sustain spontaneous repetitive firing due to depolarization block. A, Summary of the proportion of wild-type SCA3 tg/− and SCA3 tg/tg neurons exhibiting each pattern of spontaneous firing. Numbers above the bars represent the number of neurons exhibiting each pattern of firing. B, Wild-type Purkinje neurons exhibit tonic repetitive firing (average frequency, 35.8 ± 3.1 Hz; n = 21). C–E, In contrast, SCA3 tg/− neurons exhibit tonic repetitive firing (average firing frequency, 46.3 ± 4.7 Hz; n = 20; p = 0.065; C), membrane depolarization with a loss of repetitive firing (membrane potential, −48.5 ± 1.4 mV; n = 19; D), or burst firing (E). F, The passive membrane properties of SCA3 tg/− Purkinje neurons are similar to wild-type neurons. SCA3 tg/tg neurons exhibit a lower capacitance, indicating a reduction in cell size and/or dendritic arborization (*p < 0.01). G, H, Wild-type neurons (G) and depolarized SCA3 tg/−neurons (H) can sustain similar high rates of repetitive firing in response to depolarizing current injection from a negative holding potential. I, Injection of depolarizing current evokes more spikes in SCA3 tg/− neurons than in wild-type neurons. SCA3 tg/− neurons that have retained repetitive firing at rest also display more spikes in response to similar amounts of injected current compared to wild-type neurons. J, A smaller amount of injected depolarizing current in SCA3 tg/− neurons is needed to induce depolarization block (*p < 0.001).
Like wild-type neurons, SCA3 Purkinje neurons were capable of sustaining high rates of repetitive firing in response to depolarizing current injections from a holding potential of −90 mV (Figs. 2G,H). Thus, the aberrant firing properties of SCA3 tg/− neurons are not due to a nonspecific loss of membrane integrity. For the same amount of injected current, SCA3 tg/− Purkinje neurons also supported a greater number of spikes than wild-type neurons (F = 8.4; p < 0.01; Fig. 2I). This was true even for SCA3 tg/− neurons that at baseline displayed apparently normal spontaneous repetitive firing (n = 20; F = 9.5; p < 0.01; Fig. 2I). These results indicate that SCA3 Purkinje neurons are hyperexcitable in response to injected current.
The apparent discrepancy between the lack of spontaneous firing in many SCA3 tg/− neurons and the increased spiking in response to depolarizing current injection could be explained by an net increase in a depolarizing conductance, as this would increase excitability yet also result in depolarization block of intrinsic firing. To test this possibility, we assessed the amount of injected current required to induce depolarization block of repetitive firing (Fig. 2J). Indeed, much less injected current was required to produce depolarization block in SCA3 tg/− Purkinje neurons still displaying repetitive firing (0.59 ± 0.02 nA; n = 36) than in neurons from littermate controls (0.83 ± 0.03 nA; n = 18; p < 0.001), confirming a net increase in a depolarizing conductance in SCA3 tg/− Purkinje neurons. A difference in series resistance cannot explain these findings: the series resistance was 17 ± 0.5 MΩ for SCA3 and 16.3 ± 1 MΩ for wild-type Purkinje neurons (p = 0.46). In SCA3 tg/tg neurons, depolarization block was induced by 0.59 ± 0.09 nA (n = 9) of depolarizing current injection, similar to hemizygous animals. Changes in passive membrane properties cannot explain this observed difference because the input resistance and total cell capacitance were similar between SCA3 tg/− and wild-type neurons (Fig. 2F). Consistent with the thinning of the molecular layer, Purkinje neurons of SCA3 tg/tg mice had a reduced cell capacitance suggesting a reduction in cell size and/or dendritic arborization. This was not, however, accompanied by a significant increase in the input resistance of the cell (Fig. 2F).
As an additional control, we assessed the firing properties of Purkinje neurons from transgenic mice expressing human ataxin-3 with a nonpathogenic repeat of 15 glutamine residues (Cemal et al., 2002). These mice have no reduction in the thickness of the cerebellar molecular layer at 6–8 weeks, and no deficits on the balance beam at 6 and 24 weeks (Fig. 3A–C). All tested Purkinje neurons from these mice exhibited normal spontaneous firing (n = 9; Fig. 3D), and in response to current injections underwent depolarization block to a similar amount of injected depolarizing current as did wild-type neurons (p = 0.44; Fig. 3E,F).
Figure 3.

Transgenic mice overexpressing nonpathogenic ataxin-3 exhibit normal Purkinje neuron physiology. A, Calbindin immunostaining of cerebellar sections from 6- to 8-week-old wild-type (left), and SCA3 15Q tg/− (right) mice show similar cerebellar molecular layer thickness. Scale bars: 100 μm. B, Summary of data in A. C, SCA3 15Q tg/− mice show no deficits on the balance beam. D, Purkinje neurons from SCA3 15Q tg/− mice show normal spontaneous firing. E, This same neuron required 1.2 nA of injected current to undergo depolarization block of repetitive firing. F, In response to depolarizing current injections, SCA3 15Q tg/− Purkinje neurons underwent depolarization block to current injections at 0.89 ± 0.09 nA, a result indistinguishable from wild-type neurons. The previously shown (Fig. 2) response of wild-type and SCA3 84Q tg/− neurons to injected current is shown for comparison on the left. *p < 0.001.
Depolarization block in SCA3 Purkinje neurons is due to a net increase in depolarizing conductances
The membrane depolarization normally occurring between spikes in Purkinje neurons results from tetrodotoxin (TTX)-sensitive resurgent sodium current (Raman and Bean, 1999). One possibility is that the depolarized membrane potential in SCA3 tg/− neurons stems from a net increase of depolarizing sodium conductances over repolarizing potassium conductances. To test this directly, we determined the true resting membrane potential in the presence of TTX. In wild-type neurons, the gradual application of TTX initially reduced firing frequency before eliminating spikes altogether (Fig. 4A), as described previously (Raman and Bean, 1999). The resultant resting membrane potential of TTX-treated wild-type neurons was similar to TTX-treated SCA3 tg/− neurons. This was also true for those SCA3 tg/− neurons that were already depolarized at baseline (mean membrane potential of −48.8 ± 1.7 mV; n = 3), in which slow perfusion of TTX resulted in brief sets of evoked action potentials followed by hyperpolarization to −62.6 ± 0.9 mV (Fig. 4B). To further confirm a net imbalance of ionic membrane conductances favoring membrane depolarization, we determined the effect of increasing net membrane hyperpolarization in Purkinje neurons from SCA3 tg/− mice by injecting a small amount of negative current. In six of nine depolarized SCA3 tg/− neurons, small amounts of hyperpolarizing current evoked spikes. As shown in Figure 4C, injecting negative current into SCA3 tg/− neurons recapitulated the full spectrum of firing properties, including burst firing and tonic repetitive firing.
Figure 4.

SCA3 Purkinje neurons display normal resting membrane potential. A, Wild-type Purkinje neurons fire spontaneously and repetitively (top). Slow perfusion of 500 nm TTX, an inhibitor of voltage-activated sodium channels, causes a reduction in firing frequency (middle) followed by cessation of firing (bottom; resting membrane potential of −62.6 ± 1.3 mV; n = 3). B, Depolarized SCA3 tg/− neurons (top) exhibit membrane hyperpolarization with brief runs of spikes (middle) following slow perfusion of 500 nm TTX. Following equilibration of the perfused TTX with the bath (bottom), the resting membrane potential of SCA3 neurons is similar to wild-type neurons (resting potential of −62.1 ± 0.7 mV; n = 5; p = 0.76). C, Injecting a depolarized SCA3 tg/− Purkinje neuron (top) with 0.03 nA of hyperpolarizing current causes burst firing (middle). Tonic repetitive firing in this neuron can be restored with injection of −0.2 nA of hyperpolarizing current (bottom).
The amount of depolarizing current needed to induce depolarization block in the presence of inhibitors of excitatory transmission [10 μm DNQX and 5 μm CPP ((±)-3-(2-carboxypiperazin-4-yl) propyl-1-phosphonic acid) to block ionotropic glutamate receptors] or of all synaptic transmission (50 μm picrotoxin to block GABAA receptors in addition to DNQX and CPP) was 0.63 ± 0.05 nA, virtually indistinguishable from results obtained in the absence of synaptic inhibition (n = 10; p = 0.51). Moreover, the spontaneous activity of SCA3 tg/− neurons was not influenced by the presence of synaptic inhibitors. The membrane potential of depolarized SCA3 tg/− Purkinje neurons (n = 7) remained unchanged in the presence of inhibitors of synaptic transmission, suggesting that the increased excitability of SCA3 tg/− neurons reflects a change in intrinsic membrane excitability.
Thus, the increased susceptibility of SCA3 tg/− neurons to depolarization block reflects a relative increase in TTX-sensitive depolarizing sodium currents over currents that repolarize the membrane. These results suggest that pharmacologically reducing depolarizing sodium currents or increasing membrane repolarization by negative current injection could in principle restore intrinsic firing.
Promoting membrane hyperpolarization with a potassium channel activator improves Purkinje neuron intrinsic firing and motor function in SCA3 mice
As shown previously, injection of hyperpolarizing current elicits bursts of spikes in depolarized SCA3 tg/− Purkinje neurons (Fig. 4). Thus, we surmised that increasing the effective hyperpolarizing potassium current might restore repetitive firing to SCA3 tg/− neurons even if an underlying defect in inactivation of Kv currents was not directly corrected. To test this, we applied a recently developed small-conductance calcium-activated potassium (SK) channel activator, SKA-31 (Sankaranarayanan et al., 2009) to SCA3 cerebellar slices. SK channels have been demonstrated previously to regulate the firing frequency of adult Purkinje neurons by contributing to hyperpolarization between spikes (Womack and Khodakhah, 2003). The effect of 10 μm SKA-31 was determined on Purkinje neurons from SCA3 tg/− or tg/tg mice. In three of four depolarized neurons, intrinsic firing was partially restored (Fig. 5A), and in two burst firing neurons, SKA-31 converted firing into a tonic repetitive pattern. SKA-31 also increased the injected current needed to achieve depolarization block from 0.55 to 0.73 nA (n = 8; p < 0.05; Fig. 5B). These results indicate that SK channel activators can promote membrane hyperpolarization in SCA3 Purkinje neurons.
Figure 5.

An activator of small-conductance calcium-activated potassium channels improves motor function in SCA3 mice. A, Following perfusion of 10 μm SKA-31, a depolarized SCA3 neuron cycles through a 30 s trimodal pattern of firing with initial small spikes followed by brief bursts of action potentials, membrane hyperpolarization, and subsequent depolarization and resumption of the cycle. SKA-31 was applied 10 min before recording the trace. B, SKA-31 ameliorates the depolarization block of repetitive firing in SCA3 tg/tg neurons. In treated Purkinje neurons from SCA3 tg/tg mouse, the same amount of current no longer induces depolarization block (left). Significantly more current injection is required to induce depolarization block (n = 3, *p < 0.05; right). C, Compared to vehicle (black circles), short-term administration of SKA-31 at a dose of 10 mg/kg (red circles) improves the motor performance of SCA3 tg/tg mice on the balance beam. SKA-31 has no effect on the performance of wild-type mice (SKA-31, red squares; placebo, black squares).
We next sought to determine whether the observed changes in cerebellar neuron physiology contribute to the motor phenotype in SCA3 mice. To test this, we used SCA3 tg/tg mice because they have a more robust motor phenotype than SCA3 tg/− mice (Fig. 1). Because SKA-31 rapidly improved depolarization block in cerebellar slices, we tested whether short-term administration of SKA-31 could improve motor deficits in SCA3 tg/tg mice. Animals were tested over 4 d on the balance beam, with the first 2 d establishing baseline performance before SKA-31 or vehicle was administered on the final 2 d. The performance on days 1 and 2 versus days 3 and 4 was pooled for analysis. SKA-31 (10 mg/kg) significantly improved the performance of SCA3 tg/tg mice on the balance beam (p = 0.003; Fig. 5C). Whereas symptomatic SCA3 tg/tg mice typically traverse the beam by dragging their hind limbs, 5 of 10 mice treated with SKA-31 showed visible and dramatic improvement in their ability to balance on the rod and support themselves on their hind limbs. A higher dose of SKA-31 (50 mg/kg dose) caused sedation and an inability of the animals to hold onto the beam. This sedation lasted for 2–3 h, consistent with pharmacokinetic data suggesting that systemically administered SKA-31 has a limited half-life in the brain (Sankaranarayanan et al., 2009). Importantly, SKA-31 did not significantly affect the balance beam performance of wild-type littermate controls (Fig. 5C).
Kv channels undergo increased current inactivation in SCA3 tg/− neurons
What is the mechanism for the increased excitability of SCA3 Purkinje neurons? We wished to determine which alterations in ionic currents underlie the observed changes in Purkinje neuron firing. Due to the large size of Purkinje neurons, it is not possible to measure currents in intact neurons. To overcome this limitation, we used somatic and nucleated patch recordings to measure the kinetics of ion channel currents directly.
Since the interspike depolarization that drives repetitive firing in Purkinje neurons is mediated by a voltage-dependent sodium current, we tested whether the biophysical properties of voltage-activated sodium currents were altered in SCA3 tg/− Purkinje neurons. In outside-out giant nucleated patches, current activation (Fig. 6A,C), inactivation (B,C), and the fraction of slowly inactivating current (D) proved to be similar in wild-type and SCA3 tg/− Purkinje neurons.
Figure 6.

SCA3 neurons have normal voltage-activated sodium (Nav) channel biophysics. A, Successive 5 mV voltage steps were used to elicit voltage-activated sodium currents in nucleated patches from Purkinje neurons (left). Rapidly activating inward sodium currents were elicited in wild-type (middle) and SCA3 tg/− neurons (right). B, A two-step protocol was used to assess Nav channel inactivation (left). Currents elicited during the second step to 0 mV are shown for wild-type (middle) and SCA3 tg/− (right) neurons. C, Activation and inactivation parameters for wild-type and SCA3 neurons were fit to a Boltzman curve. The V1/2 of activation values were −40.6 and −40.4 mV, respectively, for wild-type and SCA3 tg/− neurons (n = 5 wild-type, 7 SCA3 patches). The V1/2 of inactivation values were −68.5 and −68.7 mV in wild-type and SCA3 neurons, respectively (p > 0.05). D, Slow inactivation was assessed as the amount of current present after 25 ms of depolarization at different voltages (in the protocol shown in A). The fraction of slowly inactivating current is similar in wild-type and SCA3 tg/− neurons.
Because we could not identify a change in the major depolarizing conductance, we hypothesized that the increase in excitability of SCA3 Purkinje neurons might be due to a reduction in a repolarizing potassium conductance. Pharmacologic inhibition of voltage-activated Kv currents has been shown to accelerate slow depolarization in Purkinje neurons (Etzion and Grossman, 2001). Thus, in SCA3 a reduction in the effective hyperpolarization mediated by Kv currents could be responsible for the depolarization block in Purkinje neurons. To test this, we used excised outside-out somatic patches to record potassium currents from Purkinje cells. Patches generated in the presence of the potassium channel inhibitors, TEA, and 2 mm barium revealed no residual current (data not shown), confirming that, under our recording conditions, all the current in somatic patches is carried by Kv channels. Successive, depolarizing voltage steps from a holding potential of −80 mV were used (Fig. 7A) to activate potassium currents in somatic patches from Purkinje neurons. The V1/2 of activation was similar in wild-type and SCA3 neurons (Fig. 7B). Patches from wild-type and SCA3 tg/− neurons displayed voltage-dependent activation of a potassium current which inactivated slowly during the 100 ms step (Fig. 7A). The mean peak amplitude of the potassium current at 40 mV was similar in wild-type and SCA3 tg/− neurons (494 and 594 pA, respectively; p > 0.05). Importantly, however, SCA3 tg/− Purkinje neurons showed significantly greater reduction of current amplitude at the end of the 100 ms voltage step (Fig. 7A,C). As in SCA3 tg/− mice, there was significantly greater inactivation of Kv currents in somatic patches from SCA3 tg/tg mice than from littermate controls (n = 6; F = 13.1; p < 0.01). Although the degree of inactivation was similar in SCA3 tg/− and SCA3 tg/tg animals (data not shown), the inactivation across patches was significantly less variable in homozygous mice: the mean standard deviation of the fractional inactivation was 0.2 ± 0.0 in SCA3 tg/− (n = 11) versus 0.13 ± 0.01 in SCA3 tg/tg mice (n = 6; p < 0.001). These results demonstrate enhanced voltage-dependent potassium current inactivation in SCA3 Purkinje neurons.
Figure 7.

Voltage-activated potassium currents undergo increased inactivation in SCA3 neurons. A, Step protocol with successive 10 mV voltage steps to activate Kv currents in Purkinje neuron somatic patches (left). Patches from wild-type Purkinje neurons display voltage-dependent activation of Kv currents that inactivate to about 70% of the peak current at the end of the 100 ms step (middle). Somatic patches from SCA3 tg/− neurons exhibit increased current inactivation at the end of the 100 ms voltage step (right). B, Kv current activation of wild-type and SCA3 tg/− neurons is similar with V1/2 of activation values of −16.1 and −17.5 mV, respectively. C, SCA3 neurons have greater fractional inactivation at the end of a 100 ms step (n = 11 patches each; F = 4.8; p < 0.05). D, Summary of Kv current deactivation time constants in wild-type and SCA3 tg/− neurons. E, The two-step protocol to determine current deactivation is shown on the left. Nearly all of the current in both wild-type (middle) and SCA3 (right) neurons is rapidly deactivating.
Purkinje neurons express a variety of Kv channel isoforms (Gruol et al., 1989; Goldman-Wohl et al., 1994; Weiser et al., 1994; McKay et al., 2005; Haghdoust et al., 2007). A change in inactivation could result either from an increase in the expression of rapidly inactivating potassium channels (for example, accessory Kvβ subunits that increase Kv1 inactivation) or from a change in the intrinsic biophysical properties of Kv channels normally expressed in Purkinje neurons. Purkinje neurons express potassium channels that close either rapidly (Kv3 channels) or slowly (Kv1 channels) following repolarization, with most of the current being rapidly deactivating (Martina et al., 2007). In both wild-type and SCA3 tg/− neurons, deactivation at −50 mV (a voltage where no current activates) could be fit to a single exponential with a decay time constant of ∼2 ms (Fig. 5D). This rapid deactivation is similar to that of cloned Kv3 channels and an order of magnitude faster than that described for the more slowly deactivating Kv1 channels (Grissmer et al., 1994), suggesting that the Kv current in somatic patches is largely carried by Kv3 channels. Even when we fit the data to two exponentials to address the possibility that a minor component of the current is carried by a more slowly deactivating current, at most only 4% of the Kv current is supplied by slowly deactivating Kv channels (n = 6 wild-type and 5 SCA3 tg/− neurons; Fig. 7D,E). Together, these results indicate that the depolarization block in SCA3 tg/− Purkinje neurons is associated with a reduction in repolarizing Kv channel current due to increased Kv3 current inactivation.
Reduction of α-dendrotoxin-sensitive potassium currents predisposes wild-type Purkinje neurons to depolarization block
We next investigated whether increased inactivation of delayed rectifier potassium channels leads to altered spike morphology in SCA3 tg/− neurons. Spikes in Purkinje neurons from SCA3 tg/− mice, whether occurring in spontaneously firing neurons or evoked by current injection in depolarized neurons, were indistinguishable from spikes in wild-type neurons (Fig. 8A). Spike width in wild-type and SCA3 neurons was also similar (Fig. 8B). Calcium-activated potassium channels help regulate firing frequency and contribute to the AHP (Cingolani et al., 2002; Walter et al., 2006). We determined the amplitude of the AHP at a firing frequency between 40 and 50 Hz in spike trains evoked by depolarizing current injection. The amplitude of Purkinje neuron AHP did not differ between wild-type and SCA3 tg/− mice (Fig. 8C), suggesting that changes in calcium-activated potassium currents do not contribute to the observed depolarization block in SCA3 Purkinje neurons.
Figure 8.

Depolarization block in SCA3 neurons is not accompanied by changes in spike width, AHP amplitude, or levels of Kv channel subunit expression. A, Overlay of spikes evoked by depolarizing current injection from wild-type (blue) and SCA3 (red) Purkinje neurons. B, Spike width from spontaneously firing SCA3 tg/− neurons and spikes evoked by current injection are similar to those from wild-type neurons. C, The amplitude of the AHP from spikes evoked from a holding potential of −90 mV is similar in Purkinje neurons from wild-type and SCA3 tg/− mice. D, Expression levels of Kv1.1, Kv1.2, Kv3.1, Kv3.3 and the accessory subunits Kvβ1 and Kvβ2 are similar in cerebellar membrane fractions from wild-type and SCA3 tg/− mice. Cadherin represents a loading control. E, Wild-type neurons sustain high rates of firing in response to depolarizing current injection in the presence of synaptic inhibitors (top), but upon application of 200 nm α-dendrotoxin (bottom), the same amount of injected current induces depolarization block (injected current for depolarization block changed from 0.84 ± 0.06 to 0.66 ± 0.09 nA; n = 4; p < 0.05).
Since recordings from nucleated and somatic patches do not reliably measure alterations in the amplitude of ionic currents in intact neurons, we applied a 1 s, 100 Hz spike waveform to dissociated postnatal day 14–18 Purkinje neurons to estimate the amplitude of currents that activated during spiking. Recordings were performed in an external solution with 50 mm NaCl to improve the quality of voltage clamp. The amplitudes of the outward and inward currents that activated during the action potential were similar in Purkinje neurons from wild-type and SCA3 tg/− mice (Fig. 9A–C,F). We next determined the total TEA-sensitive potassium current. As demonstrated previously for wild-type Purkinje neurons (Raman and Bean, 1999), the outward current activating during the spike was entirely TEA sensitive and was similarly TEA sensitive in SCA3 tg/− neurons (Fig. 9D–F). The amplitude of the TEA-sensitive current activating during the spike was also similar in wild-type and SCA3 tg/− neurons (Fig. 9G–I). Unfortunately, dissociated Purkinje neurons from 6- to 8-week-old animals are not viable; thus we cannot directly measure whole-cell ionic currents in 6- to 8-week-old neurons.
Figure 9.

Inward and outward currents in dissociated Purkinje neurons in wild-type and SCA3 tg/− mice are similar. A–C, A 1 s, 100 Hz action potential train waveform was applied to isolated Purkinje neurons to activate currents during the spike in wild-type (B) and SCA3 tg/− (C) mice. D–F, The stimulus waveform, applied in the presence of 1 mm TEA, demonstrates that the outward current is entirely TEA sensitive in Purkinje neurons from wild-type (E) and SCA3 tg/− mice (n = 3 each; F). G–I, The same stimulus waveform was applied to obtain the TEA-sensitive current in wild-type (H) and SCA3 tg/− mice (I). J, Summary of the inward and outward currents that activate in isolated Purkinje neurons during the applied action potential train waveform in wild-type and SCA3 tg/− mice.
We also determined the levels of different pore-forming and accessory potassium channel subunits in membrane fractions of cerebellar lysates from 8-week-old wild-type and SCA3 tg/− animals. There was no apparent change in protein levels for the Kv channels, Kv1.1, Kv1.2, Kv3.1, and Kv3.3, or for two accessory subunits that promote inactivation, Kvβ1 and Kvβ2 (Fig. 9D). These results suggest that the observed changes in SCA3 Purkinje neuron physiology are not accompanied by widespread changes in ion channel expression. Instead, changes in Kv channel kinetics likely cause the observed depolarization block in SCA3 tg/− neurons.
To test the idea that depolarization block can be induced without affecting spike width, we determined the effect of α-dendrotoxin, a Kv1 inhibitor, on spiking in wild-type Purkinje neurons. When 200 nm α-dendrotoxin was applied to wild-type Purkinje neurons, the amount of injected current needed to induce depolarization block was reduced by 20% (n = 4; p = 0.01; Fig. 9E), without any change in spike width (0.28 ms before and 0.30 ms after perfusion of α-dendrotoxin; p > 0.05). These results support the view that altered Kv channel kinetics can explain the depolarization block of repetitive firing in SCA3 tg/− neurons.
Polyglutamine expanded ataxin-3 influences the kinetics of Kv1 and Kv3 channels
Our results with somatic patches from SCA3 tg/− Purkinje neurons revealed an increase in inactivation of Kv3 currents. We next sought to determine whether ataxin-3 could directly affect the properties of Kv channels. In addition to Kv3 channels, Kv1 channels play an important role in Purkinje neuron repetitive spiking (McKay et al., 2005), and since these channels were not adequately represented in our somatic patches, we tested whether ataxin-3 directly affects both Kv3 and Kv1 channel kinetics. We chose Kv1.2 and Kv3.1 as representative Kv1 and Kv3 channels as these channels are known to be expressed in Purkinje neurons (Sheng et al., 1994; Sacco et al., 2006), and cell lines expressing these channels are available for heterologous expression studies (Grissmer et al., 1994). In fibroblast cell lines stably expressing Kv1.2 or Kv3.1, we determined the effect of coexpressing GFP alone, GFP-tagged nonpathogenic ataxin-3(Q28), or GFP-tagged pathogenic ataxin-3(Q84). Ataxin-3(Q84) increased the inactivation of Kv3.1 by shifting steady state inactivation by 10 mV to more hyperpolarizing membrane potentials (Fig. 10). In contrast, ataxin-3(Q28) has less of an effect on Kv3.1 inactivation, which did not reach statistical significance (p = 0.06). Current activation was unaffected by expression of ataxin-3(Q84) (data not shown). Ataxin-3 expression also affected biophysical properties of Kv1.2: ataxin-3(Q84), but not ataxin-3(Q28), shifted the activation of Kv1.2 to more positive potentials (Fig. 10) without affecting its inactivation properties (data not shown). These results indicate that ataxin-3 affects the kinetics of both Kv3 and Kv1 channels in a polyglutamine length-dependent manner. Although the magnitude of the change in Kv3 inactivation in this heterologous expression system is much less than that seen in Purkinje neurons in SCA3 mice (Fig. 7), the result suggests that the alterations in firing properties of intact SCA3 Purkinje neurons may be explained in part by an alteration in the kinetics of Kv channels.
Figure 10.
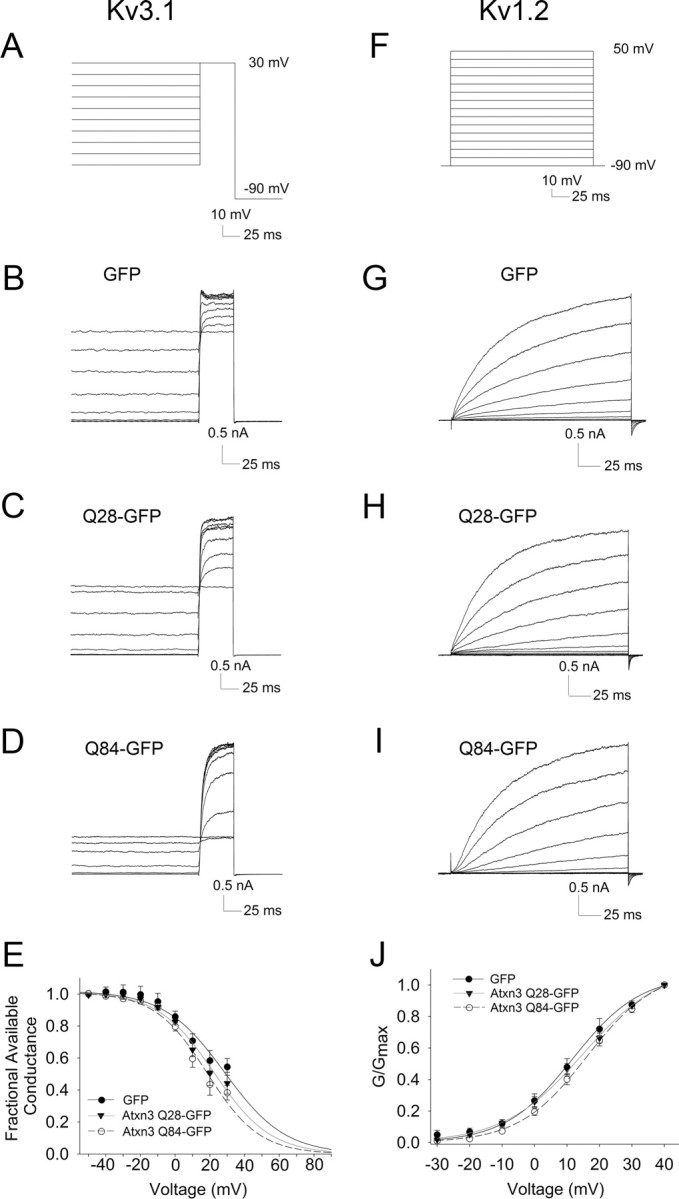
In transfected cells, polyglutamine-expanded ataxin-3 increases inactivation of Kv3.1 and shifts activation Kv1.2 to more depolarized membrane potentials. A–D, Two-step protocol (A) to evaluate Kv3.1 current inactivation in the presence of coexpressed GFP alone (B), GFP-tagged ataxin-3(Q28) (C), or GFP-tagged ataxin-3(Q84) (D). E, Inactivation curves of Kv3.1 demonstrate increased inactivation in the presence of polyglutamine-expanded ataxin-3 (F = 11.6; p = 0.0009 compared to GFP alone). F–I, Step protocol (F) used to activate Kv1.2 currents with coexpression of GFP alone (G), GFP-tagged ataxin-3(Q28) (H), or GFP-tagged ataxin-3(Q84) (I). J, Activation curves of Kv1.2 demonstrate a shift of the activation curve to the right in the presence of polyglutamine-expanded ataxin-3 (F = 11.16; p = 0.0012 compared to GFP alone).
Discussion
Our studies in SCA3 mice reveal an early and robust change in Purkinje neuron firing that precedes the onset of Purkinje cell loss and is associated with altered kinetics of voltage-activated potassium channels. Our results also support the view that these changes in physiology contribute to the motor phenotype. The observed changes in potassium channel physiology represent a novel potential mechanism contributing to disease pathogenesis in SCA3 and perhaps other degenerative ataxias.
Our results add to the growing literature implicating potassium channels as central players in the pathophysiology of ataxia. Human mutations in voltage-activated potassium channels are known to cause episodic ataxia type 1 (Kv1.1) (Browne et al., 1994) and the progressive spinocerebellar ataxia, SCA13 (Kv3.3) (Waters and Pulst, 2008). Alterations in potassium channels have also been associated with mouse models of ataxia. For example, a reduction in SK channels in the deep cerebellar nuclei causes profound ataxia (Shakkottai et al., 2004), and knockout of large-conductance calcium-activated potassium channels in mice results in Purkinje neuron depolarization block and ataxia (Sausbier et al., 2004). The changes observed here in SCA3 mice underscore the importance of potassium channels in cerebellar physiology and in the pathogenesis of certain ataxias.
Do the observed changes in the kinetics of Kv channels explain the altered firing properties of SCA3 Purkinje neurons? Although the effect of ataxin-3 on the kinetics of Purkinje neuron Kv3 channels is modest, relatively small changes in the activation and deactivation kinetics of Kv3.3 are known to result in a developmental and degenerative phenotype in another degenerative ataxia (SCA13) similar to SCA3 (Waters and Pulst, 2008). We demonstrate that mutant ataxin-3 also alters slightly the kinetics of Kv1.2 channels in a heterologous system. Kv1 channels in the cerebellum exist as heteromers of different pore-forming subunits (Koch et al., 1997). A recently described ataxia-causing mutation of Kv1.2 has much greater effects on the biophysical properties of the channel when coexpressed with Kv1.1 (Xie et al., 2010). Thus, it is possible that polyglutamine-expanded ataxin-3 has additional effects on other classes of Kv channels or heteromeric channels not tested here. Changes in other conductances may also contribute to the alterations in SCA3 Purkinje neuron firing. A reduction of calcium currents (leading to reduced activation of calcium-activated potassium currents) or a direct reduction in calcium-activated potassium currents could also contribute to Purkinje neuron depolarization block. In addition, alterations in conductances in the axon initial segment or dendrites, which are not adequately represented in the somatic patches and dissociated neurons used in our study, could also alter Purkinje neuron firing.
What is the mechanism underlying the change in potassium channel biophysics in SCA3? Our data suggest that ataxin-3 alters the kinetics of Kv channels in a polyglutamine length-dependent manner. Posttranslational modification of potassium channels can alter their biophysical properties. Sumoylation of Kv1.5 and Kv2.1 increases their inactivation (Benson et al., 2007; Dai et al., 2009), and Kv3.3 inactivation is modulated by phosphorylation by protein kinase C (Desai et al., 2008). Ataxin-3 is a deubiquitinating enzyme; thus, exploring the relationship between its enzymatic activity and its effects on the kinetics of Kv will be of considerable interest. Functional Kv channels assemble as tetramers of pore-forming α subunits. Interestingly, ataxin-3 associates with a putative ubiquitin ligase, the potassium channel tetramerization domain-10 (KCTD10) protein (Sowa et al., 2009). KCTD proteins contain a domain that is most similar to the tetramerization domain (T1) of Kv channels (Bayon et al., 2008). Further exploring the possible interaction between ataxin-3 and Kv channels may help explain the vulnerability of the affected neurons in SCA3.
The dominantly inherited SCAs are characterized primarily by degenerative changes in the cerebellum and brainstem. The most common of these ataxias, including SCA3, SCA1, SCA2, and SCA6, are caused by expansion of a polyglutamine domain in the respective disease proteins. Neuronal loss clearly cannot explain all the phenotypic changes in these degenerative disorders; particularly in early stages of disease, symptoms likely reflect neuronal dysfunction more than neuronal cell loss. For example, SCA1 knock-in mice show a neurological phenotype without significant neuronal cell loss (Lorenzetti et al., 2000), and transgenic SCA1 mice become symptomatic before the onset of significant neurodegeneration (Clark et al., 1997). Purkinje neurons of SCA1 transgenic mice also demonstrate altered electrophysiologic properties: delayed spike onset to depolarizing current injections. Both expanded ataxin-2 and ataxin-3 have been shown to associate with the type 1 inositol 1,4,5-trisphosphate receptor, an intracellular calcium release channel, and to alter intracellular calcium handling (Chen et al., 2008; Liu et al., 2009). Our results reveal a specific defect in potassium channels as an early element of neuronal dysfunction and a novel potential therapeutic target in SCA3. It would be interesting to determine whether other polyglutamine ataxias share these changes in potassium channel physiology.
Despite prominent cerebellar dysfunction in SCA3, neuronal loss in the cerebellar cortex in SCA3 autopsy samples is variable, with many cases showing <25% Purkinje cell loss (Durr et al., 1996; Schols et al., 2004). Although massive loss of Purkinje neurons is not a prominent feature in most cases of SCA3, our findings indicate that aberrant Purkinje cell function likely underlies some disease symptoms. Significant neurological signs indicating brainstem and spinal cord involvement also occur in SCA3, with prominent loss of neurons in the brainstem and deep cerebellar nucleus (Durr et al., 1996; Schols et al., 2004). Since polyglutamine-expanded ataxin-3 has a direct effect on the biophysical properties of Kv channels, and Kv channels are widely expressed in the brain, changes in the intrinsic excitability of other neurons might also occur in SCA3. It will be important to explore electrophysiologic changes in other affected neurons in SCA3 and determine whether the changes in Kv channel biophysics observed in Purkinje neurons also pertain to other neuronal populations. A similar change in potassium channel kinetics in other neurons could result in increased firing frequency and a corresponding propensity for calcium-mediated excitotoxicity. Systemically administered SKA-31 in our mouse model may therefore also have had additional effects on other affected neurons in SCA3. Changes in neurons other than Purkinje cells could also have contributed to the improved motor phenotype. Delineating the role of altered physiology and subsequent neuronal loss in SCA3 and other degenerative ataxias may reveal new, shared therapeutic targets for these currently untreatable neurodegenerative disorders.
The functional importance of the observed physiologic changes in Purkinje neurons is demonstrated by the improved motor phenotype in SCA3 mice after short-term administration of the selective calcium-activated potassium channel activator, SKA-31. SKA-31 was developed using the U.S. Food and Drug Administration-approved agent riluzole as a template. Unlike SKA-31, riluzole also inhibits voltage-activated sodium channels at the same concentration at which it activates SK channels (Sankaranarayanan et al., 2009). This sodium channel-blocking effect of riluzole would be expected to inhibit spiking and negate the membrane hyperpolarization resulting from activation of calcium-activated potassium channels. Accordingly, we anticipate that riluzole would be less effective than pure SK channel activators in correcting aberrant Purkinje neuron physiology in SCA3. Despite this theoretical limitation, a recent short-term clinical trial found that riluzole effectively reduced ataxia symptoms in patients with various forms of ataxia (Ristori et al. 2010). The results of this clinical trial, which did not include patients with SCA3, underscore the potential importance of aberrant channel physiology to a wide range of ataxias. Our findings in SCA3 mice emphasize the need to develop next-generation activators of SK channels with improved ability to cross the blood–brain barrier as potential therapeutic agents for SCA3 and other forms of ataxia.
Footnotes
This work was supported by the National Ataxia Foundation (V.G.S.), NIH Grants 1K08NS072158 (V.G.S.) and RO1 NS038712 (H.L.P.), and the Fauver Family Fund (H.L.P., V.G.S.). We thank Drs. Geoffrey Murphy and William Dauer for helpful discussions and suggestions. We are grateful to Natasha Kennedy for expert technical help.
References
- Aizenman CD, Linden DJ. Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol. 1999;82:1697–1709. doi: 10.1152/jn.1999.82.4.1697. [DOI] [PubMed] [Google Scholar]
- Bayon Y, Trinidad AG, de la Puerta ML, Del Carmen Rodriguez M, Bogetz J, Rojas A, De Pereda JM, Rahmouni S, Williams S, Matsuzawa S, Reed JC, Crespo MS, Mustelin T, Alonso A. KCTD5, a putative substrate adaptor for cullin3 ubiquitin ligases. FEBS J. 2008;275:3900–3910. doi: 10.1111/j.1742-4658.2008.06537.x. [DOI] [PubMed] [Google Scholar]
- Benson MD, Li QJ, Kieckhafer K, Dudek D, Whorton MR, Sunahara RK, Iniguez-Lluhi JA, Martens JR. SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc Natl Acad Sci U S A. 2007;104:1805–1810. doi: 10.1073/pnas.0606702104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–140. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- Cemal CK, Carroll CJ, Lawrence L, Lowrie MB, Ruddle P, Al-Mahdawi S, King RH, Pook MA, Huxley C, Chamberlain S. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum Mol Genet. 2002;11:1075–1094. doi: 10.1093/hmg/11.9.1075. [DOI] [PubMed] [Google Scholar]
- Chai Y, Shao J, Miller VM, Williams A, Paulson HL. Live-cell imaging reveals divergent intracellular dynamics of polyglutamine disease proteins and supports a sequestration model of pathogenesis. Proc Natl Acad Sci U S A. 2002;99:9310–9315. doi: 10.1073/pnas.152101299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Tang TS, Tu H, Nelson O, Pook M, Hammer R, Nukina N, Bezprozvanny I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J Neurosci. 2008;28:12713–12724. doi: 10.1523/JNEUROSCI.3909-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Gymnopoulos M, Boccaccio A, Stocker M, Pedarzani P. Developmental regulation of small-conductance Ca2+-activated K+ channel expression and function in rat Purkinje neurons. J Neurosci. 2002;22:4456–4467. doi: 10.1523/JNEUROSCI.22-11-04456.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, Orr HT. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai XQ, Kolic J, Marchi P, Sipione S, Macdonald PE. SUMOylation regulates Kv2.1 and modulates pancreatic beta-cell excitability. J Cell Sci. 2009;122:775–779. doi: 10.1242/jcs.036632. [DOI] [PubMed] [Google Scholar]
- Desai R, Kronengold J, Mei J, Forman SA, Kaczmarek LK. Protein kinase C modulates inactivation of Kv3.3 channels. J Biol Chem. 2008;283:22283–22294. doi: 10.1074/jbc.M801663200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durr A, Stevanin G, Cancel G, Duyckaerts C, Abbas N, Didierjean O, Chneiweiss H, Benomar A, Lyon-Caen O, Julien J, Serdaru M, Penet C, Agid Y, Brice A. Spinocerebellar ataxia 3 and Machado-Joseph disease: clinical, molecular, and neuropathological features. Ann Neurol. 1996;39:490–499. doi: 10.1002/ana.410390411. [DOI] [PubMed] [Google Scholar]
- Duvick L, Barnes J, Ebner B, Agrawal S, Andresen M, Lim J, Giesler GJ, Zoghbi HY, Orr HT. SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron. 2010;67:929–935. doi: 10.1016/j.neuron.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzion Y, Grossman Y. Highly 4-aminopyridine sensitive delayed rectifier current modulates the excitability of guinea pig cerebellar Purkinje cells. Exp Brain Res. 2001;139:419–425. doi: 10.1007/s002210100788. [DOI] [PubMed] [Google Scholar]
- Fierro L, Llano I. High endogenous calcium buffering in Purkinje cells from rat cerebellar slices. J Physiol. 1996;496:617–625. doi: 10.1113/jphysiol.1996.sp021713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Wohl DS, Chan E, Baird D, Heintz N. Kv3.3b: a novel Shaw type potassium channel expressed in terminally differentiated cerebellar Purkinje cells and deep cerebellar nuclei. J Neurosci. 1994;14:511–522. doi: 10.1523/JNEUROSCI.14-02-00511.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- Gruol DL, Dionne VE, Yool AJ. Multiple voltage-sensitive K+ channels regulate dendritic excitability in cerebellar Purkinje neurons. Neurosci Lett. 1989;97:97–102. doi: 10.1016/0304-3940(89)90146-8. [DOI] [PubMed] [Google Scholar]
- Haghdoust H, Janahmadi M, Behzadi G. Physiological role of dendrotoxin-sensitive K+ channels in the rat cerebellar Purkinje neurons. Physiol Res. 2007;56:807–813. doi: 10.33549/physiolres.931041. [DOI] [PubMed] [Google Scholar]
- Heng MY, Tallaksen-Greene SJ, Detloff PJ, Albin RL. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington's disease. J Neurosci. 2007;27:8989–8998. doi: 10.1523/JNEUROSCI.1830-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeub M, Herbst M, Spauschus A, Fleischer H, Klockgether T, Wuellner U, Evert BO. Potassium channel dysfunction and depolarized resting membrane potential in a cell model of SCA3. Exp Neurol. 2006;201:182–192. doi: 10.1016/j.expneurol.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci. 2003;23:4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch RO, Wanner SG, Koschak A, Hanner M, Schwarzer C, Kaczorowski GJ, Slaughter RS, Garcia ML, Knaus HG. Complex subunit assembly of neuronal voltage-gated K+ channels. Basis for high-affinity toxin interactions and pharmacology. J Biol Chem. 1997;272:27577–27581. doi: 10.1074/jbc.272.44.27577. [DOI] [PubMed] [Google Scholar]
- Liu J, Tang TS, Tu H, Nelson O, Herndon E, Huynh DP, Pulst SM, Bezprozvanny I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci. 2009;29:9148–9162. doi: 10.1523/JNEUROSCI.0660-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzetti D, Watase K, Xu B, Matzuk MM, Orr HT, Zoghbi HY. Repeat instability and motor incoordination in mice with a targeted expanded CAG repeat in the Sca1 locus. Hum Mol Genet. 2000;9:779–785. doi: 10.1093/hmg/9.5.779. [DOI] [PubMed] [Google Scholar]
- Magistretti J, Mantegazza M, Guatteo E, Wanke E. Action potentials recorded with patch-clamp amplifiers: are they genuine? Trends Neurosci. 1996;19:530–534. doi: 10.1016/s0166-2236(96)40004-2. [DOI] [PubMed] [Google Scholar]
- Martina M, Metz AE, Bean BP. Voltage-dependent potassium currents during fast spikes of rat cerebellar Purkinje neurons: inhibition by BDS-I toxin. J Neurophysiol. 2007;97:563–571. doi: 10.1152/jn.00269.2006. [DOI] [PubMed] [Google Scholar]
- McKay BE, Molineux ML, Mehaffey WH, Turner RW. Kv1 K+ channels control Purkinje cell output to facilitate postsynaptic rebound discharge in deep cerebellar neurons. J Neurosci. 2005;25:1481–1492. doi: 10.1523/JNEUROSCI.3523-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz E, Rey MJ, Mila M, Cardozo A, Ribalta T, Tolosa E, Ferrer I. Intranuclear inclusions, neuronal loss and CAG mosaicism in two patients with Machado-Joseph disease. J Neurol Sci. 2002;200:19–25. doi: 10.1016/s0022-510x(02)00110-7. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riess O, Rub U, Pastore A, Bauer P, Schols L. SCA3: neurological features, pathogenesis and animal models. Cerebellum. 2008;7:125–137. doi: 10.1007/s12311-008-0013-4. [DOI] [PubMed] [Google Scholar]
- Ristori G, Romano S, Visconti A, Cannoni S, Spadaro M, Frontali M, Pontieri FE, Vanacore N, Salvetti M. Riluzole in cerebellar ataxia: a randomized, double-blind, placebo-controlled pilot trial. Neurology. 2010;74:839–845. doi: 10.1212/WNL.0b013e3181d31e23. [DOI] [PubMed] [Google Scholar]
- Sacco T, De Luca A, Tempia F. Properties and expression of Kv3 channels in cerebellar Purkinje cells. Mol Cell Neurosci. 2006;33:170–179. doi: 10.1016/j.mcn.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan A, Raman G, Busch C, Schultz T, Zimin PI, Hoyer J, Kohler R, Wulff H. Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol. 2009;75:281–295. doi: 10.1124/mol.108.051425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, Korth M, Shipston MJ, Knaus HG, Wolfer DP, Pedroarena CM, Storm JF, Ruth P. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci U S A. 2004;101:9474–9478. doi: 10.1073/pnas.0401702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. doi: 10.1016/S1474-4422(04)00737-9. [DOI] [PubMed] [Google Scholar]
- Shakkottai VG, Chou CH, Oddo S, Sailer CA, Knaus HG, Gutman GA, Barish ME, LaFerla FM, Chandy KG. Enhanced neuronal excitability in the absence of neurodegeneration induces cerebellar ataxia. J Clin Invest. 2004;113:582–590. doi: 10.1172/JCI20216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Tsaur ML, Jan YN, Jan LY. Contrasting subcellular localization of the Kv1.2 K+ channel subunit in different neurons of rat brain. J Neurosci. 1994;14:2408–2417. doi: 10.1523/JNEUROSCI.14-04-02408.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Waters MF, Pulst SM. Sca13. Cerebellum. 2008;7:165–169. doi: 10.1007/s12311-008-0039-7. [DOI] [PubMed] [Google Scholar]
- Weiser M, Vega-Saenz de Miera E, Kentros C, Moreno H, Franzen L, Hillman D, Baker H, Rudy B. Differential expression of Shaw-related K+ channels in the rat central nervous system. J Neurosci. 1994;14:949–972. doi: 10.1523/JNEUROSCI.14-03-00949.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K. Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar Purkinje neurons. J Neurosci. 2003;23:2600–2607. doi: 10.1523/JNEUROSCI.23-07-02600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Harrison J, Clapcote SJ, Huang Y, Zhang JY, Wang LY, Roder JC. A new Kv1.2 channelopathy underlying cerebellar ataxia. J Biol Chem. 2010;285:32160–32173. doi: 10.1074/jbc.M110.153676. [DOI] [PMC free article] [PubMed] [Google Scholar]