

Abstract
TGF-β and activin induce the phosphorylation and activation of Smad2 and Smad3, but how these proteins stimulate gene transcription is poorly understood. We report that TGF-β receptor phosphorylation of Smad3 promotes its interaction with the paralogous coactivators CBP and p300, whereas CBP/p300 binding to nonphosphorylated Smad3 or its oligomerization partner Smad4 is negatively regulated by Smad–intramolecular interactions. Furthermore, p300 and TGF-β receptor-phosphorylated Smad3 synergistically augment transcriptional activation. Thus, CBP/p300 are important components of activin/TGF-β signaling and may mediate the antioncogenic functions of Smad2 and Smad4.
Keywords: CBP, p300, phosphorylation, Smad protein, TGF-β, transcription
Initiation of gene transcription by RNA polymerase II is dependent on general initiation and specific transcription factors (Roeder 1996). Although general initiation factors such as the TATA-box binding protein are in close proximity to the polymerase holoenzyme and mediate basal gene activity, efficient gene transcription requires the binding of (activated) specific transcription factors to DNA promoter/enhancer elements. Specific transcription factors may establish contacts with the basal transcription machinery on their own but often need bridging coactivators such as CBP (CREB binding protein) and p300.
The paralogous proteins CBP and p300 were originally identified as interaction partners for the cAMP response element binding protein (CREB) and the adenoviral E1A protein, respectively (Chrivia et al. 1993; Eckner et al. 1994). Subsequently, a variety of different DNA-binding transcription factors and coactivators have been shown to rely on CBP/p300 for their function in vivo and in in vitro transcription assays (Janknecht and Hunter 1996; Nakajima et al. 1997; Shikama et al. 1997). In addition, CBP/p300 were identified as histone acetyltransferases, suggesting that they potentiate transcription by acetylation-dependent loosening of the chromatin structure (Bannister and Kouzarides 1996; Ogryzko et al. 1996). However, CBP/p300 can also acetylate and thereby enhance the DNA-binding activity of the tumor suppressor p53 (Gu and Roeder 1997), indicating that acetylation of specific transcription factors could contribute to the mechanism of coactivation by CBP/p300.
In humans, loss of one CBP allele apparently causes Rubinstein–Taybi syndrome, which is characterized by abnormal pattern formation and mental retardation (Petrij et al. 1995). This phenotype was partially reproduced in hemizygous CBP+/− mice, whereas homozygous CBP−/− as well as p300−/− mice are not viable (Tanaka et al. 1997; Yao et al. 1998). These data suggest that CBPand p300 are limiting cofactors within the cell. Accordingly, competition for CBP/p300 can explain the phenomenon of squelching—the interference of transcription factor activity by other factors not binding to the same gene promoter (Janknecht and Hunter 1996; Kamei et al. 1996).
Signaling by transforming growth factor-β (TGF-β) superfamily members recently has been shown to rely on Smad proteins (Heldin et al. 1997). Pathway-restricted Smad proteins become phosphorylated at their extreme carboxyl terminus by ligand-bound TGF-β receptor family members and then translocate to the nucleus and activate gene transcription by either associating with DNA-bound factors (X. Chen et al. 1997) or direct binding to DNA promoter elements (Kim et al. 1997; Yingling et al. 1997). Specific Smad proteins are required for different signals (Heldin et al. 1997): Smad1 and Smad5 mediate signaling in response to bone morphogenetic proteins, whereas activin and TGF-β utilize both Smad2 and Smad3. All pathway-restricted Smad proteins are able to oligomerize with the common mediator Smad4, which is identical to the DPC4 tumor suppressor that is often missing or mutated in pancreatic cancers, thus preventing the antiproliferative effect of TGF-β (Hahn et al. 1996). Oligomerization with Smad4, which lacks the potential carboxy-terminal phosphorylation sites, augments transactivation by the pathway-restricted Smad proteins.
Here we demonstrate that Smad proteins utilize CBP/p300 to exert their effects. The transcription factor Smad3 binds to CBP/p300 both in vitro and in vivo, and TGF-β receptor phosphorylation of Smad3 promotes this interaction. Furthermore, CBP/p300 cooperate with Smad2–Smad4 in activating gene transcription, whereas a dominant-negative CBP can suppress Smad-dependent transcription. These results establish CBP/p300 as important cofactors in activin/TGF-β signaling pathways.
Results
In vivo interaction between Smad3 and CBP/p300
To identify novel interaction partners of CBP/p300, we undertook a yeast two-hybrid screen with CBP amino acids 1891–2175 as bait. One isolated cDNA clone encoded the carboxy-terminal amino acids 219–467 of the human Smad2 transcription factor. As a high degree of homology exists between Smad2 and Smad3, and full-length Smad3 cDNA was readily available, we focused our further research on Smad3 and its oligomerization partner Smad4.
To demonstrate an in vivo interaction between p300 and Smad3, amino-terminally Myc-tagged, full-length Smad3 (6Myc–Smad32–425) was expressed in human 293T cells alone or in the presence of HA-tagged p300. Cell extracts were prepared and immunoprecipitations were performed with α-HA antibodies followed by Western blotting utilizing α-Myc antibodies. In the presence of coexpressed p300–HA, Smad3 was coimmunoprecipitated (Fig. 1A, lane 4, top). Similarly, we detected coimmunoprecipitation of Smad3 with HA-tagged CBP (lane 8). In contrast, Smad4 (6Myc–Smad42–552) did not interact with CBP/p300 (lanes 6,9). Furthermore, a partial nuclear colocalization of endogenous Smad2/3 and endogenous p300 was observed by confocal laser microscopy (Fig. 1B): Although antibodies directed against a common epitope in Smad2 and Smad3 elicited both cytoplasmic and nuclear staining, p300 was predominantly detected in nuclear speckles that coincided with a high degree of nuclear Smad2/3 staining. Together, these results indicate that CBP/p300 and Smad3 interact in vivo.
Figure 1.
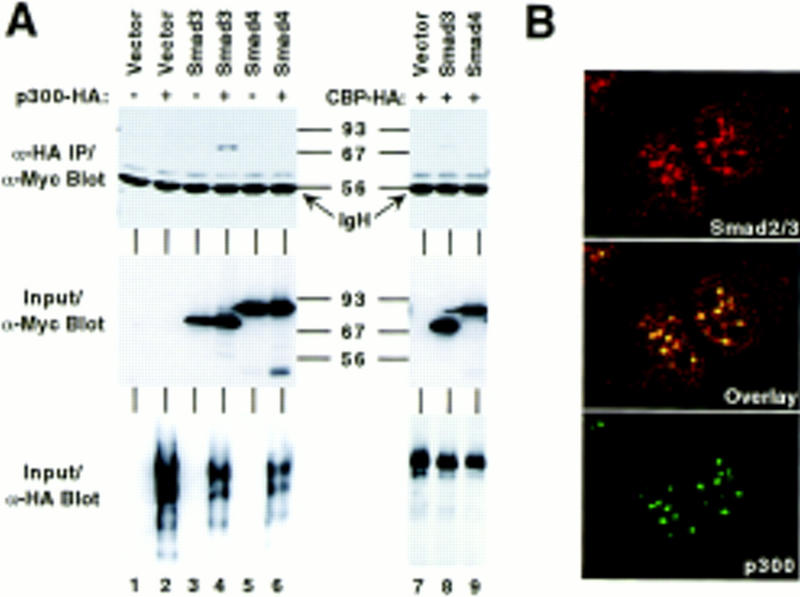
In vivo interaction of Smad3 and CBP/p300. (A) An empty Myc-tag expression vector or those encoding Smad3 (6Myc–Smad32–425) and Smad4 (6Myc–Smad42–552) was cotransfected with HA-tagged p300 or HA-tagged CBP as indicated into 293T cells. Immunoprecipitations were performed with α-HA mAbs, and coimmunoprecipitated Smad proteins detected by Western blotting with α-Myc mAbs (top). (Middle and bottom) Expression analysis of the Myc- and HA-tagged proteins, respectively, by Western blotting of cell extracts. (B) Confocal laser microscopy. Nontransfected, serum-starved, formaldehyde-fixed Mv1Lu cells were challenged with α-Smad2/3 (N-19, Santa Cruz) and α-p300 mAbs and stained with secondary antibodies coupled to Texas Red (top) and FITC dyes (bottom), respectively. Colocalization is apparent by yellow staining in the composite middle panel.
Mapping of interaction domains
To define the CBP/p300 interaction domain within Smad3, a panel of Myc-tagged Smad3 truncations were constructed. As shown in Figure 2A, deletion of Smad3 carboxy-terminal amino acids led to a markedly higher degree of interaction with p300 than observed with the full-length protein (cf. 2–342 to 2–425). Similarly, deletion of amino-terminal amino acids (see 210–425) stabilized the interaction of Smad3 and p300, whereas amino acids 2–210 failed to bind p300. A combined amino- and carboxy-terminal truncation (210–342) bound to p300 comparably as well as the 2–342 and 210–425 molecules, indicating that the carboxyl and amino termini of Smad3 can only negatively regulate binding of Smad3 to p300 when the other terminus is still present. Thus, intramolecular interactions between the amino and carboxyl termini, as reported for Smad2 and Smad4 (Hata et al. 1997), most likely restrict the access of CBP/p300 to Smad3 amino acids 210–342. This may explain why the amino terminus of Smad3 represses the transactivation function residing in the carboxy-terminal half (Heldin et al. 1997).
Figure 2.


Mapping of interaction domains. α-HA coimmunoprecipitations of Myc-tagged Smad3 (A) or Smad4 (B) proteins with HA-tagged p300. (Input) Approximately 2% of Myc-tagged proteins in the cell extracts. Numbers at top refer to amino acids. (C) GST pull-down assays. Bound Myc-tagged Smad proteins were revealed by α-Myc Western blotting. (D) GST pull-down assay with 6HisT7–Smad3210–425 followed by α-T7-tag Western blotting. (E) α-HA coimmunoprecipitations of Myc-tagged Smad2 proteins with p300–HA.
Interestingly, the Smad4 truncation 2–510 interacted with p300 (Fig. 2B), which also held true for Smad4270–510, whereas deletion of the amino-terminal half of Smad4 (see 270–552) was not sufficient to cause a detectable interaction. Thus, Smad4 is capable of interacting with p300 when carboxy-terminal amino acids are deleted. However, although binding of full-length Smad4 to CBP/p300 was not detected, this may not preclude a low affinity binding in vivo.
To localize the Smad interaction domain within CBP, we used GST–CBP fusion proteins in pull-down experiments (Fig. 2C). No binding could be detected with full-length Smad3 (2–425) under the experimental conditions employed, presumably because of the weakness of this interaction, or with the truncation 2–210. Smad3210–342 bound to CBP amino acids 1891–2175, which interestingly also interact with coactivators of the nuclear steroid receptors (H. Chen et al. 1997; Spencer et al. 1997). This binding was specific, as none was observed with GST itself, CBP regions 451–721 and 2175–2441 (Fig. 2C), or with other regions of CBP (data not shown). Similarly, Smad4270–510 specifically bound to CBP1891–2175 (data not shown). Furthermore, binding of Smad3 to CBP was direct, as purified GST–CBP1891–2175, but not GST, interacted with bacterially expressed, purified Smad3210–425 (Fig. 2D).
Finally, we analyzed whether Smad2 could also coimmunoprecipitate with p300. To that end, Myc-tagged, full-length Smad22–467 was coexpressed with p300-HA and α-HA immunoprecipitations performed. Full-length Smad2 coimmunoprecipitated with p300 (Fig. 2E), as did Smad2225–467 (Fig. 2E). As observed for the homologous Smad3, deletion of the Smad2 amino terminus resulted in a higher degree of interaction with p300.
Coactivation by Smad and CBP/p300 proteins
Next, we examined whether CBP/p300 could coactivate transcription from the TGF-β responsive plasminogen activator inhibitor-1 reporter gene construct 3TP–lux (Lagna et al. 1996) in 293T cells. p300 enhanced Smad2 (data not shown)- and Smad3 (Fig. 3A)-mediated transcription. Transcription in the absence of overexpressed Smad was much less affected (Fig. 3A) and could be caused by either endogenous Smad proteins or AP-1 transcription factors, which both are capable of binding to the 3TP–lux construct (Yingling et al. 1997). Similar results were obtained when CBP was employed in place of p300 (data not shown). Surprisingly, enhancement of Smad4-dependent transcription by p300 also occurred (Fig. 3A), implying that Smad4 and p300 might interact in vivo. Furthermore, coexpression of Smad3, Smad4, and p300 resulted in a cooperative activation of the 3TP–lux construct (Fig. 3A), suggesting that these three different proteins form a transcriptionally active complex.
Figure 3.



Coactivation mediated by p300 and Smad proteins. (A) 6Myc–Smad32–425 (0.1 μg), 6Myc–Smad42–552 (0.3 μg), and p300–HA (1 μg) were cotransfected, with the 3TP–lux reporter construct into 293T cells. Relative luciferase levels are depicted. (B) One microgram of 6Myc–Smad32–425, 6Myc–Smad42–552, or the empty expression vector was cotransfected with or without 0.7 μg of p300–HA into 293T cells. Relative luciferase activities derived from the cotransfected SBE4–luc reporter are presented. (C) Mv1Lu cells were transfected with the 3TP–lux reporter construct and additionally with 3 μg of p300–HA, 0.1 μg of Myc–Smad3, or 0.6 μg of Smad4–HA expression vectors. Relative luciferase activities are given. (D) Twenty-five nanograms of Myc–Smad3 expression plasmid or the empty vector pcDNA3 was cotransfected with combinations of 40 ng of TβRI–T204D, 1 μg of pEV3S–CBP1891–2175, and 0.1 μg of p300–HA plasmids into 293T cells. Luciferase activity derived from the cotransfected 3TP–lux reporter construct is depicted. (E) U2 OS cells were transfected with 100 ng of Myc–Smad3 plasmid, or pcDNA3, and the 3TP–lux reporter gene construct. Where indicated, 6 μg of pEV3S–CBP1891–2175 was cotransfected.
We also employed a luciferase reporter construct (SBE4–luc) driven by consensus binding sites for Smad3 and Smad4 (Zawel et al. 1998). Again, p300 enhanced Smad3- and Smad4-dependent transcription, whereas luciferase activity in the absence of overexpressed Smad proteins was unaffected (Fig. 3B). In addition, elevation of Smad3- or Smad4-dependent transcription by p300 did not occur with a CMV or a PGK promoter (data not shown), indicating that the observed effects were promoter specific.
We also investigated whether the Smad proteins functionally interacted with CBP/p300 in another cell line (Mv1Lu) that is responsive to TGF-β and contains endogenous Smad2/3/4 (Nakao et al. 1997). In the absence of overexpressed Smads, the 3TP–lux reporter construct was stimulated by the overexpression of p300 in serum-starved cells (Fig. 3C). Upon TGF-β1 stimulation the reporter gene activity was superinduced by coexpression of p300, which may be explained by a coactivation mediated by endogenous Smad2/3 proteins and overexpressed p300. Similarly, p300 coactivated transcription with overexpressed Smad3 both in the presence and absence of TGF-β1. Overexpressed Smad4 also cooperated with p300, albeit less pronouncedly. Thus, CBP/p300 functionally cooperate with both Smad2 and Smad3 and the common mediator Smad4 in different cell lines under various growth conditions.
CBP/p300 possess amino- and carboxy-terminal activation domains as well as a central acetyltransferase function, which may all be required for coactivation (Janknecht and Hunter 1996; Shikama et al. 1997). Thus, the truncation CBP1891–2175, which solely constitutes the docking region for Smad3 and Smad4, might compete with endogenous CBP/p300 for Smad proteins and thereby interfere with Smad activity. To test this hypothesis, we overexpressed CBP1891–2175 and studied the impact on basal and induced transcription mediated by Smad3 in 293T cells. Basal transcription was not significantly changed, although TβRI–T204D (a constitutively active TGF-β type I kinase receptor)-induced transcription was significantly reduced (Fig. 3D). Similarly, transcription induced by TβRI–T204D in the absence of overexpressed Smad3, which is likely to be mediated by endogenous Smad proteins, was diminished. Overexpression of p300 abolished the repressive effect of CBP1891–2175, indicating that CBP1891–2175 competitively inhibits CBP/p300 from coactivating with Smad3. As a control, we also studied the impact of CBP1891–2175 on the c-fos serum response element. Here, transcription was nearly unaffected by CBP1891–2175 in the presence or absence of Smad3 and/or TβRI–T204D, and a β-actin promoter construct was even activated by CBP1891–2175 (data not shown). Furthermore, even basal 3TP–lux activity was reduced by CBP1891–2175 in the presence and absence of overexpressed Smad3 in U2 OS cells (Fig. 3E). Collectively, these data suggest that CBP1891–2175 acts as a dominant-negative molecule in different cell lines and that Smad3 normally relies on CBP/p300 for its function.
Phosphorylation-dependent cooperation between Smad3 and CBP/p300
Because Smad3 becomes phosphorylated on the carboxy-terminal SSVS motif upon TGF-β stimulation (Liu et al. 1997), we examined whether phosphorylation at the carboxyl terminus affects binding to CBP/p300. Although no coimmunoprecipitation of Myc-tagged Smad3 with endogenous p300 or CBP was detectable (Fig. 4A, lanes 1,5), coexpression of TβRI–T204D resulted in robust binding of Myc-tagged Smad3 to endogenous p300 and CBP in vivo (lanes 2,6). In contrast, coexpression of TβRI–T204D did not lead to coimmunoprecipitation of Smad4, which lacks the extreme carboxy-terminal phosphorylation sites, with endogenous p300 or CBP (lanes 4,8). Mutation of the carboxy-terminal SSVS motif to AAVA in Smad3 (C3A mutant) suppressed the TβRI–T204D-dependent in vivo interaction with endogenous CBP (Fig. 4B, left). These results were reproduced in vitro with GST–CBP1891–2175 (Fig. 4B, right). Thus, phosphorylation of Smad3 at its extreme carboxyl terminus promotes binding to CBP/p300.
Figure 4.


Phosphorylation-dependent cooperation between Smad3 and CBP/p300. (A) 6Myc–Smad32–425 or 6Myc–Smad42–552 was coexpressed with TβRI–T204D where indicated. Immunoprecipitations were performed with α-p300 or α-CBP antibodies and coimmunoprecipitated Smad proteins revealed by α-Myc Western blotting. (B) Wild-type (wt) or the triple alanine mutant C3A of 6Myc–Smad32–425 was coexpressed with TβRI–T204D in 293 T cells, α-CBP immunoprecipitations of cell extracts were performed and Smad3 proteins were revealed by α-Myc Western blotting (lanes 1–4). Lanes 5–8 depict a corresponding in vitro pull-down experiment employing GST–CBP1891–2175. (C) Wild-type or C3A Myc–Smad3 expression plasmid (0.5 μg) was transfected with 6 μg of p300–HA and 0.5 μg TβRI–T204D vector into HeLa cells. Activation of the cotransfected 3TP–lux reporter gene is shown.
We then analyzed whether phosphorylation of Smad3 and coexpression of p300 could jointly enhance Smad3-dependent transcription. To that end, wild-type or Smad3–C3A were coexpressed with p300 and/or TβRI–T204D in HeLa cells (Fig. 4C). Smad3-dependent 3TP–lux activation was raised by ∼160% or ∼115% by p300 or TβRI–T204D, respectively, whereas joint coexpression of p300 and TβRI–T204D increased luciferase activity by ∼475%, indicating that p300 and TβRI–T204D synergize to activate Smad3. No such synergism was observed upon overexpression of Smad3–C3A or in the absence of overexpressed Smad3. Thus, enhanced interaction of CBP/p300 with Smad3 upon TGF-β triggered phosphorylation causes elevation of transcriptional activity.
Discussion
We have established a link between the nuclear coactivators CBP/p300 and the activin/TGF-β-regulated Smad proteins. Transcription mediated by Smad2 or Smad3, as well as by the common mediator Smad4, was enhanced by CBP/p300. Importantly, we demonstrated that the Smad-binding domain within CBP (amino acids 1891–2175) can act as a suppressor of Smad3-dependent transcription, indicating that Smad3 is normally dependent on CBP/p300 for mediating full activation of gene transcription. Also, coactivation by CBP/p300 occurred in different cell lines and, as demonstrated with the Mv1Lu cells, under different growth conditions: in cells continuously growing in medium supplemented with 10% serum (data not shown) and in serum-starved cells before and after TGF-β1 stimulation (Fig. 3C). This suggests that coactivation of CBP/p300 and Smad3, and probably also Smad2 and Smad4, is a general phenomenon.
In vertebrates, activin is involved in the induction of dorsal mesoderm or erythroid differentiation and TGF-β can induce growth of the extracellular matrix or suppress mitogenicity and immune reactions (Heldin et al. 1997). Furthermore, Smad2 and Smad4 are indispensable for many steps during embryogenesis (Sirard et al. 1998; Waldrip et al. 1998; Yang et al. 1998). Therefore, as partners of Smad proteins, CBP/p300 are strongly implicated to play an important role in various aspects of development, as indicated by the abnormal pattern formation in Rubinstein–Taybi syndrome caused by CBP haploinsufficiency (Petrij et al. 1995).
Utilization of CBP/p300 as a coactivator places the activin/TGF-β-regulated Smad proteins in a network of transcription factors that all compete for the limiting amounts of CBP/p300 and may thus influence each other’s activity by squelching (Janknecht and Hunter 1996). For instance, ligand-bound nuclear receptors of the steroid hormone superfamily can inhibit the function of the phorbol-ester-activated AP-1 transcription factor by depletion of the available CBP/p300 pool, or cAMPmediated stimulation of CREB can suppress gene activation by retinoic acid (Kamei et al. 1996). Similarly, a carboxy-terminal truncation of Smad3 that leaves the CBP/p300-binding domain intact can efficiently inhibit cAMP or phorbol-ester-stimulated gene transcription (Mucsi and Goldberg 1997), presumably by competing with CREB and AP-1 for CBP/p300.
Phosphorylation of the SSVS motif of Smad3 is essential for its transactivation function (Liu et al. 1997) and is thought to result in a conformational change (Heldin et al. 1997), which may expose the CBP/p300 docking region. Smad3 had a dramatically enhanced affinity toward CBP/p300 after phosphorylation by TGF-β type I kinase receptor at its SSVS motif, or after amino- or carboxy-terminal truncation. Thus, intramolecular interactions between the amino terminus and the nonphosphorylated carboxyl terminus of Smad3 most likely prevent an efficient binding to CBP/p300, and similarly the Smad4 carboxyl terminus, which contains no TGF-β receptor phosphorylation sites, exerts an inhibitory effect on binding to CBP/p300. The enhanced interaction of Smad3 with CBP/p300 upon TGF-β receptor-mediated phosphorylation translates into increased activity of Smad3, as indicated by the fact that coexpression of p300 and TβRI–T204D synergistically potentiated Smad3-dependent 3TP–lux activation. Thus, enhancement of cooperation with CBP/p300 represents another way in which activin/TGF-β receptor-mediated phosphorylation of Smad3 promotes gene transcription.
CBP/p300 have been suspected to be tumor suppressors, as they can suppress transformation by the adenoviral E1A protein (Smits et al. 1996). Our study furthers this notion, as cooperation between CBP/p300 and the tumor suppressors Smad2 and Smad4 may be a necessary basis for safeguarding against tumorigenesis. Consistent with this, development of colorectal carcinomas is associated with mutations in p300 (Muraoka et al. 1996), Smad2 (Eppert et al. 1996), and Smad4 (Thiagalingam et al. 1996). Finally, disruption of the interplay between CBP/p300 and Smad2/Smad4 oligomers may render colorectal carcinomas unresponsive to the growth-inhibitory effect of TGF-β (Zhou et al. 1998).
Materials and methods
Plasmids
Mammalian expression vectors for TβRI–T204D (Hoodless et al. 1996), carboxy-terminally HA-tagged human Smad4, the 3TP–lux reporter construct (Lagna et al. 1996), the SBE4–luc plasmid (Zawel et al. 1998), and CBP constructs (Janknecht and Nordheim 1996) were as published. Myc-tagged versions of human Smad2, rat Smad3, and human Smad4 were generated by PCR and verified by DNA sequencing. For expression of His/T7-tagged Smad3210–425, an appropriate cDNA fragment was cloned into pET-28a(+) (Novagen).
Transient transfection assays
Human embryonal kidney 293T, mink lung Mv1Lu, human cervix carcinoma HeLa, or human osteogenic sarcoma U2OS cells were grown to 25% confluency on 6-cm dishes and then transfected by the calcium phosphate coprecipitation method. Cells were kept in medium containing 10% fetal calf serum except 12 hr before and during the 16-hr induction with 5 ng/ml human recombinant TGF-β1 (R&D Systems) when the serum concentration was lowered to 0.2%. Luciferase activity was determined as described (Janknecht and Nordheim 1996).
Coimmunoprecipitations
One microgram of Myc-tagged Smad and, where indicated, 0.5 μg of TβRI–T204D and 6 μg of HA-tagged p300 or CBP expression plasmids were transfected into 293T cells. Cells were lysed in 600 μl of 0.5× lysis buffer (1×:10 mm Tris, 30 mm Na4P2O7, 50 mm NaCl, 50 mm NaF, 1% Triton X-100 at pH 7.1), 0.2 mm DTT, and inhibitor cocktail (20 μg/ml aprotinin, 20 μg/ml leupeptin, 0.5 mm PMSF, 0.5 mm Na3VO4) at 4°C. Immunoprecipitations were performed with the mouse mAb 12CA5 (α-HA) or Santa Cruz rabbit polyclonal antibodies A-22 (α-CBP) or C-20 (α-p300) and 20 μl of protein A beads (Repligen). Proteins were revealed after SDS-PAGE and Western blotting with the ECL detection kit (Amersham).
GST pull-down assays
GST fusion proteins were produced in Escherichia coli, purified as described previously (Janknecht and Nordheim 1996), and bound to glutathione–agarose (Sigma). Protein extracts from 293T cells, transfected with 3 μg of Myc-tagged Smad3 variant expression plasmids (±0.5 μg TβRI–T204D), were prepared in 800 μl of 1× lysis buffer, 1 mm DTT, and inhibitor cocktail. Extract (30 μl) was incubated with 15 μl of preloaded glutathione–agarose beads in 600 μl of 0.25× lysis buffer/1 mm DTT/inhibitor cocktail and incubated for 2 hr at 4°C. After boiling in sample buffer, SDS–PAGE was performed and bound proteins were detected by α-Myc Western blotting. Alternatively, 6HisT7–Smad3210–425 was produced in E. coli, purified by Ni2+–chelate affinity chromatography, and utilized in the GST pull-down assay. Bound 6HisT7–Smad3210–425 was detected after Western blotting with mouse mAb α-T7-tag (Novagen).
Acknowledgments
We thank L. Attisano, Y. Chen, J. Massagué, B. Vogelstein, and J. Wrana for providing plasmids, G. Pao for help with confocal laser microscopy, and R. Palmer for discussions. This work was supported by scholarships from the Deutsche Forschungsgemeinschaft (R.J.) and the Wellcome Trust (N.J.W.) and U.S. Public Health Service grants CA14195 and CA39780 (T.H.). T.H. is a Frank and Else Schilling American Cancer Society Research professor.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rjanknecht@aim.salk.edu; FAX 619-457-4765.
References
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RPS, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, Livingston DM. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes & Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, Tsui L-C, Bapat B, Gallinger S, Andrulis IL, Thomsen GH, Wrana JL, Attisano L. MADR2 maps to 18q21 and encodes a TGFβ-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. 1996;86:543–552. doi: 10.1016/s0092-8674(00)80128-2. [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Hahn SA, Schutte M, Hoque ATMS, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- Hata A, Lo RS, Wotton D, Lagna G, Massagué J. Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature. 1997;388:82–87. doi: 10.1038/40424. [DOI] [PubMed] [Google Scholar]
- Heldin C-H, Miyazono K, ten Dijke P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Hoodless PA, Haerry T, Abdollah S, Stapleton M, O’Connor MB, Attisano L, Wrana JL. MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell. 1996;85:489–500. doi: 10.1016/s0092-8674(00)81250-7. [DOI] [PubMed] [Google Scholar]
- Janknecht R, Hunter T. A growing coactivator network. Nature. 1996;383:22–23. doi: 10.1038/383022a0. [DOI] [PubMed] [Google Scholar]
- Janknecht R, Nordheim A. Regulation of the c-fos promoter by the ternary complex factor Sap-1a and its coactivator CBP. Oncogene. 1996;12:1961–1969. [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin S-C, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Kim J, Johnson K, Chen HJ, Carroll S, Laughon A. Drosophila Mad binds to DNA and directly mediates activation of vestigial by Decapentaplegic. Nature. 1997;388:304–308. doi: 10.1038/40906. [DOI] [PubMed] [Google Scholar]
- Lagna G, Hata A, Hemmati-Brivanlou A, Massagué J. Partnership between DPC4 and SMAD proteins in TGF-β signalling pathways. Nature. 1996;383:832–836. doi: 10.1038/383832a0. [DOI] [PubMed] [Google Scholar]
- Liu X, Sun Y, Constantinescu SN, Karam E, Weinberg RA, Lodish HF. Transforming growth factor β-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc Natl Acad Sci. 1997;94:10669–10674. doi: 10.1073/pnas.94.20.10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucsi I, Goldberg HJ. Dominant-negative SMAD-3 interferes with transcriptional activation by multiple agonists. Biochem Biophys Res Commun. 1997;232:517–521. doi: 10.1006/bbrc.1997.6321. [DOI] [PubMed] [Google Scholar]
- Muraoka M, Konishi M, Kikuchi-Yanoshita R, Tanaka K, Shitara N, Chong J-M, Iwama T, Miyaki M. p300 gene alterations in colorectal and gastric carcinomas. Oncogene. 1996;12:1565–1569. [PubMed] [Google Scholar]
- Nakajima T, Uchida C, Anderson SF, Parvin JD, Montminy M. Analysis of a cAMP-responsive activator reveals a two-component mechanism for transcriptional induction via signal-dependent factors. Genes & Dev. 1997;11:738–747. doi: 10.1101/gad.11.6.738. [DOI] [PubMed] [Google Scholar]
- Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J-i, Heldin C-H, Miyazono K, ten Dijke P. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RCM, Masuno M, Tommerup N, van Ommen G-JB, Goodman RH, Peters DJM, Breuning MH. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Roeder RG. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci. 1996;21:327–335. [PubMed] [Google Scholar]
- Shikama N, Lyon J, La Thangue NB. The p300/CBP family: Integrating signals with transcription factors and chromatin. Trends Cell Biol. 1997;7:230–236. doi: 10.1016/S0962-8924(97)01048-9. [DOI] [PubMed] [Google Scholar]
- Sirard C, de la Pompa JL, Elia A, Itie A, Mirtsos C, Cheung A, Hahn S, Wakeham A, Schwartz L, Kern SE, Rossant J, Mak TW. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes & Dev. 1998;12:107–119. doi: 10.1101/gad.12.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits PHM, de Wit L, van der Eb AJ, Zantema A. The adenovirus E1A-associated 300 kDa adaptor protein counteracts the inhibition of the collagenase promoter by E1A and represses transformation. Oncogene. 1996;12:1529–1535. [PubMed] [Google Scholar]
- Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, McKenna NJ, Onate SA, Tsai SY, Tsai MJ, O’Malley BW. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389:194–198. doi: 10.1038/38304. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Naruse I, Maekawa T, Masuya H, Shiroishi T, Ishii S. Abnormal skeletal patterning in embryos lacking a single CBP allele: A partial similarity with Rubinstein-Taybi syndrome. Proc Natl Acad Sci. 1997;94:10215–10220. doi: 10.1073/pnas.94.19.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JKV, Markowitz S, Hamilton SR, Kern SE, Kinzler KW, Vogelstein B. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nature Genet. 1996;13:343–346. doi: 10.1038/ng0796-343. [DOI] [PubMed] [Google Scholar]
- Waldrip WR, Bikoff EK, Hoodless PA, Wrana JL, Robertson EJ. Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell. 1998;92:797–808. doi: 10.1016/s0092-8674(00)81407-5. [DOI] [PubMed] [Google Scholar]
- Yang X, Li C, Xu X, Deng C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci. 1998;95:3667–3672. doi: 10.1073/pnas.95.7.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T-P, Oh SP, Fuchs M, Zhou N-D, Ch’ng L-E, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–372. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- Yingling JM, Datto MB, Wong C, Frederick JP, Liberati NT, Wang X-F. Tumor suppressor Smad4 is a transforming growth factor β-inducible DNA binding protein. Mol Cell Biol. 1997;17:7019–7028. doi: 10.1128/mcb.17.12.7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- Zhou S, Buckhaults P, Zawel L, Bunz F, Riggins G, Dai JL, Kern SE, Kinzler KW, Vogelstein B. Targeted deletion of Smad4 shows it is required for transforming growth factor β and activin signaling in colorectal cancer cells. Proc Natl Acad Sci. 1998;95:2412–2416. doi: 10.1073/pnas.95.5.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]