

Abstract
In this population-based study, we determined the frequency and clinical characteristics of leptomeningeal disease (LMD) developing in the context of oligodendroglial tumors (oligodendrogliomas and oligoastrocytomas). LMD occurred in only 3.9% (8/204) of oligodendroglial tumors and in patients with more recurrences [mean 2.88 vs. 1.27 in LMD and non-LMD, respectively (p = 0.001)]. In contrast to LMD from systemic solid tumors, the median survival following the diagnosis of LMD in oligodendroglial tumors was surprisingly long at 22 months (95% CI 11–33 months). Treatment with oral chemotherapy seemed as effective as more aggressive treatments (e.g. repeat RT or intrathecal chemotherapy) in these patients.
Keywords: Oligodendroglial tumors, Leptomeningeal spread
Introduction
Oligodendroglial tumors (oligodendrogliomas and mixed oligoastrocytomas) represent 5–20% of gliomas [1, 2]. In oligodendroglial tumors, co-deletion of chromosomes 1p and 19q (likely a manifestation of a recurring unbalanced 1q/19p translocation) is associated with better overall prognosis [3–5] and response to treatment [6–8]. We recently encountered a series of patients with the unusual complication of leptomeningeal disease (LMD) from oligodendroglial tumors which had several unusual features (e.g. survival with LMD of up to 3.6 years in one patient) [9]. We performed this study to determine how commonly this occurred at the population level and to identify clinical features that might predict the occurrence of LMD.
Patients and methods
Ethical approval was received from the Conjoint Health Research Ethics Board of the University of Calgary to review charts for every patient diagnosed with an oligodendroglial tumor (oligodendrogliomas and mixed oligoastrocytomas WHO grades II and III) diagnosed in Southern Alberta (population 1.8 million) between 1 July 1991 and 31 Aug 2009. Patients were identified through the Alberta Cancer Registry and their individual records were reviewed.
Of the 208 identified patients, four were excluded because they were lost to follow-up without data available. In our institution 1p/19q status was evaluated in the primary tumor by either polymerase chain reaction (PCR) or fluorescent in situ hybridization (FISH) techniques.
The criteria for the diagnosis of leptomeningeal disease (LMD) were any two or more of the following [9]: (1) neurological symptoms or signs consistent with LMD, (2) MRI changes consistent with LMD (i.e., leptomeningeal, subependymal, or subarachnoid spread), or (3) a cytological diagnosis of LMD on examination of cerebrospinal fluid (CSF).
Statistics
Results were analyzed with SPSS Version 16.0 for Microsoft Windows and were two-tailed. For continuous variables, Spearman’s rank correlation test was used to identify associations. For categorical variables, Chi-square or Fisher’s exact tests were used. When comparing the means of continuous variables between groups, an unpaired two group t-test was used.
Progression-free survival (PFS) was defined as the interval between initial diagnosis and evidence of recurrence/progression, death, or lost to follow up. Overall survival (OS) was calculated from date of initial diagnosis to death or lost to follow-up. Time to first onset of LMD was calculated from initial tumor diagnosis.
The Kaplan–Meier method was used to estimate distributions for PFS and OS. The log-rank test was used to assess differences between these distributions with respect to the occurrence of clinical variables. Cox multivariate analysis (forward stepwise Wald) was done to assess their independent predictive value. All tests were two-tailed and p-values less than 0.05 were considered statistically significant.
Results
Population-based analysis of oligodendroglial tumors and risk of LMD
From July 1991 to August 2009, 208 patients in Southern Alberta were diagnosed with oligodendroglial grade II and III tumors; 204 were included in this analysis. This corresponds to an incidence of 1/100,000/year. Their characteristics are summarized in Table 1. For the entire group, mean age at diagnosis was 42 years (range 18–78) and 120 patients (59%) were male. None of the 204 patients had oligodendroglial tumors originated from the posterior fossa. Four patients had bifrontal tumors and the rest were distributed homogeneously in the hemispheres with 108/200 (54%) in the left hemisphere. 137/204 (67%) of the patients had tumors involving the frontal lobes. Biomarker data was available on 161 patients (79%); 93 (58%) were co-deleted for 1p and 19q.
Table 1.
Clinical characteristics of patients with oligodendroglial tumors that developed leptomeningeal disease compared with 196 that did not
| Total Population (n = 204) | LMD (n = 8)a | No LMD (n = 196)b | Significance (p)a vs. b | |
|---|---|---|---|---|
| Mean age in years (range) | 42 (18–78) | 40 (28–50) | 42 (18–78) | 0.636 |
| Median KPS (range) | 85 (50–100) | 85 (70–90) | 90 (50–100) | 0.477 |
| Debulking (%) | 173 (85) | 7 (87.5) | 166 (85) | 0.828 |
| Pathology at diagnosis (%) | 0.079 | |||
| • Oligo. Gr. II | 62 (30.5) | 1 (12.5) | 61 (31.1) | |
| • Oligo. Gr. III | 49 (24) | 5 (62.5) | 44 (22.4) | |
| • Mixed OA Gr. II | 52 (25.5) | 1 (12.5) | 51 (26) | |
| • Mixed OA Gr. III | 41 (20) | 1 (12.5) | 40 (20.4) | |
| 1p/19q | 0.127 | |||
| • Not tested (%) | 43 (21) | 1/7 (14) | 42 (21) | |
| • Codeletedc (% of tested) | 93/161 (58) | 6/7 (86) | 87/154 (56.4) | |
| Initial treatment (%) | 0.785 | |||
| • Observation | 96 (47) | 5 (62.5) | 91 (46) | |
| • RT alone | 35 (17) | 1 (12.5) | 34 (17.3) | |
| • ChT alone | 18 (9) | 1 (12.5) | 17 (9) | |
| • SeqRTChT | 26 (13) | 0 | 26 (13.3) | |
| • ConcRTChT | 29 (14) | 1 (12.5) | 28 (14.3) | |
| Mean number of recurrences | 1.35 | 2.88 | 1.27 | 0.001 |
| PFS (months) (mean; 95% CI) | 67 (59–75) | 47 (22–72) | 68 (59–77) | 0.176 |
| OS (months) (mean; 95% CI) | 130 (118–142) | 107 (82–132) | 133 (120–146) | 0.063 |
LMD leptomeningeal disease, Oligo oligodendroglioma, OA oligoastrocytoma, RT radiotherapy, ChT chemotherapy, SeqRTChT sequential RT followed by ChT, ConcRTChT concurrent RT and ChT, PFS progression free survival, OS overall Survival
aPatients with leptomeningeal disease
bPatients without leptomeningeal disease
cIncludes patients with only 1p loss
With respect to initial treatment, 85/114 (75%) patients with low grade tumors were observed (i.e., received no tumor treatment beyond surgery) while 78/90 (87%) with anaplastic tumors received immediate postoperative treatment that consisted of radiotherapy (RT) alone (14%), chemotherapy (ChT) alone (24%), RT followed by ChT (32%) or concurrent RT and ChT (30%).
We analyzed clinical factors associated with OS and found that published prognostic factors were relevant in our population. Univariate analysis for OS showed that females lived longer (Log Rank test; p = 0.036). Among grade III tumors, patients with pure oligodendrogliomas lived a mean of 124 months compared to 84 months for those with mixed tumors (p < 0.0001). Patients with WHO grade II histology lived longer than those with anaplastic (grade III) tumors (p < 0.0001) and co-deletion of 1p/19q was associated with better overall survival (p = 0.004). Extent of initial resection did not impact OS (even when controlling for pathology, WHO grade, or genetic subtype). Tumors recurred in 122 patients and 73 had further surgery. Surgery at first recurrence was associated with longer OS (mean OS of 140 vs. 97 months; p = 0.001).
Comparison of clinical and pathological characteristics in patients with LMD
Only 3.9% (8 of 204) of patients with oligodendroglial tumors developed LMD during the 18 year study period (Table 1). The only factor statistically associated with LMD was the number of tumor recurrences that were higher in the LMD group (mean of 2.88 recurrences) as compared to the no-LMD group (1.27 recurrences; p = 0.001). There was no significant difference in patients with or without LMD in terms of age, KPS, surgical resection, pathology [pure vs. mixed; p = 0.47) or low grade vs. high grade (p = 0.073)], co-deletion of 1p/19q (p = 0.127), or initial postoperative treatment (p = 0.785).
Outcomes in patients with LMD
The mean time from the initial diagnosis of a brain tumor until the diagnosis of LMD was 77 months (95% CI 51–102 months). Surprisingly, in contrast to LMD from systemic solid tumors or GBM which have a universally poor prognosis, the progression-free and overall survivals were not statistically different between patients with and without LMD (Table 1) although there was a trend for both to be shorter in patients with LMD (Fig. 1). The mean survival following the diagnosis of LMD mean was 22 months (95% CI 11–33 months) but this varied considerably from 1.8 to 42 months (Fig. 2).
Fig. 1.
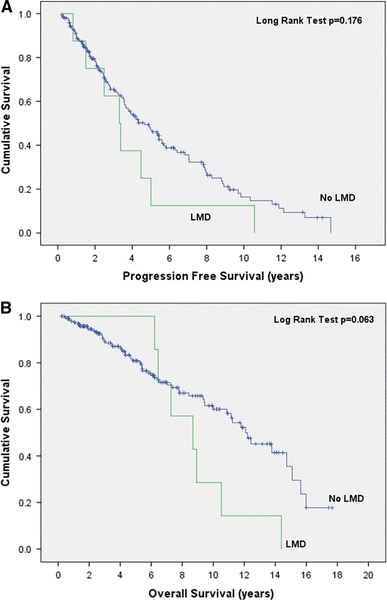
Progression free survival (a) and overall survival (b) of patients with oligodendroglial tumors with and without LMD measured from the initial pathological diagnosis
Fig. 2.
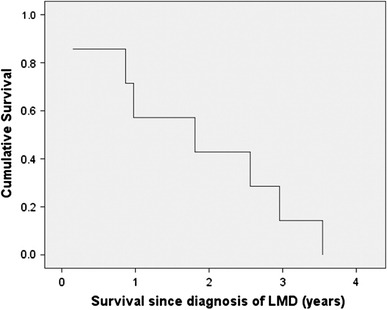
Overall survival of 8 patients with oligodendroglial tumors from diagnosis of LMD to death
Subanalysis in patients with 1p/19q co-deletion
Considering the subjectivity and evolution of pathologic criteria for the diagnosis of pure and mixed oligodendroglial tumors, we thought that genetic subtyping would provide a more objective classification. We included a total of 93 patients that had 1p/19q co-deletion or 1p loss. In this subgroup again the number of recurrences was associated with development of LMD with a mean of 1.1 and 2.8 recurrences for patients without and with LMD, respectively (p = 0.004). 0.2% (1/53) and 12.5% (5/40) of patients with low and high grade tumors developed LMD, respectively (p = 0.04). In this subgroup the median survival was 107 months (95% CI 101–113 months) for patients that developed LMD. This was significantly shorter than 177 months (95% CI 155–199 months) for patients without LMD (p = 0.033).
Discussion
To our knowledge, this is the first population-based study of the rare complication of LMD in oligodendroglial tumors. Other population-based studies of oligodendroglioma [10, 11] have not reported this complication. We found that LMD occurred in 3.9% of patients with oligodendroglial tumors, usually later in the natural history of the disease. The only clinical factor statistically associated with its development was the number of tumor recurrences. This could also be a surrogate for multiple surgeries but incomplete information on treatment of recurrences precludes a definitive conclusion. There was a trend for LMD to develop in patients with anaplastic tumors with 1p19q co-deletion and in those who were observed initially rather than treated with chemotherapy or radiotherapy immediately after diagnosis. In patients harboring 1p/19q codeletion, the diagnoses of LMD implied shorter OS. Survival following the diagnosis of LMD varied widely but on average was almost 2 years. This contrasts with rapidly fatal LMD in those with GBM or systemic solid tumors [12, 13].
Our subgroup analysis of anaplastic oligodendrogliomas found a trend for patients who did not receive postoperative treatment with radio- or chemotherapy to develop LMD.
We are uncertain about optimal treatment in this setting. Our patients tolerated various therapies, including intrathecal thiotepa, but we found no complete or partial responses. We now avoid potentially toxic therapies in these patients with longer survivals and use oral temozolomide, a DNA methylating agent that penetrates the CNS and is well tolerated.
Our study has limitations which may have falsely raised or lowered the true incidence of LMD. A higher rate might reflect our longstanding interest in this disease or tendency to investigate with neuro-imaging and lumbar puncture seemingly minor symptoms or signs. Conversely, the incidence might be spuriously low because craniospinal axis MRI/routine CSF examination were not routinely performed in all oligodendroglial tumor patients. We are also limited in the conclusions we can draw because of the very small number of patients. Therefore only the strongest risks of LMD were identified and trends we observed (e.g. the association of 1p and 19q co-deletion) may be confirmed in larger studies.
Finally, this study is also limited to classical clinical and pathological variables gleaned from cancer registry data and chart review. Central pathology review was not performed for our study, and the classification of the tumors might have varied between pathologists. Furthermore, diagnostic practices may have changed over the course of the study period that could impact the classification of tumors as pure oligodendroglioma or mixed oligodendrogliomas. We were unable to evaluate the association of ventricle opening during surgery to the development of LMD. Aside from some data on 1p and 19q status, our study does not further interrogate genomic/molecular factors that could either influence the classification of oligodendroglial tumors or contribute to the risk of LMD. We are now using genomic technologies in archival formalin-fixed paraffin-embedded tumor samples in patients from this study to determine if we can identify genomic signatures associated with this rare complication.
Acknowledgments
We thank the Families of Clark H. Smith and Kathleen Lorette for their generous support of Neuro-oncology. JGC holds the Alberta Cancer Foundation Chair in Brain Tumor Research. JC holds a Clinician Investigator Award from Alberta Innovates Health Solutions.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Engelhard HH, Stelea A, Mundt A. Oligodendroglioma and anaplastic oligodendroglioma: clinical features, treatment and prognosis. Surg Neurol. 2003;60:443–456. doi: 10.1016/S0090-3019(03)00167-8. [DOI] [PubMed] [Google Scholar]
- 2.Bromberg J, Van den Bent MJ. Oligodendrogliomas: molecular biology and treatment. The Oncologist. 2009;14:155–163. doi: 10.1634/theoncologist.2008-0248. [DOI] [PubMed] [Google Scholar]
- 3.Cairncross G, Berkey B, Shaw E, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24:2707–2714. doi: 10.1200/JCO.2005.04.3414. [DOI] [PubMed] [Google Scholar]
- 4.van den Bent MJ, Carpentier AF, Brandes AA, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organization for Research and Treatment of Cancer phase III trial. J Clin Oncol. 2006;24:2715–2722. doi: 10.1200/JCO.2005.04.6078. [DOI] [PubMed] [Google Scholar]
- 5.Jenkins RB, Blair H, Ballman KV, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66:9852–9861. doi: 10.1158/0008-5472.CAN-06-1796. [DOI] [PubMed] [Google Scholar]
- 6.Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473–1479. doi: 10.1093/jnci/90.19.1473. [DOI] [PubMed] [Google Scholar]
- 7.Bauman GS, Ino Y, Ueki K, et al. Allelic loss of chromosome 1p and radiotherapy plus chemotherapy in patients with oligodendrogliomas. Int J Radiat Oncol Biol Phys. 2000;48:825–830. doi: 10.1016/S0360-3016(00)00703-3. [DOI] [PubMed] [Google Scholar]
- 8.Ino Y, Betensky RA, Zlatescu MC, Sasaki H, et al. Molecular subtypes of anaplastic oligodendroglioma: implications for patient management at diagnosis. Clin Cancer Res. 2001;7:839–845. [PubMed] [Google Scholar]
- 9.Roldán G, Scott J, George D, Parney I, Easaw J, Cairncross G, Forsyth P, Yan E. Leptomeningeal disease from oligodendroglioma: clinical and molecular analysis. Can J Neurol Sci. 2008;35:204–209. doi: 10.1017/s0317167100008647. [DOI] [PubMed] [Google Scholar]
- 10.Nielsen MS, Christensen HC, Kosteljanetz M, Johansen C. Incidence of an survival from oligodendroglioma in Denmark, 1943–2002. Neuro Oncol. 2009;11:311–317. doi: 10.1215/15228517-2008-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005;64:479–489. doi: 10.1093/jnen/64.6.479. [DOI] [PubMed] [Google Scholar]
- 12.Oechsle K, Lange-Brock V, Kruell A, Bokemeyer C, de Wit M. Prognostic factors and treatment options in patients with leptomeningeal metastases of different primary tumors: a retrospective analysis. J Cancer Res Clin Oncol. 2010;136:1729–1735. doi: 10.1007/s00432-010-0831-x. [DOI] [PubMed] [Google Scholar]
- 13.Clarke JL, Perez HR, Jacks LM, Panageas KS, Deangelis LM. Leptomeningeal metastases in the MRI era. Neurology. 2010;74:1449–1454. doi: 10.1212/WNL.0b013e3181dc1a69. [DOI] [PMC free article] [PubMed] [Google Scholar]