

Abstract
Oncogene activation increases susceptibility to apoptosis. Thus, tumorigenesis must depend, in part, on compensating mutations that protect from programmed cell death. A functional screen for cDNAs that could counteract the proapoptotic effects of the myc oncogene identified two related bHLH family members, Twist and Dermo1. Both of these proteins inhibited oncogene- and p53-dependent cell death. Twist expression bypassed p53-induced growth arrest. These effects correlated with an ability of Twist to interfere with activation of a p53-dependent reporter and to impair induction of p53 target genes in response to DNA damage. An underlying explanation for this observation may be provided by the ability of Twist to reduce expression of the ARF tumor suppressor. Thus, Twist may affect p53 indirectly through modulation of the ARF/MDM2/p53 pathway. Consistent with a role as a potential oncoprotein, Twist expression promoted colony formation of E1A/ras-transformed mouse embryo fibroblasts (MEFs) in soft agar. Furthermore, Twist was inappropriately expressed in 50% of rhabdomyosarcomas, a tumor that arises from skeletal muscle precursors that fail to differentiate. Twist is known to block myogenic differentiation. Thus, Twist may play multiple roles in the formation of rhabdomyosarcomas, halting terminal differentiation, inhibiting apoptosis, and interfering with the p53 tumor-suppressor pathway.
Keywords: Twist, oncogenes, apoptosis, tumorigenesis, Dermo1
A defining characteristic of tumor cells is the escape from regulatory mechanisms that normally restrain cell proliferation. This is accomplished through the accumulation of multiple genetic alterations. Among these are the inactivation of key tumor suppression pathways and the activation of oncogenes (for review, see Vogelstein and Kinzler 1998).
The products of cellular oncogenes such as ras and myc are components of normal growth control pathways. These form part of the program that promotes entry into the division cycle in response to appropriate environmental cues. However, in tumor cells, the normal function of these genes is subverted to provide hyperactive proliferative signals. It is becoming increasingly clear that normal cells respond to inappropriate growth signals by activating homeostatic growth control pathways that protect multicellular organisms from tumor formation.
Constitutive activation of Ras promotes transformation of some immortalized cells. However, Ras activation in normal cells provokes cellular senescence. This irreversible growth arrest probably negates the ability of these cells to contribute to tumor formation (Serrano et al. 1997; Lin et al. 1998). In contrast, other cellular and viral oncogenes sensitize cells to undergo programmed cell death on exposure to stimuli that might normally cause a reversible growth arrest. For example, Myc induces apoptosis on removal of serum survival factors from primary cells or Rat1 fibroblasts (Evan et al. 1992; Hermeking and Eick 1994; Wagner et al. 1994). Similarly, primary mouse embryo fibroblasts (MEFs) that express E1A are sensitized to programmed cell death in response to contact inhibition, growth factor withdrawal, and DNA damage (Debbas and White 1993; Lowe and Ruley 1993; Lowe et al. 1993, 1994).
The growth inhibitory properties of many oncogenes dictate that tumorigenesis requires the cooperation of different classes of genes. For example, transformation by Ras can proceed only in the presence of additional mutations that prevent Ras-induced senescence (Serrano et al. 1997). Furthermore, the ability of Myc or E1A to promote tumorigenesis requires that transformed cells be protected from the proapoptotic effects of these oncoproteins. An understanding of oncogene cooperation requires both a knowledge of the mechanisms by which oncogene activation provokes homeostatic responses and a clarification of the routes through which cooperating oncogenic events defeat these protective controls.
It is clear that oncogene-transformed cells require additional genetic alterations that render them resistant to apoptotic stimuli. Such genetic changes are likely to be obligate for oncogenesis. However, the full spectrum of these events has proven difficult to elucidate, because protective genes are not likely to be discovered via the cellular transformation assays that have proven a rich source of new oncogenes. For example, bcl-2 synergizes with myc in the generation of Burkitt lymphoma but does not cooperate with myc in transformation of mouse fibroblasts in vitro (Vaux et al. 1988). As an approach to the discovery of potential oncogenes that might elude conventional methods, we have undertaken a search for cellular genes that can counter the proapoptotic effects of myc activation.
Results and Discussion
A genetic screen for antiapoptotic proteins
Direct, functional selection of genetic alterations that evoke specific phenotypes has provided a powerful method for the dissection of numerous biological pathways in genetically tractable eukaryotes such as yeasts, Caenorhabditis elegans, and Drosophila. These approaches have been extended to cultured mammalian cells by several groups (e.g., Deiss and Kimchi 1991; Gudkov et al. 1994; Rayner and Gonda 1994; Wong et al. 1994; for review, see Gudkov and Roninson 1997; Kimchi 1998). Building on these prior studies, we have developed a suite of tools that streamlines the process of complementation screening in mammalian cells (Sun et al. 1998; Hannon et al. 1999).
We have designed a series of modified, replication-deficient retrovirus vectors (MaRX) and packaging cell lines (LinX) that allow high-efficiency gene transfer to a wide range of cell types. Recovery of integrated MaRX proviruses from selected cell populations is facilitated by two key modifications. First, the MaRX provirus contains, within the LTR, target sequences for a site-specific recombinase. Second, the MaRX provirus contains an optimized bacterial replicon. Treatment of genomic DNA from infected cells with the appropriate recombinase enzyme results in excision of a circular plasmid comprising the integrated virus that can be propagated in bacterial cells. Despite the fact that this plasmid contains only a single LTR, it is capable of producing infectious retrovirus on transfection into the LinX packaging cells. Thus, by this approach we can recover cDNAs that confer a specific phenotype and transfer them directly into new recipient cells without intervening cloning steps.
The myc oncogene can predispose a wide variety of cell types to programmed cell death. However, the design of a genetic selection for cDNAs that can counter the proapoptotic effects of myc requires the use of cells that die with high efficiency. Rat1/MycER cells undergo apoptosis on simultaneous Myc activation and growth factor withdrawal (Evan et al. 1992). However, even on delivery of a strong proapoptotic stimulus, a significant number of cells survive (∼0.1%–1% of the population). For this reason, we could not select directly for cells carrying protective cDNAs through a single round of treatment. Instead, we designed a genetic screen in which cell populations would be exposed to multiple, iterative rounds of killing and rescue with the hope that protective cDNAs would be continuously enriched, whereas neutral cDNAs would be counter selected (Fig. 1).
Figure 1.
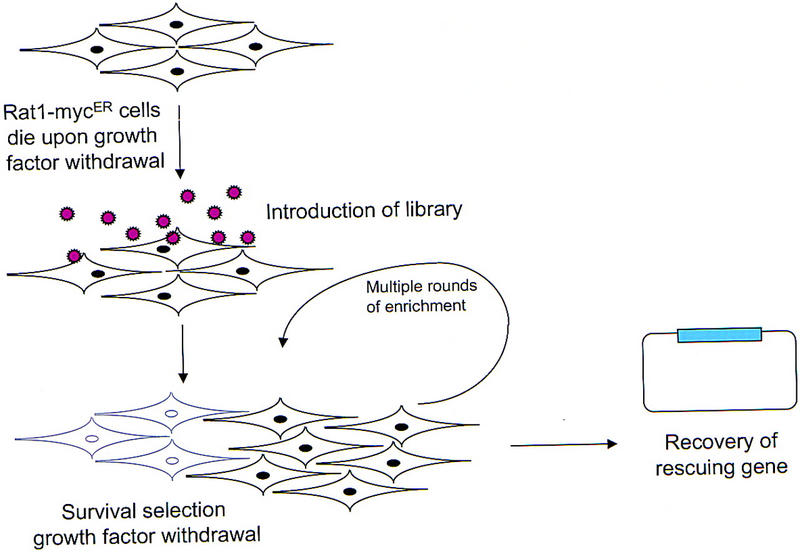
Schematic outline of the screen designed to identify genes that confer protection from Myc-induced apoptosis.
To test our approach, we reconstructed the screen using a well-characterized antiapoptotic gene, bcl-2. Rat1/MycER cells were coinfected with retroviruses that direct the expression of Bcl-2 and LacZ. These marked (LacZ-positive), Bcl-2-expressing cells were mixed in varying proportions with unmarked control cells, and the mixtures were subjected to multiple cycles of killing by exposure to proapoptotic conditions followed by rescue and expansion of resistant cells under normal growth conditions. The proportion of LacZ/Bcl-2 cells was monitored following each round, and after four cycles, β-galactosidase staining indicated that Bcl-2-expressing cells had been enriched ∼50,000-fold from a frequency of 1/105 to a frequency of 1/2. Because we reasoned that other antiapoptotic genes might not be as effective as bcl-2, we designed the selection procedure to allow recovery of genes that were enriched a minimum of 1,000-fold by four killing cycles.
Populations of Rat1/MycER cells were infected with a retroviral cDNA library that had been prepared from cells that were committed to apoptosis. Individual plates containing ∼5 × 106 cells were infected with pools containing from 10,000 to 100,000 distinct cDNAs. This insured that each individual cDNA was represented multiple times in the starting cell population. Infected cell pools were subjected to four cycles of enrichment by the apoptosis/rescue protocol that we had established using Bcl-2 as a model. Proviruses were recovered from populations that had been enriched for resistant cells, and highly represented genes were identified by fingerprinting 50 cDNAs from each population. Most of the pools contained cDNA clones that had been clearly enriched by the procedure.
The genetic strategy was validated by the isolation of cDNAs encoding proteins that had been shown previously to protect from apoptosis. For example, we isolated many (9) independent cDNAs encoding Mcl-1 a member of the Bcl-2 family that prevents cell death and promotes differentiation in hematopoietic cells (Kozopas et al. 1993). Furthermore, we identified as a protective protein glutathione peroxidase, an enzyme that can counteract apoptosis induced by reactive oxygen species (Hockenbery et al. 1993). We also obtained two independent clones of Ha-ras. Expression of ras has been shown recently to prevent apoptosis in Rat1/MycER cells via activation of the AKT pathway (Kauffmann-Zeh et al. 1997). All three of these genes protected Rat1/MycER cells from apoptosis under our experimental conditions (not shown).
Twist and Dermo1 prevent Myc-dependent cell death
In addition to known antiapoptotic genes, we also isolated a large number of potentially protective cDNAs that had not been shown previously to antagonize cell death. Among these were multiple independent isolates encoding two closely related proteins, Twist and Dermo1. twist was represented by two independent cDNAs whereas dermo1 was isolated four times.
Twist was originally identified in Drosophila as a protein involved in establishing dorso–ventral polarity (Thisse et al. 1987). Dermo1 emerged from a two-hybrid screen for tissue-specific factors that could interact with the ubiquitous bHLH protein, E12. Twist and Dermo1 belong to the basic–helix–loop–helix (bHLH) family of transcription factors and are quite similar (>90% identity) in the bHLH and carboxy-terminal domains. The amino termini are less closely related; Dermo1 lacks a glycine-rich region that is present in Twist (Li et al. 1995). Although specific transcriptional targets of Twist and Dermo1 have not yet been identified in mammals, expression patterns in Drosophila, Xenopus, and mouse suggest an involvement in the regulation of diverse developmental processes, particularly in the formation of mesoderm (Futchbauer et al. 1995; Li et al. 1995; Gitelman 1997).
The enrichment of two such closely related proteins during the genetic selection and the isolation of each from multiple, independent cell pools prompted a deeper investigation of the possibility that Twist and Dermo1 could interfere with oncogene-induced apoptosis. Expression of either Twist or Dermo1 reduced the number of apoptotic cells that were observed following either Myc activation or serum withdrawal to ∼50% of the number observed in control (LacZ-infected) or uninfected populations (Fig. 2). Similar levels of protection were evident from an analysis of viable cells that remain following serum depletion (Fig. 2). As a complement to these assays, we quantified a biochemical marker of apoptosis following Myc activation. Twist expression reduced levels of active CCP32 (caspase-3) to about one-half of those detected in control, LacZ-expressing cells (data not shown). In aggregate, these results demonstrate that both Twist and Dermo1 can protect from oncogene-induced apoptosis.
Figure 2.

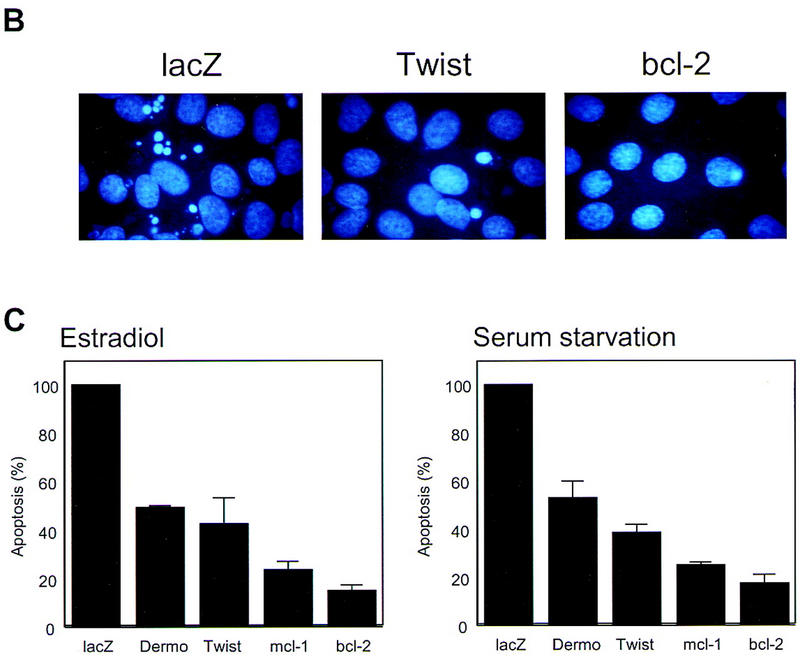
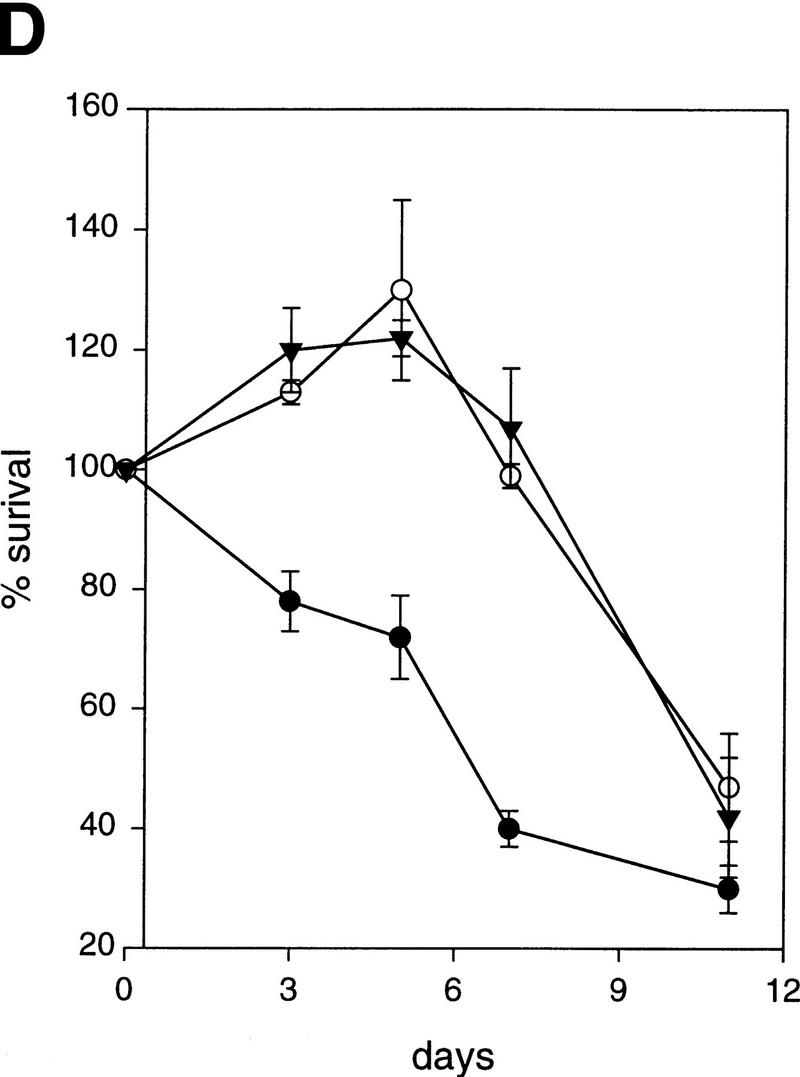
Twist protects from Myc-induced apoptosis. (A) Rat-1/MycER cells were infected with retroviral vectors (pHygroMarXII) that direct the expression of LacZ (a,d,g), Twist (b,e,h), or Bcl-2 (c,f,i), respectively. All cells were grown initially in 10% FBS. Cells were then exchanged into either 0.1% FBS plus 0.1 μm estradiol for 24 hr (d–f) or into 0.1% FBS for 8 days (g–i). (B) Protection from Myc-induced apoptosis was quantified by counting the number of apoptotic bodies (stained with Hoechst 33342) in cells that had been grown in 0.1% FBS plus 0.1 μm estradiol for 24 hr. Representative fields are shown. (C) For quantitation of apoptosis, cells that had been plated in 10% FBS were shifted into either 0.1% FBS plus 0.1 μm estradiol for 24 hr (left) or into 0.1% FBS for 72 hr (right). Twist- and Dermo1-infected populations are shown in comparison with lacZ-infected cells. The extent of apoptosis in each cell line is given with reference to the number of apoptotic bodies counted in the LacZ-infected population (100%). Bcl-2 and Mcl-1-infected cells were included as controls. Values represent means ± s.e.m. of three independent experiments. (D) Apoptosis of Rat-1/MycER cells following serum withdrawal was also quantified by counting viable cells. At the indicated times, adherent and nonadherent populations were collected and stained with trypan blue. Numbers of viable cells are given with reference to the starting populations. (●) Gal; (○) Twist; (▾) Dermo1. Values represent means ± s.d. of three independent experiments.
In addition to its ability to protect from acute apoptotic stimuli, Twist also conferred long-term protection. Both Rat1 and Rat1/MycER cells die on prolonged (∼3 weeks) serum starvation. Expression of Twist not only delayed the appearance of apoptotic cells but also prevented cell death in a significant percentage of infected cells (Fig. 2; data not shown). In longer term assays, Twist was indistinguishable from Bcl-2 in the ability to prevent cell death. Dermo1 was similar to Twist in its ability to protect from both acute and long-term proapoptotic stimuli.
Twist and Dermo1 could potentially protect from programmed cell death through a variety of mechanisms. Because Twist and Dermo1 share features of transcription factors, we asked whether ectopic expression of these proteins affected the abundance of known antiapoptotic proteins. We found no evidence for changes in the levels of several members of the bcl-2 family, nor did we observe altered expression of the MycER protein that provided the proapoptotic stimulus in these cells. Twist and other bHLH transcription factors have been implicated in the control of diverse developmental processes. We therefore tested the possibility that ectopic expression of Twist or Dermo1 might alter the identity of fibroblasts in a manner that increased resistance to myc-induced cell death. However, Twist-infected fibroblasts maintained their original morphology and retained the expression of a constellation of markers that is characteristic of the fibroblastic lineage (data not shown).
Twist antagonizes p53
The tumor suppressor p53 plays a critical role in regulating cell death in response to a variety of stimuli. In fact, Myc-induced cell death has a clearly demonstrated dependence on p53 in a number of experimental systems (Hermeking and Eick 1994; Wagner et al. 1994). Thus, we examined the possibility that Twist might protect from apoptosis by damping the p53 response. Although Rat1/MycER cells express wild-type p53, the proapoptotic role of p53 in these cells is not well established. We therefore used a cell line in which apoptosis has a demonstrated dependence on p53 function.
MEFs that express both E1A and Ha-RasV12 (C8 MEF; Lowe et al. 1993,1994) execute a cell death program in response to a variety of insults; among these are DNA damage, growth factor deprivation, and contact inhibition. This apoptosis is strictly p53-dependent, because analogously engineered MEFs derived from p53-null mice do not die under identical conditions (Lowe et al. 1993). As was also observed for Bcl-2, ectopic expression of Twist dramatically delayed apoptosis following adriamycin treatment of C8 MEFs (Fig. 3B). Moreover, Twist–C8 MEF resisted serum starvation and contact inhibition; a significant population of Twist–C8 MEF survived in the long term (∼2 weeks), whereas essentially none of the LacZ-infected control cells survived (Fig. 3A). Similar levels of protection were afforded by ectopic expression of Dermo1 (Fig. 3).
Figure 3.




Twist antagonizes p53. (A) E1A-Ras expressing MEFs (C8 MEFs) were infected with a retroviral vector (pBabe-puro) encoding LacZ, Twist, Dermo1, or Bcl-2, as indicated. Cells were plated in the presence of 10% FBS and after 24 hr were shifted to 0.1% FBS for 5 days. The result of a representative experiment is shown. Similar results were obtained after adriamycin-treatment (0.2 μg/ml) and after contact inhibition. (B) C8 MEFs infected with retroviruses that direct the expression of either Twist (shaded bars) or β-gal (solid bars) were treated with 0.2 μg/ml adriamycin for the indicated times. Viable cell numbers were determined by trypan blue staining and normalized to the starting cell number for each culture (100%). Values represent means ± s.d. for four independent experiments from two independent infections. (C) MEF-A3s were infected with retroviral vectors (pBabe-puro) that direct the expression of LacZ, Twist, or Bcl-2. After drug selection, cells were plated at low density and shifted to the permissive temperature of 32°C. Colony formation was monitored after 10 days. (D) C8 MEFs were infected with a control, LacZ (G) virus, or with viruses that direct the expression of Myc-tagged Twist (T) or Dermo1 (D). Protein expression was verified by Western blotting with a monoclonal antibody to the Myc tag (9E10).
Considered as a whole, our data suggested that the antiapoptotic effects of Twist and Dermo1 may result, at least in part, from antagonism of the p53 pathway. We therefore asked whether Twist could interfere with other aspects of p53 function. Specifically, we probed the effect of Twist on p53-mediated growth arrest. Embryo fibroblasts from a p53-null mouse were engineered to express a temperature-sensitive version of p53 (p53Val135; MEF-A3). MEF-A3 can be continuously grown at 39°C, a temperature at which p53 assumes a nonfunctional conformation. However, at the permissive temperature of 32°C, the ectopically expressed p53 assumes a wild-type conformation, and cells reversibly arrest in the G1 phase of the cell cycle (Michalovitz et al. 1990). MEF-A3s were infected either with a retrovirus that directs Twist expression or with a control (LacZ) virus. Upon shift to 32°C, control cells arrested and failed to form colonies, whereas a significant percentage of Twist-infected cells continued to proliferate and formed colonies (Fig. 3C). The penetrance of this bypass was similar to that seen on expression of a highly effective p53 antisense RNA (A. Carnero, D. Beach, and G. Hannon, unpubl.). In contrast, MEF-A3 cells infected with a Bcl-2 virus did not form colonies (Fig. 3C). This suggests a specific antagonism of p53 by Twist rather than rescue of colony formation as a secondary consequence of the ability of Twist to protect from apoptosis.
The ability of p53 to transactivate target genes is key for efficient induction of growth arrest and apoptosis (Attardi et al. 1996; Chen et al. 1996). Therefore, we tested whether Twist interfered with the ability of p53 to function as a transcriptional activator. Increasing amounts of a Twist expression construct were transfected into p53-null MEFs in combination with fixed amounts of a p53 expression vector and a p53-dependent reporter. In a dose-dependent manner, Twist suppressed transcription from a synthetic, p53-responsive promoter (PG-13) but had no effect on nonresponsive promoters (Fig. 4A; data not shown). Similar effects were observed in U2OS cells wherein the synthetic p53-responsive promoter depends on endogenous p53 for its activity (Fig. 4B).
Figure 4.



Twist interferes with the induction of p53-target genes. (A) p53 −/−MEFs were transfected with 1 μg of the p53-reporter plasmid (PG-13), a p53-expression plasmid (2 ng), and increasing amounts of Twist expression vector (pCDNA3–Twist). As an internal control, 0.5 μg of a LacZ-expressing plasmid was included in each transfection. At 24 hr post-transfection, cells were lysed and luciferase and β-galactosidase activities were measured (Promega). Values represent means ± s.e.m. of luciferase activities (normalized to β-galactosidase activities) from three independent transfections. (B) U2OS cells were transfected with 1 μg of the p53-reporter plasmid (PG-13), Twist-expressing vector (pCDNA3–Twist) as indicated, and 0.5 μg of LacZ plasmid. At 24-hr post-transfection, cells were processed and analyzed as described above. (C) C8 cells infected with either a Twist (T) or a control (lacZ, G) retrovirus were treated with adriamycin (adria, 0.2 μg/ml) for 10 hr. Total RNA was prepared from these and from parallel, untreated cultures (nt). Transcripts corresponding to p21, MDM2, and Bax were visualized by Northern blotting. For MDM2, only the major (3.3 kb) transcript is shown. The minor transcript (1.8 kb) showed the same relative response. All blots were probed with β-actin to control for RNA loading (a representative is shown).
Because Twist could interfere with the transcription of a p53-dependent reporter, we probed the effects of ectopic Twist expression on the induction of p53 target genes. For these experiments, we used C8 cells in which Twist had been shown to antagonize p53-dependent apopotsis. Control cells, infected with a β-galactosidase retrovirus, induce p21, bax, and MDM2 mRNAs on treatment with adriamycin, a DNA damaging agent that provokes a p53 response (Fig. 4C, lanes G). In contrast, Twist-expressing cells fail to induce p21 and induce MDM2 to a lesser extent than do control cells (Fig. 4C, lanes T). Induction of bax is also impaired in Twist expressing cells; however, effects on this gene are more subtle than are those observed for other targets. In these same cells, neither the basal level of p53 protein or mRNA nor the extent to which p53 protein was induced following adriamycin treatment was influenced by ectopic Twist expression (data not shown).
A potential mechanism underlying regulation of p53 by Twist
Recent evidence suggests that oncogenes such as myc and E1A sensitize cells to p53-dependent cell death, at least in part, through effect on the ARF tumor suppressor (de Stanchina et al. 1998; Zindy et al. 1998). ARF is an upstream regulator of p53 that acts through effects on the localization and activity of MDM2 (Honda and Yasuda et al. 1998; Zhang et al. 1998; Weber et al. 1999; Zhang and Xiong 1999). Expression of either E1A or Myc in primary MEFs provoked substantial increases in ARF mRNA levels (de Stanchina et al. 1998; Zindy et al. 1998), leading, in turn, to activation of the p53 pathway and to consequent induction of downstream targets such as p21 and MDM2. The p53 pathway failed to respond to E1A or Myc in ARF-null cells, placing ARF as a key mediator of homeostatic responses to oncogene expression. Therefore, we asked whether Twist expression had any effect on ARF.
C8 cells that have been engineered to ectopically express Twist show a dramatic reduction in ARF mRNA as compared with control (LacZ-expressing) cells (Fig. 5). This down-regulation is striking considering that loss of p53 function such as is observed in the Twist-expressing cells normally results in substantial increases in the abundance of the ARF transcript (Quelle et al. 1995).
Figure 5.

Twist down-regulates ARF. RNA was prepared from C8 cells infected with retroviruses that direct the expression of either Twist (T) or LacZ (G). ARF transcript was visualized by Northern blotting with an ARF-specific probe (exon 1β). For comparison, RNA was also prepared from BALBc 3T3 cells (B) that lack ARF expression and from primary MEFs (M) that are known to express ARF at high levels. The same blot was also probed for mouse β-actin as a control. Identical results are obtained from multiple independent infections and with cells plated under a variety of different conditions. Induction of apoptosis by treatment with adriamycin had no effect on the reduction of ARF mRNA by Twist.
Down-regulation of ARF provides a potential mechanism by which Twist may affect p53 function. ARF-null MEFs are resistant to p53-induced growth arrest, and ectopic expression of p53 in these cells does not activate the p21 promoter (Kamijo et al. 1998). Furthermore, MEFs lacking ARF exhibit a reduced apoptotic response to myc and E1A. Disruption of ARF led to an ∼50% reduction in cell death in myc-expressing MEFs that had been deprived of serum survival factors (Zindy et al. 1998). Resistance was not as complete as was achieved on disruption of p53 itself, indicating that ARF-independent mechanisms also contribute to factor-dependant survival. Similarly, ARF disruption in E1A-expressing MEFs led to a decreased sensitivity to adriamycin (de Stanchina et al. 1998). In both cases, the response of p53 targets was attenuated, although not all were affected to the same degree. Thus, cells that express Twist ectopically share many features with ARF-null MEFs. However, in Twist-expressing cells, some ARF mRNA persists. Furthermore, Twist is undoubtedly a multifunctional protein. Therefore, the consequences of ARF loss and ectopic Twist expression are unlikely to perfectly overlap.
Relatively little is known about the regulation of ARF expression. It is still unclear whether induction of ARF by oncogenes such as myc and E1A is direct or results from secondary effects on transcriptional regulators such as E2F-1 that also modulate ARF transcription (DeGregori et al. 1997; Bates et al. 1998; Robertson and Jones 1998). Similarly, we do not yet know whether down-regulation of ARF by Twist is mediated through an effect on the ARF promoter or through an indirect route.
Although decreases in ARF may be sufficient to explain the observed effects of Twist on p53, we cannot exclude that additional mechanisms may also contribute. The activity of p53 is tightly controlled by a complex series of pathways that are interconnected by feedback loops. Both the synthesis and degradation of the p53 protein are regulated in response to inducing stimuli (for review, see Ko and Prives 1996; Agarwal et al. 1998). In addition, post-translational modifications such as acetylation and phosphorylation as well as an association with cofactors regulate the stability and the specific activity of this transcription factor (Haupt et al. 1997; Kubbutat et al. 1997; Shieh et al. 1997; Siciliano et al. 1997). In particular, the activity of p53 can be modulated by its interaction with the coactivator p300/CBP. p300/CBP is an acetyltransferase that can modify p53 and alter its ability to bind target sequences in vitro (Avantaggiati et al. 1997; Gu et al. 1997; Lill et al. 1997). Moreover, through its interaction with MDM2, p300 has been linked to p53 degradation (Grossman et al. 1998). Twist has been shown recently to interact physically with p300 and to inhibit acetyltransferase activity in an in vitro assay (Hamamori et al. 1999). Thus, we cannot rule out the possibility that Twist may also modulate p53 activity through effects on p300/CBP and related partners.
twist is a candidate oncogene product for rhabdomyosarcoma
Increased resistance to programmed cell death, disruption of the INK4/ARF locus, and loss of p53 activity are common characteristics of human tumor cells. Expression of Twist can prevent apoptosis, down-regulate ARF, and interfere with p53 function. We therefore examined the possibility that twist might have properties that are characteristic of oncogene products.
Loss of anchorage dependence is a hallmark of tumor cells, and the ability to promote anchorage-independent growth is a common property of oncogenes. Some transformed cells, such as fibroblasts that express either a combination of Ras and Myc or a combination of Ras and E1A (e.g., C8 MEFs), show a low efficiency of anchorage-independent growth. This failure is probably due to a predisposition to apoptosis because inhibition of programmed cell death through loss of p53, inactivation of the apoptotic machinery, or expression of protective oncoproteins such as Bcl-2 can promote colony formation (Nikiforov et al. 1996; Soengas et al. 1999). Therefore, we tested whether expression of either Twist or Dermo1 could allow the growth of C8 cells in soft agar. In accord with previous reports, C8 cells infected with a control, LacZ retrovirus form a few small colonies in semisolid media (Fig. 6). In contrast, expression of either Twist or Dermo1 stimulates formation of robust colonies in soft agar. Similar results are obtained on expression of either Bcl-2 (Fig. 6) or a dominant, interfering allele of p53 (Nikiforov et al. 1996). These results indicate that twist and dermo1 share one property of oncogene products, the ability to promote anchorage-independent growth.
Figure 6.

Twist and Dermo1 promote colony formation in soft agar. C8 MEFs were infected with retroviruses that direct the expression of LacZ, Twist, Dermo1, or Bcl-2. Infected cells were plated in soft agar, and colony formation was assessed after 2 weeks (as indicated). The LacZ-expressing cells form only a few, small colonies. Cells infected with Twist, Dermo1, or Bcl-2 form large colonies similar to those that are observed on expression of dominant-interfering alleles of p53. Expression of Twist or Dermo1 enhances colony formation by approximately five to sevenfold.
To assess the possibility that aberrant twist expression might be a feature of human cancers, a variety of human tissue and tumor samples (archivally preserved primary patient material) were tested for the abundance of Twist protein. Consistent with studies on mouse embryos (Futchbauer 1995; Gitelman 1997), antibodies raised against either the amino or the carboxyl terminus of Twist gave a specific nuclear staining pattern in human embryonic mesenchymal tissues (not shown). Twist protein was not detectable in a panel of common epithelial tumors such as those of the breast, colon, ovary, and lung. However, high-level Twist expression was seen in ∼50% of rhabdomyosarcomas (8/15 cases analyzed). In Twist-positive tumors, antisera directed against either the amino or carboxyl terminus decorated a percentage of neoplastic cells ranging between 60% and 90%. Surrounding normal tissues and Twist-negative rhabdomyosarcomas displayed no detectable Twist immunoreactivity (Fig. 7).
Figure 7.
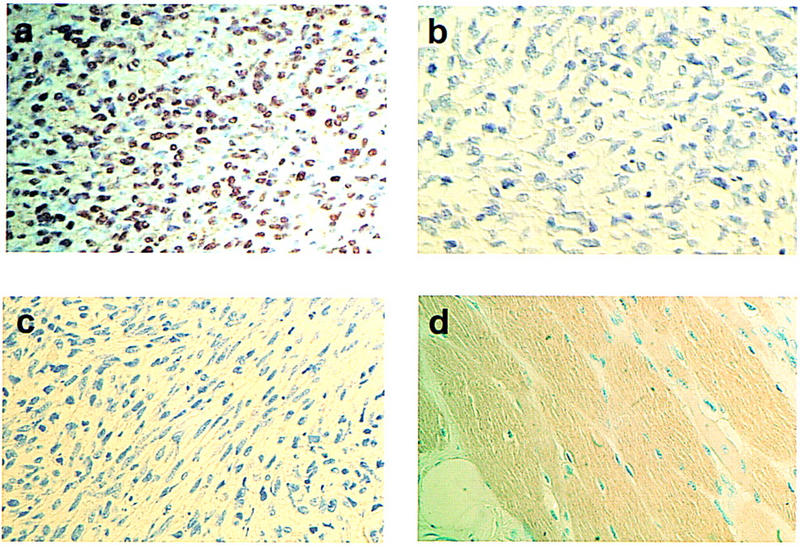
Twist is highly expressed in rhabdomyosarcomas. (a) Formalin-fixed histologic sections of a rhabdomyosarcoma were stained with a Twist-specific antibody (SC-6070). Most of the neoplastic cells show nuclear accumulation of Twist protein. (b) Specificity of the signal was confirmed by loss of Twist immunoreactivity after preincubation of Twist antibody with the antigenic peptide. (c) A representative stain of a Twist-negative rhabdomyosarcoma is shown. (d) The lack of Twist immunoreactivity in differentiated skeletal muscle. Original magnification 400×.
Rhabdomyosarcomas constitute a heterogeneous group of malignant tumors, mainly affecting children, that originate from undifferentiated mesenchymal cells. In rhabdomyosarcoma cells, skeletal muscle differentiation is arrested at an early stage despite the expression of myogenic markers, such as myoD and myogenin (Pappo 1996). It has been well established that Twist is excluded from the developing myotome and is not expressed in differentiated skeletal muscle (Fig. 7d; Futchbauer 1995; Gitelman 1997). These observations led to in vitro experiments that demonstrated the ability of Twist to block myogenic differentiation in cultured cells (Spicer et al. 1996; Hamamori et al. 1997; Hebrok et al. 1997). Therefore, our finding of inappropriate Twist expression in rhabdomyosarcomas suggests that Twist may have multiple roles in the formation of these tumors. First, Twist expression might halt the developmental program that leads to terminal differentiation and withdrawal of muscle cell precursors from the division cycle. Second, Twist expression might antagonize apoptosis. Third, Twist may interfere with the p53 tumor suppressor pathway, the loss of which is one of the most common genetic alterations in human tumors.
Although inappropriate expression of Twist may have a role in the genesis of some tumors, reduction of Twist activity can also have dramatic consequences for a developing organism. In Drosophila, Xenopus, and mouse, twist is essential for mesoderm formation (Chen and Behringer 1995; Futchbauer 1995; Thisse et al. 1995; Gitelman 1997). twist-null mice die at day 11.5 postcoitum. Just prior to death, these animals show a massive wave of apoptosis in the developing somites, a site in which Twist is normally expressed (Chen and Behringer 1995). Alterations in Twist activity have also been linked to developmental abnormalities in humans. Mutations in the twist gene have been causatively linked to Saethre–Chotzen syndrome (el Gouzzi et al. 1997; Howard et al. 1997) , a hereditary disorder characterized by a variety of limb and craniofacial anomalies. Of these, craniosynostosis is the most striking. This malformation of the skull is caused by premature fusion of cranial sutures. It has long been proposed that many craniosynostosis syndromes result from local perturbation of apoptotic programs that are essential for proper timing of suture fusion (Bourez et al. 1997). Considered together, the phenotype of organisms with altered Twist activity is consistent with a role for Twist in regulating apoptosis during development. Rhabdomyosarcoma cells may exploit this normal function of Twist to counteract the proapoptotic stimuli that result from oncogene activation.
Materials and methods
Cells
Rat-1/MycER cells that express an estrogen-inducible myc gene (Evan et al. 1992) were maintained at 5% CO2 in DMEM without phenol red (wDMEM), supplemented with 10% FBS. C8 MEF cells (mouse embryo fibroblasts that express E1A and H-RasVal12) (Lowe et al. 1993) and the ecotropic packaging cell line, LinX (Hannon et al. 1999), were maintained in 5% CO2 in DMEM, supplemented with 0.01% Na pyruvate and 10% FBS. MEF-A3 cells were produced by infecting MEFs derived from p21/p53-null mice with a retroviral vector that directs the expression of a temperature-sensitive mutant of p53, p53val135. A resulting clonal cell line (MEF-A3) that rapidly arrested after p53 induction at the permissive temperature (32°C) was used for colony formation analysis. MEF-A3s were grown in 5% CO2 in DMEM supplemented with 0.01% Na pyruvate and 10% FBS at 39°C.
Library construction and screen strategy
Poly(A)+ RNA was extracted by the use of Triazol reagent (GIBCO–BRL), from Rat-1/MycER cells committed to apoptosis by 6 hr of serum starvation. Oligo(dT)-primed cDNA was produced with the Stratagene ZapII cDNA synthesis kit (Hannon et al. 1993). Fragments were cloned into the retroviral expression vector pHygroMaRXII at the EcoR1 and XhoI sites (Hannon et al. 1999). The library was divided into 100 independent DNA plasmid pools, each with a complexity of 104–105 clones. Each plasmid pool was used to transfect LinX E packaging cells by the calcium phosphate method. At 72-hr post-transfection, viral supernatants were collected, filtered, supplemented with 4 μg/ml Polybrene, and used to infect Rat-1/MycER cells. After infection, cells were selected with hygromicin and then subjected to an apoptosis/rescue schedule as described: A total of 5 × 105 Rat-1/MycER cells were plated onto 10-cm dishes. At 24-hr postplating, apoptosis was induced by adding fresh medium supplemented with 0.1% FBS plus 0.1 μm estradiol for 3 days, followed by 2 days of culture in serum starvation without estradiol. Cells were then refed with medium containing 10% FBS. Rescued cells were then replated at low density and subjected to another cycle of killing as above. Cycles of killing/rescue were repeated four times.
Recovery of viral genomes and analysis of the recovered plasmids
Genomic DNA was extracted from cell populations that had been enriched for resistance to apoptosis by a standard proteinase K/SDS method. Five micrograms of genomic DNA were treated with CRE recombinase, phenol extracted, ethanol precipitated, and used to transform electrocompetent bacteria. Fifty recovered plasmids were analyzed from each pool by restriction digestion. Those plasmids that represented >5% of the recovered plasmid species were sequenced. A subset of these was again introduced into Rat1/MycER cells and tested for the ability to protect from apoptosis.
Quantitation of apoptosis in Rat-1/MycER cells
Rat-1/MycER cells infected with pHygroMarX retroviral vectors that direct the expression of the Twist, Dermo1, Bcl-2, and LacZ, respectively, were plated at low density (105 cells/well) onto acid-treated coverslips in 6 well plates. Twenty-four hours after plating, cells were washed twice with PBS and then induced to apoptose. Myc-induced apoptosis was triggered by treatment with 0.1 μm estradiol in 0.1% FBS. Apoptosis was monitored 24 hr post-induction. Growth factor deprivation-induced apoptosis was triggered by replacing the culture medium with fresh DMEM supplemented with 0.1% FBS. The extent of apoptosis was quantified at 72 hr post-treatment by Hoechst staining (Attardi et al. 1996). Briefly, cells were directly stained with 4 μ/ml of Hoechst 33342 for 10 min, washed with PBS, and mounted. At least 100 fields/slide were analyzed and the number of apoptotic bodies was evaluated blind by two independent observers.
Moreover, protection from apoptosis was also analyzed by trypan blue exclusion. Cells were seeded into six-well plates (105/ well) 24 hr prior to serum withdrawal. At various times, adherent and nonadherent cells were pooled and viability assessed by trypan blue exclusion.
Apoptosis in C8 MEFs
MEFs that express E1A and H-RasVal12 (C8 MEFs) were infected with retroviral vectors (pBABE-Puro) that drive the expression of LacZ, Bcl-2, Twist, Dermo1, or Myc-tagged versions of Twist and Dermo1. In all assays, Myc-tagged Twist and Dermo1 were indistinguishable from the untagged proteins. Twist and Dermo1 protein expression in C8 MEFs was confirmed by Western blotting with a monoclonal antibody to the Myc tag (9E10).
After selection, cells were plated at low density and maintained in complete media to monitor the cell–cell contact-triggered apoptosis, subjected to serum starvation (0.1% FBS) for 5 days, or treated with adriamycin (0.2 μg/ml) for 2 days.
Cell viability after adriamycin treatment was assessed by trypan blue exclusion. Briefly, cells were seeded into six-well plates (105/ well) 48 hr prior to drug treatment (adriamycin, 0.2 μg/ml). At various times, adherent and nonadherent cells were pooled and a trypan blue exclusion test performed.
RNA extraction and Northern blot analysis
Total RNA was extracted from C8 MEFs infected with pBABE-lacZ and pBABE-Twist expression vectors in normal growth conditions and after induction of apoptosis by adriamycin treatment (0.1 μg/ml ) for 10 hr. Triazol reagent (GIBCO-BRL) was used according to the manufacturer’s instructions. An additional final precipitation in LiCl was performed to further purify RNA from contaminant DNA. Briefly, after Triazol extraction the RNA pellet was resupended in 5 vol of 100 mm HEPES (pH 7.5) and the same volume of 5 m LiCl was added drop-wise. Precipitation was performed at −20°C for at least 4 hr.
For Northern blots, 10 μg of total RNA was loaded per lane and fractionated in a 1% agarose/formaldehyde gel. After transfer onto Hybond N+ membrane (Amersham), blots were hybridized with 32P-labeled probes specific for mouse p21, mdm2, p19ARF (exon Iβ), and human bax genes. A probe specific for mouse β-actin was used to confirm equal loading. Membranes were hybridized overnight at 65°C in 0.2 m NaPO4, 1 mm EDTA, 7% SDS, and 1% BSA in the presence (mouse probes) or absence (human bax probe) of 15% formamide. Membranes were washed twice in 0.1% SDS, 0.2× SSC and once in 0.1× SSC at 60°C, followed by autoradiography.
Bypass of p53-induced growth arrest
MEF-A3 cells that express a temperature-sensitive version of p53 (p53val135) were infected with pBABE-Puro vectors that drive the expression of LacZ, Twist, or Bcl-2, respectively. After selection, 5 × 104 cells were plated in quadruplicate in 10 cm plates. On the following day, two plates were shifted to the permissive temperature (32°C). The remaining two plates were used as controls for plating efficiency. After 10–15 days, colony formation was scored by crystal violet staining.
Cell transfections and transcription assays
Transfections of MEF p53−/− and U2OS cells were performed according to the calcium phosphate precipitation protocol as described (Hamamori et al. 1997). A total of 9 μg of plasmid DNA per 6-cm-diam. dish was used. As an internal control, 0.5 μg of a LacZ-expressing plasmid was included in each transfection. At 20 hr post-transfection, cells were refed, incubated for 2 additional days, and harvested for reporter gene assays. Luciferase and β-galactosidase activities were measured by a MLX microtiter plate luminometer (Dynex, Chantilly, VA). Values represent means ± s.e.m. of luciferase activities (normalized to β-galactosidase) from at least three independent transfections performed in duplicate.
Anchorage-independent growth in C8 MEFs
C8 MEFs infected with retroviral vectors (pBABE-Puro) that drive the expression of LacZ, Twist, Dermo1, or Bcl-2, were analyzed for anchorage-independent growth in semi-solid medium. Approximately 105 cells were plated in 0.3% low-melting-point agarose/growth medium onto 60-mm dishes with a 0.5% agarose underlay. Colonies were photographed after 2 weeks.
Immunohistochemistry
A series of common human tumors including 10 gastric and colorectal carcinomas, 6 breast, 10 lung, and 4 ovarian carcinomas, 2 Kaposi’s sarcomas, 3 melanomas, 8 leiomyosarcomas, and 15 rhabdomyosarcomas were analyzed by immunohistochemistry for Twist expression with an avidin–biotin–peroxidase complex (ABC) technique. Formalin-fixed histologic sections were incubated with an affinity-purified goat polyclonal antibody raised against a peptide corresponding to an amino acid sequence at the amino terminus of human Twist (SC-6070, Santa Cruz Biotechnology; dilution, 0.2 μg/ml) or with an affinity-purified goat polyclonal antibody raised against a peptide corresponding to an amino acid sequence mapping at the carboxyl terminus of human Twist (SC-6269, Santa Cruz Biotechnology, dilution: 0.2 μg/ml). The primary antibodies were incubated at +4°C overnight. Immunoreaction was visualized with a biotin-conjugated anti-goat antiserum followed by peroxidase–streptavidin and DAB chromogen development. The specificity of the SC-6070 antiserum was also confirmed by an adsorption test. Peptide sc 6070p (SantaCruz) corresponding to the amino acid sequence of the amino terminus of human Twist (1 μg/ml) was incubated with the anti-Twist goat polyclonal antibody (0.2 μg/ml) for 2 hr at room temperature before immunostaining. The percentage of immunoreactive cells was evaluated by scanning sections and counting at least 1000 neoplastic cells.
Acknowledgments
R.M. was supported by a grant from the Italian Association for Cancer Research (AIRC). L.K. is supported in part by a grant from the National Institutes of Health (NIH). Y.H. is supported by an Initial Investigatorship (1104-FI1) and V.S. by a grant-in-aid (1060-G1) from the American Heart Association of Greater Los Angeles. D.B. is the Hugh and Catherine Stevenson Chair in Cancer Biology. G.J.H. is supported by grants from the US Army (DAMD 17-96-1-6053), the NIH, and the Stewart Trust and is a Pew Scholar in the Biomedical Sciences. We thank Chris McCollough, Sara Piccinin, and Martina Fabris for their help and support, Bert Vogelstein for providing the PG-13 reporter construct, and Scott Lowe and Linda Penn for C8 MEF and Rat-1/MycER cells, respectively.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL hannon@cshl.org; FAX (516) 367-8874.
References
- Agarwal ML, Taylor WR, Chernov MV, Chernova OB, Stark GR. The p53 network. J Biol Chem. 1998;273:1–4. doi: 10.1074/jbc.273.1.1. [DOI] [PubMed] [Google Scholar]
- Attardi LD, Lowe SW, Brugarolas J, Jacks T. Transcriptional activation by p53, but not induction of the p21 gene, is essential for oncogene-mediated apoptosis. EMBO J. 1996;15:3693–3701. [PMC free article] [PubMed] [Google Scholar]
- Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–1184. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH. p14ARF links the tumor suppressors RB and p53. Nature. 1998;395:124–125. doi: 10.1038/25867. [DOI] [PubMed] [Google Scholar]
- Bourez RL, Mathijssen IM, Vaandrager JM, Vermeij-Keers C. Apoptotic cell death during normal embryogenesis of the coronal suture: Early detection of apoptosis in mice using annexin V. J Craniofac Surg. 1997;8:441–445. doi: 10.1097/00001665-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Chen X, Ko LJ, Jayaraman L, Prives C. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes & Dev. 1996;10:2438–2451. doi: 10.1101/gad.10.19.2438. [DOI] [PubMed] [Google Scholar]
- Chen ZF, Behringer RR. twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes & Dev. 1995;9:686–699. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes & Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Leone G, Miron A, Jokoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiss LP, Kimchi A. A genetic tool used to identify thioredoxin as a mediator of a growth inhibitory signal. Science. 1991;252:117–120. doi: 10.1126/science.1901424. [DOI] [PubMed] [Google Scholar]
- De Stanchina E, McCurrach ME, Zindy F, Shieh S-Y, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. E1A signaling to p53 involves the p19ARF tumor suppressor. Genes & Dev. 1998;12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Ghouzzi V, Le Merrer M, Perrin-Schmitt F, Lajeunie E, Benit P, Renier D, Bourgeois P, Bolcato-Bellemin AL, Munnich A, Bonaventure J. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet. 1997;15:42–46. doi: 10.1038/ng0197-42. [DOI] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Fuchtbauer EM. Expression of M-twist during postimplantation development of the mouse. Dev Dyn. 1995;204:316–322. doi: 10.1002/aja.1002040309. [DOI] [PubMed] [Google Scholar]
- Gitelman I. Twist protein in mouse embryogenesis. Dev Biol. 1997;189:205–214. doi: 10.1006/dbio.1997.8614. [DOI] [PubMed] [Google Scholar]
- Grossman SR, Perez M, Kung AL, Joseph M, Mansur C, Xiao Z, Kumar S, Howley PM, Livingston DM. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell. 1998;2:405–415. doi: 10.1016/s1097-2765(00)80140-9. [DOI] [PubMed] [Google Scholar]
- Gu W, Shi XL, Roeder RG. Synergistic activation of transcription by CBP and p53. Nature. 1997;387:819–823. doi: 10.1038/42972. [DOI] [PubMed] [Google Scholar]
- Gudkov AV, Roninson IB. Isolation of genetic suppressor elements (GSEs) from random fragment cDNA libraries in retroviral vectors. Methods Mol Biol. 1997;69:221–240. doi: 10.1385/0-89603-383-x:221. [DOI] [PubMed] [Google Scholar]
- Gudkov AV, Kazarov AR, Thimmapaya R, Axenovich SA, Mazo IA, Roninson IB. Cloning mammalian genes by expression selecion of genetic suppressor elements: Association of kinesin with drug resistance and cell immortalization. Proc Natl Acad Sci. 1994;91:3744–3748. doi: 10.1073/pnas.91.9.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamori Y, Hung-Yi W, Sartorelli V, Kedes L. The basic domain of myogenic basic helix-loop-helix (bHLH) proteins is the novel target for direct inhibition by another bHLH protein, Twist. Mol Cell Biol. 1997;17:6563–6573. doi: 10.1128/mcb.17.11.6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamori Y, Sartorelli V, Ogryzko V, Puri PL, Wu HY, Wang JY, Nakatani Y, Kedes L. Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein twist and adenoviral E1A. Cell. 1999;96:405–413. doi: 10.1016/s0092-8674(00)80553-x. [DOI] [PubMed] [Google Scholar]
- Hannon GJ, Demetrick D, Beach D. Isolation of the Rb-related p130 through its interaction with CDK2 and cyclins. Genes & Dev. 1993;7:2378–2391. doi: 10.1101/gad.7.12a.2378. [DOI] [PubMed] [Google Scholar]
- Hannon GJ, Sun P, Carnero A, Xie LY, Maestro R, Conklin D, Beach D. Genetics in mammalian cells. Science. 1999;283:1129–1130. doi: 10.1126/science.283.5405.1129. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Hebrok M, Fuchtbauer A, Fuchtbauer EM. Repression of muscle-specific gene activation by the murine Twist protein. Exp Cell Res. 1997;232:295–303. doi: 10.1006/excr.1997.3541. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- Honda R, Yasuda H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999;18:22–27. doi: 10.1093/emboj/18.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard TD, Paznekas WA, Green ED, Chiang LC, Ma N, Ortiz de Luna RI, Garcia Delgado C, Gonzalez-Ramos M, Kline AD, Jabs EW. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15:36–41. doi: 10.1038/ng0197-36. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci. 1998;95:822–829. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signaling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Kimchi A. DAP genes: Novel apoptotic genes isolated by a functional approach to gene cloning. Biochim Biophys Acta. 1998;1377:F13–F33. doi: 10.1016/s0304-419x(98)00002-x. [DOI] [PubMed] [Google Scholar]
- Ko LJ, Prives C. p53: Puzzle and paradigm. Genes & Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci. 1993;90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Voudsen KH. Regulation of p53 stability by Mdm2. Nature. 1997;278:261–263. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Li L, Cserjesi P, Olson EN. Dermo-1: A novel twist-related bHLH protein expressed in the developing dermis. Dev Biol. 1995;172:280–292. doi: 10.1006/dbio.1995.0023. [DOI] [PubMed] [Google Scholar]
- Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Binding and modulation of p53 by p300/CBP coactivators. Nature. 1997;387:823–827. doi: 10.1038/42981. [DOI] [PubMed] [Google Scholar]
- Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes & Dev. 1998;12:3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes & Dev. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Jacks T, Housman DE, Ruley HE. Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc Natl Acad Sci. 1994;91:2026–2030. doi: 10.1073/pnas.91.6.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalovitz D, Halevy O, Oren M. Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell. 1990;62:671–680. doi: 10.1016/0092-8674(90)90113-s. [DOI] [PubMed] [Google Scholar]
- Nikiforov MA, Hagen K, Ossovskaya VS, Connor TM, Lowe SW, Deichman GI, Gudkov AV. p53 modulation of anchorage independent growth and experimental metastasis. Oncogene. 1996;13:1709–1719. [PubMed] [Google Scholar]
- Pappo AS. Rhabdomyosarcoma and other soft tissue sarcomas in children. Curr Opin Oncol. 1996;8:311–316. doi: 10.1097/00001622-199607000-00008. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Rayner JR, Gonda TJ. A simple and efficient procedure for generating stable expression libraries by cDNA cloning in a retroviral vector. Mol Cell Biol. 1994;14:880–887. doi: 10.1128/mcb.14.2.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD, Jones PA. The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Mol Cell Biol. 1998;18:6457–6473. doi: 10.1128/mcb.18.11.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Siciliano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA damage induces phosphorylation of the amino terminus of p53. Genes & Dev. 1997;11:3471–3481. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 1999;284:156–159. doi: 10.1126/science.284.5411.156. [DOI] [PubMed] [Google Scholar]
- Spicer DB, Rhee J, Cheung WL, Lassar AB. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science. 1996;272:1476–1480. doi: 10.1126/science.272.5267.1476. [DOI] [PubMed] [Google Scholar]
- Sun P, Dong P, Dai K, Hannon GJ, Beach D. p53-independent role of MDM2 in TGF-β resistance. Science. 1998;282:2270–2272. doi: 10.1126/science.282.5397.2270. [DOI] [PubMed] [Google Scholar]
- Thisse B, el Messal M, Perrin-Schmitt F. The twist gene: Isolation of a Drosophila zygotic gene necessary for the establishment of dorsoventral pattern. Nucleic Acids Res. 1987;15:3439–3453. doi: 10.1093/nar/15.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. The genetic basis of human cancer. New York, NY: McGraw-Hill; 1998. [Google Scholar]
- Wagner AJ, Kokontis JM, Hay N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1/cip1. Genes & Dev. 1994;23:2817–2830. doi: 10.1101/gad.8.23.2817. [DOI] [PubMed] [Google Scholar]
- Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- Wong BY, Chen H, Chung SW, Wong PM. High-efficiency identification of genes by functional analysis from a retroviral cDNA library. J Virol. 1994;68:5523–5531. doi: 10.1128/jvi.68.9.5523-5531.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y. Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol Cell. 1999;3:579–591. doi: 10.1016/s1097-2765(00)80351-2. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MR. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]