

Abstract
Studies with sPLA2 Group X, and cPLA2α gene‐targeted mice suggest that absence of sPLA2 Group X results in protection from ischemia/reperfusion (I/R) injury in the heart, and absence of cPLA2α Group IV is protective in the brain. Although latter studies might suggest a similar deleterious role for cPLA2α in I/R injury in the heart, the pathophysiology of stroke is intricately related to excitotoxicity and cannot necessarily be extrapolated to the heart. We report here that unlike findings in the brain, cPLA2α(−/−) mice have exaggerated injury following I/R in vivo. In contrast, there is no difference in injury induced by simulated ischemia in cardiomyocytes isolated from cPLA2α(−/−) versus cPLA2α(+/+) mice. This suggests that cPLA2α does not have an important cardiomyocyte autonomous effect on ischemic injury. Prostaglandin E2 (PGE2) levels are significantly reduced in the hearts of the cPLA2α(−/−) mice, and the enhanced injury is ameliorated by treatment with the PGE analog, misoprostol. We demonstrate that cPLA2α is cardioprotective in vivo, and this is likely via cPLA2α‐mediated production of cardioprotective eicosanoids. These studies are the first to identify a protective role for cPLA2 in I/R injury in any organ and raise concerns over long‐term inhibition of cPLA2. Clin Trans Sci 2011; Volume 4: 236–242
Keywords: ischemia‐reperfusion injury, cytosolic phospholipase A2alpha, prostaglandin E2, cell death
Introduction
Phospholipases A2 catalyze the release of arachidonic acid (AA) from the sn‐2 position of phospholipids, thereby producing free AA and lysophospholipids. 1 , 2 , 3 , 4 There are three classes of these enzymes based on structure, cofactor requirements, and function. Secretory phospholipases A2 (sPLA2) are low molecular weight enzymes that contain an N‐terminal secretion signal peptide. They are believed to play important roles in inflammatory processes, but roles in the heart are less clear. DeWindt et al. reported no effect of deletion of sPLA2 IIA on ischemic injury in an isolated heart model. 5 However, Nijmeijer et al. reported that sPLA2s bound to ischemic cardiomyocytes and contributed to cell death via both direct cytotoxic effects and by facilitating inflammatory responses. 6 In addition, antibodies directed against sPLA2 have been reported to be effective against ischemia/reperfusion (I/R) injury. 7 Recently, Fujioka et al. have clarified the role of one sub‐group of sPLA2, Group X, in I/R. Deletion of the gene encoding sPLA2 Group X, which is exclusively expressed in neutrophils, significantly decreased infarct size following I/R. 8 Thus sPLA2 X may mediate many of the pathologic consequences of neutrophil infiltration into the infarct zone. Recently, two clinical trials with a nonselective sPLA2 inhibitor, varespladib, have been completed, one in patients with acute coronary syndromes and the other in patients going to percutaneous coronary intervention. 9 , 10 Neither of these trials were positive. A third trial is recruiting patients with vaso‐occlusive crisis from sickle cell disease. Results of a trial with varespladib examining plasma lipoproteins was recently completed and suggested usage of the agent in prevention of atherosclerosis. 11
The calcium‐independent iPLA2 family consists of two isoforms, iPLA2β and γ. iPLA2 activity has been reported to account for 80% of total PLA2 activity in the normal myocardium, and the enzyme rapidly associates with the membrane following ischemia. 12 , 13 Furthermore, the putative iPLA2‐selective inhibitor, bromo‐enol lactone, was protective in the ischemic isolated, perfused heart. 14 Of note, transgenic mice over‐expressing iPLA2β developed lethal ventricular tachyarrhythmias with ischemia, and this was associated with a marked increase in lysophospholid release. 15 These authors also reported that deletion of iPLA2γ, which is localized predominantly to mitochondria and peroxisomes, led to profound abnormalities in bioenergetic mitochondrial function that resulted in increased mortality in response to myocardial stressors, including thoracic aortic constriction. 16
The calcium‐dependent cytosolic phospholipase A2 (cPLA2) family consists of three members, α, β, and γ. The catalytic activity of the enzymes is calcium‐independent but submicromolar concentrations of intracellular calcium are necessary for the enzyme’s translocation to sites of activity (e.g., Golgi, endoplasmic reticulum and nuclear membrane), mediated by the calcium and lipid binding domain. 4 Activity is modulated by phosphorylation by members of the mitogen‐activated protein kinase family, particularly p38‐MAPK, making cPLA2s responsive to extracellular signals. 17 , 18 Based primarily on studies performed in the cPLA2α knockout mouse, cPLA2α has been implicated as central to the production of eicosanoids including prostaglandins (e.g., PGE2 and PGI2) and leukotrienes as well as platelet‐activating factor. 2 , 3 , 19 , 20 This is due in part to the fact that cPLA2s, as opposed to other PLA2s, are relatively selective for AA (the precursor of eicosanoids) at the sn‐2 position of phospholipids. cPLA2 is also known to activate neutrophils, and prostaglandin production in these cells relates to NADPH oxidase activation. 21
Reduced production of these signaling intermediates likely leads to the many phenotypes identified in the cPLA2α(−/−) mice, including enhanced recovery from allergen‐induced bronchoconstriction, reduced injury following administration of a dopaminergic‐selective neurotoxin, less severe anaphylaxis, acute lung injury, and arthritis, and reduced intestinal polyposis in a mouse model of hereditary colon cancer. 2 , 3 , 22 , 23 , 24 , 25 cPLA2α has also been demonstrated to play a role in cardiomyocyte hypertrophy induced by aortic constriction. 26 Indeed, the broad group of disease states mediated, at least in part, by cPLA2α, has made the enzyme a target of drug development, albeit with little success to date.
Of most relevance to this paper, however, the cPLA2α(−/−) mouse was markedly protected from ischemic injury of the brain. 19 , 27 Although as noted, this process is importantly driven by excitotoxicity, we reasoned that the cPLA2α KO mouse might also be protected from I/R injury in the heart. To our knowledge, no studies to date have addressed this issue in vivo, and those that have examined the issue in vitro, employed inhibitors with variable selectivity. The studies in vitro have led to the conclusion that cPLA2α inhibition is protective. 28 , 29
Herein, we examine the role of cPLA2 in I/R injury in the heart. We employ the cPLA2α(−/−) mouse to determine the response to I/R in vivo and also examine simulated ischemia in cardiomyocytes isolated from the mouse. Our studies identify a complex role for cPLA2α but, surprisingly, the net effect in vivo of cPLA2α deletion is worsened I/R injury. These studies suggest that while cPLA2 inhibitors may be of value in stroke and many inflammatory disease states, they may be detrimental in the setting of ischemic heart disease. Given the common scenario of patients with coronary artery disease and coexisting cerebro‐vascular or inflammatory diseases, our findings raise theoretical concerns about the concept of cPLA2 inhibition as a long‐term therapeutic strategy.
Methods
Animals
Experiments were carried out according to National Institutes of Health Guidelines on the Use of Laboratory Animals, and all procedures were approved by the Thomas Jefferson University Committee on Animal Care. cPLA2 gene‐targeted mice were generated and bred as previously described. 19
Determination of blood pressure in conscious, unrestrained mice
Mice were anesthetized with ketamine (50 mg/kg i.p.) and xylazine (2.5 mg/kg i.p.). A fluid‐filled catheter (DSI Instruments, St. Paul, MN, USA) was inserted into the left carotid artery, and the transducer with battery was placed in the subcutaneous layer of the subscapular region. Mice were allowed to recover and 4 days later, blood pressure was measured via telemetry in conscious, unrestrained animals on two successive days. Measurements were made in all mice at approximately the same time of day to minimize diurnal variations in blood pressure.
In vivo I/R
Surgical procedures were performed as previously described. 30 , 31 Briefly, mice (8–10 weeks old) were anesthetized with 2% isoflurane inhalation. The heart was exposed and exteriorized through a left thoracotomy at the level of the fifth intercostal space. A slipknot was made around the left anterior descending (LAD) coronary artery 1–2 mm from its origin with a 6–0 silk suture. Sham‐operated animals were subjected to the same surgical procedures except that the suture was passed under the LAD but was not tied. Following 30 minutes of ischemia, the slipknot was released and the myocardium was reperfused for 24 hours. A single dose of buprenorphine (0.3 mg/kg s.c.) was administered for pain treatment. At the end of the experiment, animals were euthanized by CO2 asphyxiation. For studies with the specific PGE2 analogue, misoprostol (13820, Cayman Chemical, Ann Arbor, MI, USA) compound was administered into the peritoneum at a dose of 150 μg/kg/day (two injections of 75 μg/kg in 0.2 cc of saline) during the 3 days before and during the I/R protocol.
Determination of area at risk and infarct size
Infarct size was determined as previously described. 30 , 31 Briefly, at the end of the 24‐hour reperfusion period, mice were reanesthetized and the ligature around the LAD was retied through the previous ligation site. Following this, 2% Evans blue dye was injected, the heart was then quickly excised, and cut into five sections from apex to base. Sections were then incubated in a 1% 2,3,5‐triphenyltetrazolium chloride (TTC, Sigma, St. Louis, MO, USA) solution and digitally photographed. The area not at risk (ANAR, Evan’s blue‐stained area), and the area at risk (AR, including both TTC‐positive [noninfarct] and TTC‐negative [infarct] staining area) were measured using the computer‐based image analyzer SigmaScan Pro 5.0 (SPSS Science, Chicago, IL, USA). Myocardial infarct size was expressed as a percentage of the AR (I/AR), and the AR was expressed as the percentage of total left ventricle (AR/[AR+ANAR]).
TUNEL analysis
For quantification of cell death, we employed terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling (TUNEL); we used a kit from Chemicon (Billerica, MA, USA) according to the manufacturer’s instructions. Images were viewed with a Nikon Eclipse 80i microscope (Nikon Inc., Melville, NY, USA). NIS elements software was used to record immunofluorescence images.
Evaluation of myocardial neutrophil infiltration
Paraformaldehyde‐fixed portions of the heart were embedded in paraffin and cut into 5‐μM thick sections. The neutrophil clone 7/4 (Abcam, Cambridge, UK) primary antibody was added in blocking solution (2% BSA, 0.2% horse serum in PBS supplemented with 0.2% NP‐40) and incubated overnight. Vectastin Elite ABC kit (Vector Laboratories, Burlingame, CA, USA) followed by DAB Plus Kit (Invitrogen, Carlsbad, CA, USA) were used. NIS elements software was used to record images. For determination of number of neutrophils in the infarct area, neutrophils were counted by an observer blinded to the genotype.
Immunoblotting
Heart tissue lysates were matched for protein concentration, loaded on SDS‐Page (10–15%) and transferred to nitrocellulose membranes (Whatman, Kent, UK). The membranes were then blocked with 5% nonfat milk for 1 hour and incubated overnight with primary antibodies as indicated. The following day, membranes were washed three times and incubated with appropriate secondary antibody for 1 hour at room temperature. Protein expression levels were quantified by determining band density with Quantity One analysis application from Bio‐Rad (Hercules, CA, USA). The signals were normalized to that of glyceraldehyde 3‐phosphate dehydrogenase to correct for potential differences in loading.
Simulated ischemia in isolated adult mouse cardiomyocytes
Adult mouse myocytes were isolated from wild‐type (WT) and cPLA2α(−/−) mice as previously described. 32 , 33 Simulated ischemia (termed metabolic inhibition with anoxia) was provoked by incubating the cells in hypoxia buffer (modified Krebs buffer [137‐mM NaCl, 3.8‐mM KCl, 0.49‐mM MgCl2, 0.9‐mM CaCl2, 4.0‐mM Hepes] supplemented with 20‐mM sodium lactate, 2.75‐mM 2‐deoxyglucose, 12‐mM KCl, 1‐mM sodium dithionate at pH 6.7) for 45 minutes. 34 , 35 Thereafter, cells were incubated in normal growth medium for rest of the experiment. Cell death was evaluated by measuring adenylate kinase release 16 hours following the hypoxia.
Measurement of PGE2 levels
After surgery, hearts were divided into control and ischemic regions, and snap frozen in liquid nitrogen. Harvested heart tissues were homogenized in cold PBS, centrifuged 5 minutes at 300 rpm and supernatants were used for protein analysis and PGE2 assays. PGE2 assay was conducted according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA). Assay was performed by using 10 μg of protein per reaction, which was in the range providing the maximum sensitivity for the assay. Each assay sample as well as the PGE2 standards were measured in triplicates. PGE2 values were determined by using the standard curve.
Statistical analysis
All values in the text and figures are presented as mean ± standard error of mean of independent experiments from given n‐sizes. Statistical significance of multiple treatments was determined by Student t‐test, one‐way or two‐way analysis of variance followed by the Bonferroni post‐hoc test when appropriate. P values < 0.05 were considered significant.
Results
Telemetry in conscious animals
We first asked whether there were differences in basal heart rate or blood pressure between cPLA2α(−/−) mice and littermate WT cPLA2α(+/+) mice. These studies had previously been performed in anesthetized mice and had shown no differences. We measured these parameters via radio‐telemetry in conscious, unrestrained mice with indwelling pressure transducers and, again, found no differences in either systolic blood pressure (cPLA2α(+/+): 125 ± 10 mmHg; cPLA2α(−/−): 120 ± 15 mmHg) or heart rate (cPLA2α(+/+): 466 ± 10; cPLA2α(−/−): 472 ± 52; n= 3 mice per genotype).
Determination of area at risk and infarct size
We then subjected cPLA2α(−/−) and littermate cPLA2α(+/+) mice to 30 minutes of ischemia followed by 24 hours of reperfusion. While area at risk, as determined by Evans Blue dye injection, was not different between the genotypes ( Figure 1A ), infarct size, as determined by TTC staining, was significantly increased in the cPLA2α(−/−) mice (37.9 ± 7.3% vs. 52.2 ± 9.8%, Figure 1B , P < 0.001).
Figure 1.
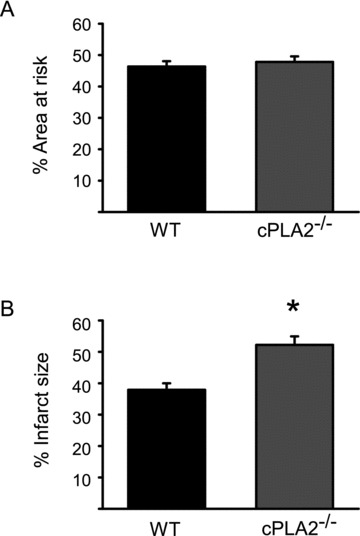
Deletion of cPLA2α increases cardiac I/R injury in vivo. WT and cPLA2α(−/−) mice were subjected to I/R as described in the Materials and Methods section. Area at risk was then determined by Evans Blue dye and infarct size by TTC staining. (A) Quantification of area at risk. (B) Quantification of infarct size expressed as a percent of area at risk. n= 12 WT and 13 cPLA2α(−/−). *p < 0.001.
Analysis of cell death and neutrophil infiltration
Cell death, as determined by TUNEL staining, was increased in the ischemic zone of left ventricles in both genotypes at 6 hours post‐I/R, but, consistent with the infarct size data, the number of TUNEL‐positive cells was higher in cPLA2α(−/−) animals ( Figure 2A , P < 0.05). Of note (and not surprisingly, given the known role of cPLA2α in neutrophil function), enhanced neutrophil infiltration into the ischemic zone did not appear to be the cause of the increased injury since neutrophil number was reduced by 29% in the ischemic zone of hearts of cPLA2α(−/−) mice compared to the cPLA2α(+/+) mice ( Figure 2B ).
Figure 2.
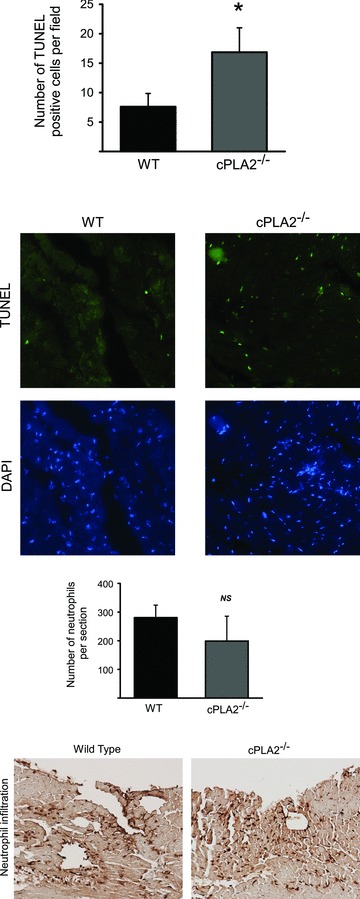
I/R‐induced apoptosis and neutrophil infiltration in the WT versus cPLA2α(−/−) mouse. WT and cPLA2α(−/−) mice were subjected to I/R injury as described in the Materials and Methods. At 6 hours of reperfusion, hearts were collected and processed for (A) TUNEL or (B) labeling for infiltrating neutrophils as described in Materials and Methods. n= 9 WT and 7 cPLA2α(−/−) hearts. Between three and seven fields per heart were analyzed, and values are expressed as the average of the number of positive cells per field. Representative images are shown below each graph. *p < 0.05 as determined by Mann‐Whitney test. NS = nonsignificant.
Activation of intracellular signaling pathways
We next studied possible involvement of intracellular signaling pathways in mediating the protective effects of cPLA2α. Protein extracts of ischemic zones from hearts of cPLA2α(−/−) and cPLA2α(+/+) mice were analyzed at 30 minutes, 90 minutes, 3 hours, 6 hours, or 24 hours following I/R injury. To our surprise, the only notable difference found was decreased extracellular signal‐regulated kinase (ERK) phosphorylation in hearts of cPLA2α(−/−) mice at 3 hours post‐I/R ( Figure 3 , P < 0.05). However, there was no difference in activity of other major protective or detrimental signaling pathways (such as Akt, Stat3, p38, or JNK) between the genotypes at any of the time points (data not shown).
Figure 3.
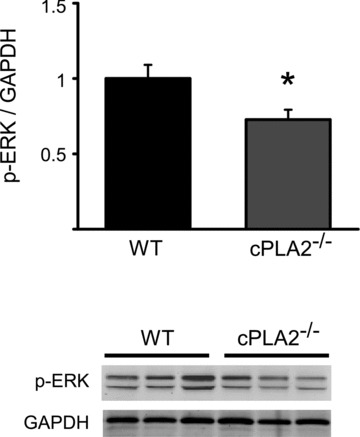
ERK phosphorylation following I/R in the WT versus cPLA2α(−/−) mouse. WT and cPLA2α(−/−) mice were subjected to I/R injury, and heart tissue from ischemic area was collected at 3 hours of reperfusion. Shown is quantification of ERK phosphorylation (upper panel) and representative western blot (lower panel). n= 5 WT and 5 cPLA2α(−/−) hearts. *p < 0.05.
Simulated ischemia in isolated adult cardiomyocytes
We then asked whether the increased injury was due to a cell‐autonomous effect of deletion of cPLA2α in cardiomyoyctes, To address this question, we isolated adult mouse cardiomyocytes from cPLA2α(+/+) and cPLA2α(−/−) hearts and exposed them to simulated ischemia for 45 minutes followed by reoxygenation. We found no difference in rates of adenylate kinase release, a measure of sarcolemmal integrity, between cPLA2α(+/+) versus cPLA2α(−/−) cardiomyocytes ( Figure 4 ). These data suggest that cPLA2α does not have a major cardiomyocyte‐autonomous role in regulating injury.
Figure 4.
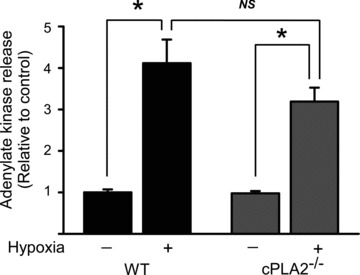
Cell death following simulated ischemia in isolated cardiomyocytes from WT versus cPLA2α(−/−) mice. Cardiomyocytes were isolated as described in Materials and Methods and then were subjected to 45 minutes of hypoxia followed by 16 hours of reperfusion. Quantification of adenylate kinase release (a marker of loss of sarcolemmal integrity) is shown. Data are from three independent experiments, n= 3–5 per experiment. *p < 0.001. NS = nonsignificant.
Analysis of PGE2 levels
Production of the eicosanoid, PGE2, is dependent on cPLA2α in a variety of settings. Ischemia activates cPLA2, AA is released, and PGE2 synthesis is increased in the heart during I/R. 4 , 36 , 37 , 38 Furthermore, PGE2 has been implicated in cardioprotection (see below). Therefore, we asked whether levels of this critical eicosanoid were reduced in the heart of the cPLA2α(−/−) mouse following ischemic stress. We subjected the cPLA2α(+/+) and cPLA2α(−/−) mice to either myocardial infarction (MI) secondary to permanent left anterior descending coronary artery occlusion or transient I/R injury. Analysis of cardiac samples showed that PGE2 levels were slightly lower in both the ischemic and nonischemic zones of the cPLA2α(−/−) versus cPLA2α(+/+) mice following permanent ligation of LAD ( Figure 5A ). With I/R injury, PGE2 levels were markedly decreased in both the ischemic and nonischemic zones of cPLA2α(−/−) as compared to cPLA2α(+/+) mice ( Figure 5B , P < 0.05).
Figure 5.
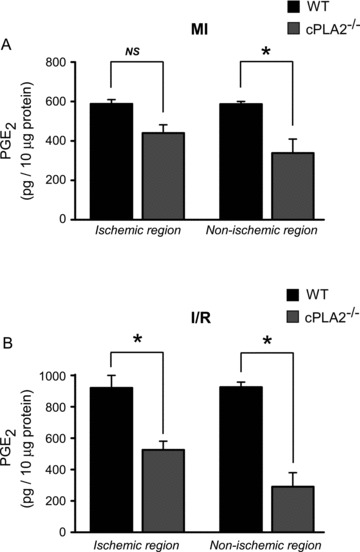
PGE2 production is decreased in the hearts of cPLA2α(−/−) in vivo. WT and cPLA2α(−/−) mice were subjected to either (A) 30‐minute ischemia without reperfusion (MI) or (B) 30‐minute ischemia followed by 20‐minute reperfusion (I/R). At the end of the experiment, the ischemic and nonischemic regions of the LV were isolated and snap‐frozen in liquid nitrogen. Shown are PGE2 levels in the ischemic and nonischemic regions of WT and cPLA2α(−/−) mice. Quantification of PGE2 production was performed as described in the Materials and Methods section. n= 3–4 per experiment, assayed in triplicate. *p < 0.05.
PGE2 analog misoprostol reduces infarct size
Our data indicate that the increased infarct size in cPLA2α(−/−) following I/R is associated with decreased PGE2 levels in the heart. We then asked whether we could reduce the infarct size in the cPLA2α(−/−) mice with exogenous administration of PGE2. Prior to inducing I/R injury, cPLA2α(−/−) and cPLA2α(+/+) mice were treated with the PGE analog, misoprostol, which interacts with both the EP3 and EP4 receptors. Misoprostol treatment had no effect on area at risk, as determined by Evans Blue dye injection ( Figure 6A ). Remarkably, treatment with misoprostol reduced the infarct size in the cPLA2α(−/−) mice to the point that the difference in infarct size between the cPLA2α(−/−) and cPLA2α(+/+) mice was abolished ( Figure 6B ). These data, taken together, are consistent with PGE2 being a critical eicosanoid product downstream of cPLA2α that mediates cardioprotection.
Figure 6.
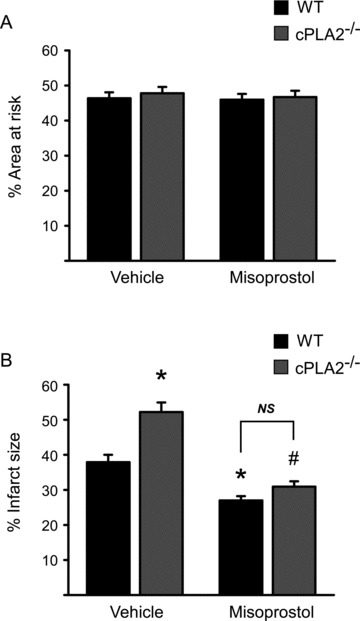
Misoprostol rescues the increased I/R injury in the cPLA2α(−/−) in vivo. WT and cPLA2α(−/−) mice were pretreated with vehicle or misoprostol as described in Materials and Methods and then were subjected to ischemia (30 minutes) and reperfusion (24 hours) prior to sacrifice for determination of area at risk (panel A) and infarct size (panel B). (A) Area at risk is similar irrespective of genotype or treatment condition. (B) Quantification of infarct size expressed as a percent of area at risk. n= 12 WT, 13 cPLA2α(−/−), 5 WT + misoprostol, and 7 cPLA2α(−/−)+ misoprostol. *p < 0.001 versus WT + Vehicle; #p < 0.001 versus cPLA2α(−/−)+ Vehicle. NS = nonsignificant. The misoprostol (−) mice are the same as those shown in Figure 1 .
Discussion
Activation of cPLA2s releases AA, which is then metabolized to various prostaglandins and to thromboxane by cyclo‐oxygenases, to leukotrienes by lipoxygenases, and to epoxytrienoic acids (EETs) and hydroxyeicosatetraenoic acids (HETEs) by cytochrome P450 enzymes. 39 , 40 Some of these metabolites (e.g., PGE2 and PGI2 as well as various EETs) are believed to be protective in the heart whereas others (e.g. HETEs) are felt to be detrimental. 39 , 40 Thus, the net effect of cPLA2 inhibition might reflect the balance of production of these various mediators of protection versus injury. Studies examining the role of cPLA2s in I/R injury have consistently reported that cPLA2 is detrimental, although as noted, many of these studies have relied on inhibitors of questionable selectivity, have been done in isolated cells, or have specifically examined I/R injury in the brain with its inherently different pathophysiology compared to the heart. Taken together, our data demonstrate (1) a protective effect of cPLA2αin vivo, (2) no protection by cPLA2α in isolated cardiomyocytes, and (3) amelioration of the deleterious effects of deletion of cPLA2αin vivo by administration of a stable PGE analog. Although misoprostol ameliorated injury in the cPLA2α(−/−) mouse, we cannot conclude that PGE2 is the only (or even the primary) mediator of cardioprotection in vivo following I/R. Rather, we believe that other eicosanoid products of cPLA2α likely contribute to cardioprotection in the cPLA2α(+/+) mouse.
How might cPLA2α mediate protection in vivo but not in the isolated cardiomyocyte model? The lack of an effect in isolated adult cardiomyocytes subjected to simulated ischemia suggests that cPLA2α does not play an important role in a cardiomyocyte‐autonomous manner. Rather, protective effects may primarily be due to effects on other cell types (e.g., endothelial cells, vascular smooth muscle cells, or even fibroblasts). Answers to this question must await cell‐type‐specific knockouts of cPLA2α. This conclusion differs from that reached by Engelbrecht and Ellis, who subjected isolated neonatal rat cardiomyocytes to hypoxia/reoxygenation (H/R). 28 There are several possible explanations for this. First, cPLA2α may play different roles in the response of neonatal rat versus adult mouse myocytes to H/R. Alternatively, these authors employed a chemical inhibitor (AACOCF3) that inhibits all three members of the cPLA2 class (and may have some inhibitory activity against other PLA2s). Thus, it is possible that cPLA2β and/or γ may be more important in cardiomyocytes, or that some activity against iPLA2s may play a role in the protection seen with AACOCF3.
cPLA2α is an essential factor in the generation of PGE2 in many cells, 19 , 41 and we confirmed that cPLA2α plays a key role in PGE2 production in vivo following ischemic injury in the heart. PGE2 has important protective effects against I/R injury since germline deletion of the receptor for PGE2 (EP4 receptor) enhanced I/R injury, and an EP4 agonist (4819‐CD) was cardioprotective. 37 ERK is one of the key signaling pathways shown to protect the heart from I/R injury. 42 , 43 , 44 PGE2 activates ERK, and the antiapoptotic actions of PGE2 are abolished by inhibition of the ERK pathway, 45 , 46 which is consistent with the noted decrease in ERK phosphorylation in ischemic regions of cPLA2α(−/−) hearts. Thus, it seems likely that the enhanced infarct size in the cPLA2α(−/−) is due, at least in part, to impaired generation of this eicosanoid. Consistent with this conclusion, treatment with the PGE2 mimetic misoprostol, which binds to both the EP4 and EP3 receptors for PGE2, rescued the phenotype of increased infarct size in the knock out animals. The protective effect of misoprostol was also noted in WT animals, though it was less marked.
The EP4 receptor is expressed in cardiomyocytes. The receptor couples to the pro‐survival phosphoinositide‐3 kinase pathway in multiple cell types. 47 We considered that either this or activation of STAT3 48 , 49 may explain the protection seen. However, we saw no significant differences in activation of these signaling pathways in the hearts of cPLA2α(−/−) versus cPLA2α(+/+), as demonstrated by no differences in phospho‐Akt or phospho‐STAT3 (data not shown). Since such studies largely reflect signaling in cardiomyocytes (the vast majority of cell mass in the heart), these data suggest that reduced PGE2/EP4 signaling in cPLA2α(−/−) cardiomyocytes is not an important mechanism of increased I/R injury in the cPLA2α(−/−). Consistent with this, prior studies have shown that cardiomyocyte‐specific deletion of the EP4 receptor resulted in no increase in MI size, in contrast to the increase in MI size noted above in the germline KO of EP4. 37 , 50 Finally, activation of the EP4 receptor by a synthetic ligand only weakly activated signaling in cardiomyocytes but strongly activated it in noncardiomyocytes. 37 These data, which suggest a limited, if any, protective role for PGE2 and the EP4 receptor in regulating ischemic injury specifically in cardiomyocytes, further support the conclusion that PGE2 exerted beneficial effects on cell types other than cardiomyocytes. Thus, we believe our data are most consistent with the detrimental effect of cPLA2α deletion being due to a reduction in PGE2 and, likely, other eicosanoid‐mediated protective effects on noncardiomyocytes in the heart.
In summary, cPLA2α is protective in vivo in the setting of I/R injury. This does not appear to be due to an important direct action of cPLA2α in cardiomyocytes. cPLA2α‐dependent production of PGE2 and, possibly, other prostaglandins (or EETs) appear to be critical to the infarct‐reducing effect seen in the knock out in vivo. Thus, inactivation of cPLA2α can produce widely divergent effects on I/R injury in various tissues, depending on the major factors driving injury. The variation likely depends, in large part, on the level of expression of cPLA2α, and the specific location of receptors for protective versus deleterious eicosanoids. In any case, our findings suggest that targeting of cPLA2α with small molecule inhibitors for any of the numerous disease states that are regulated by cPLA2α should be balanced against potential deleterious effects in patients with concomitant cardiovascular disease.
Conflict of Interest
None
Acknowledgments
This work was supported by NIH grants HL67371 and HL61688 (TF), HL58672 and HL74854 (JC), and DK054741 and DK39773 (JVB). RK was supported by grants from Emil Aaltonen Foundation, Finnish Foundation for Cardiovascular Research, Sigrid Juselius Foundation, and The Maud Kuistila Memorial Foundation. MB, DH, and KCW were supported by grants from the American Heart Association. MB was also supported by a Fonds de la Recherche en Santé du Québec scholarship. TF was supported by The Kahn Foundation and the Scarperi Family.
References
- 1. Bonventre JV. Roles of phospholipases A2 in brain cell and tissue injury associated with ischemia and excitotoxicity. J Lipid Mediat Cell Signal. 1997; 17: 71–79. [DOI] [PubMed] [Google Scholar]
- 2. Bonventre JV, Sapirstein A. Group IV cytosolic phospholipase A2 (PLA2) function: insights from the knockout mouse. Adv Exp Med Biol. 2002; 507: 25–31. [DOI] [PubMed] [Google Scholar]
- 3. Uozumi N, Shimizu T. Roles for cytosolic phospholipase A2alpha as revealed by gene‐targeted mice. Prostaglandins Other Lipid Mediat. 2002; 68–69: 59–69. [DOI] [PubMed] [Google Scholar]
- 4. Lambert IH, Pedersen SF, Poulsen KA. Activation of PLA2 isoforms by cell swelling and ischaemia/hypoxia. Acta Physiol (Oxf). 2006; 187: 75–85. [DOI] [PubMed] [Google Scholar]
- 5. De Windt LJ, Willems J, Roemen TH, Coumans WA, Reneman RS, Van Der Vusse GJ, Van BM. Ischemic‐reperfused isolated working mouse hearts: membrane damage and type IIA phospholipase A2. Am J Physiol Heart Circ Physiol. 2001; 280: H2572–H2580. [DOI] [PubMed] [Google Scholar]
- 6. Nijmeijer R, Willemsen M, Meijer CJ, Visser CA, Verheijen RH, Gottlieb RA, Hack CE, Niessen HW. Type II secretory phospholipase A2 binds to ischemic flip‐flopped cardiomyocytes and subsequently induces cell death. Am J Physiol Heart Circ Physiol. 2003; 285: H2218–H2224. [DOI] [PubMed] [Google Scholar]
- 7. Prasad MR, Popescu LM, Moraru II, Liu XK, Maity S, Engelman RM, Das DK. Role of phospholipases A2 and C in myocardial ischemic reperfusion injury. Am J Physiol. 1991; 260: H877–H883. [DOI] [PubMed] [Google Scholar]
- 8. Fujioka D, Saito Y, Kobayashi T, Yano T, Tezuka H, Ishimoto Y, Suzuki N, Yokota Y, Nakamura T, Obata JE, et al Reduction in myocardial ischemia/reperfusion injury in group X secretory phospholipase A2‐deficient mice. Circulation. 2008; 117: 2977–2985. [DOI] [PubMed] [Google Scholar]
- 9. Dzavik V, Lavi S, Thorpe K, Yip PM, Plante S, Ing D, Overgaard CB, Osten MD, Lan J, Robbins K, et al The sPLA2 inhibition to decrease enzyme release after percutaneous coronary intervention (SPIDER‐PCI) trial. Circulation. 2010; 122: 2411–2418. [DOI] [PubMed] [Google Scholar]
- 10. Rosenson RS, Hislop C, Elliott M, Stasiv Y, Goulder M, Waters D. Effects of varespladib methyl on biomarkers and major cardiovascular events in acute coronary syndrome patients. J Am Coll Cardiol. 2010; 56: 1079–1088. [DOI] [PubMed] [Google Scholar]
- 11. Rosenson RS, Elliott M, Stasiv Y, Hislop C. Randomized trial of an inhibitor of secretory phospholipase A2 on atherogenic lipoprotein subclasses in statin‐treated patients with coronary heart disease. Eur Heart J. 2011; 32: 999–1005. [DOI] [PubMed] [Google Scholar]
- 12. Ford DA, Hazen SL, Saffitz JE, Gross RW. The rapid and reversible activation of a calcium‐independent plasmalogen‐selective phospholipase A2 during myocardial ischemia. J Clin Invest. 1991; 88: 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McHowat J, Creer MH. Catalytic features, regulation and function of myocardial phospholipase A2. Curr Med Chem Cardiovasc Hematol Agents 2004; 2: 209–218. [DOI] [PubMed] [Google Scholar]
- 14. Williams SD, Gottlieb RA. Inhibition of mitochondrial calcium‐independent phospholipase A2 (iPLA2) attenuates mitochondrial phospholipid loss and is cardioprotective. Biochem J. 2002; 362: 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mancuso DJ, Abendschein DR, Jenkins CM, Han X, Saffitz JE, Schuessler RB, Gross RW. Cardiac ischemia activates calcium‐independent phospholipase A2beta, precipitating ventricular tachyarrhythmias in transgenic mice: rescue of the lethal electrophysiologic phenotype by mechanism‐based inhibition. J Biol Chem. 2003; 278: 22231–22236. [DOI] [PubMed] [Google Scholar]
- 16. Mancuso DJ, Sims HF, Han X, Jenkins CM, Guan SP, Yang K, Moon SH, Pietka T, Abumrad NA, Schlesinger PH, et al Genetic ablation of calcium‐independent phospholipase A2gamma leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J Biol Chem. 2007; 282: 34611–34622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leslie CC. Properties and regulation of cytosolic phospholipase A2. J Biol Chem. 1997; 272: 16709–16712. [DOI] [PubMed] [Google Scholar]
- 18. Choi JH, Choi EK, Park SJ, Ko HM, Kim KJ, Han SJ, Choi IW, Im SY. Impairment of p38 MAPK‐mediated cytosolic phospholipase A2 activation in the kidneys is associated with pathogenicity of Candida albicans. Immunology. 2007; 120: 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bonventre JV, Huang Z, Taheri MR, O’Leary E, Li E, Moskowitz MA, Sapirstein A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997; 390: 622–625. [DOI] [PubMed] [Google Scholar]
- 20. Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, et al Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997; 390: 618–622. [DOI] [PubMed] [Google Scholar]
- 21. Levy R. The role of cytosolic phospholipase A2‐alfa in regulation of phagocytic functions. Biochim Biophys Acta. 2006; 1761: 1323–1334. [DOI] [PubMed] [Google Scholar]
- 22. Hong KH, Bonventre JC, O’Leary E, Bonventre JV, Lander ES. Deletion of cytosolic phospholipase A(2) suppresses Apc(Min)‐induced tumorigenesis. Proc Natl Acad Sci USA. 2001; 98: 3935–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kudo I, Murakami M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002; 68–69: 3–58. [DOI] [PubMed] [Google Scholar]
- 24. Hegen M, Sun L, Uozumi N, Kume K, Goad ME, Nickerson‐Nutter CL, Shimizu T, Clark JD. Cytosolic phospholipase A2alpha‐deficient mice are resistant to collagen‐induced arthritis. J Exp Med. 2003; 197: 1297–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miyaura C, Inada M, Matsumoto C, Ohshiba T, Uozumi N, Shimizu T, Ito A. An essential role of cytosolic phospholipase A2alpha in prostaglandin E2‐mediated bone resorption associated with inflammation. J Exp Med. 2003; 197: 1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haq S, Kilter H, Michael A, Tao J, O’Leary E, Sun XM, Walters B, Bhattacharya K, Chen X, Cui L, et al Deletion of cytosolic phospholipase A2 promotes striated muscle growth. Nat Med. 2003; 9: 944–951. [DOI] [PubMed] [Google Scholar]
- 27. Tabuchi S, Uozumi N, Ishii S, Shimizu Y, Watanabe T, Shimizu T. Mice deficient in cytosolic phospholipase A2 are less susceptible to cerebral ischemia/reperfusion injury. Acta Neurochir Suppl. 2003; 86: 169–172. [DOI] [PubMed] [Google Scholar]
- 28. Engelbrecht AM, Ellis B. Apoptosis is mediated by cytosolic phospholipase A2 during simulated ischaemia/reperfusion‐induced injury in neonatal cardiac myocytes. Prostaglandins Leukot Essent Fatty Acids. 2007; 77: 37–43. [DOI] [PubMed] [Google Scholar]
- 29. Winstead MV, Lucas KK, Dennis EA. Group IV cytosolic phospholipase A2 mediates arachidonic acid release in H9c2 rat cardiomyocyte cells in response to hydrogen peroxide. Prostaglandins Other Lipid Mediat. 2005; 78: 55–66. [DOI] [PubMed] [Google Scholar]
- 30. DeGeorge BR Jr, Gao E, Boucher M, Vinge LE, Martini JS, Raake PW, Chuprun JK, Harris DM, Kim GW, Soltys S, et al Targeted inhibition of cardiomyocyte Gi signaling enhances susceptibility to apoptotic cell death in response to ischemic stress. Circulation. 2008; 117: 1378–1387. [DOI] [PubMed] [Google Scholar]
- 31. Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, Fan Q, Chuprun JK, Ma XL, Koch WJ. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010; 107: 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen X, Shevtsov SP, Hsich E, Cui L, Haq S, Aronovitz M, Kerkela R, Molkentin JD, Liao R, Salomon RN, et al The beta‐catenin/T‐cell factor/lymphocyte enhancer factor signaling pathway is required for normal and stress‐induced cardiac hypertrophy. Mol Cell Biol. 2006; 26: 4462–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Song J, Zhang XQ, Wang J, Cheskis E, Chan TO, Feldman AM, Tucker AL, Cheung JY. Regulation of cardiac myocyte contractility by phospholemman: Na+/Ca2 +exchange versus Na+ ‐K+ ‐ATPase. Am J Physiol Heart Circ Physiol. 2008; 295: H1615–H1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ho JC, Wu S, Kam KW, Sham JS, Wong TM. Effects of pharmacological preconditioning with U50488H on calcium homeostasis in rat ventricular myocytes subjected to metabolic inhibition and anoxia. Br J Pharmacol 2002; 137: 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu J, Kam KW, Zhou JJ, Yan WY, Chen M, Wu S, Wong TM. Effects of heat shock protein 70 activation by metabolic inhibition preconditioning or kappa‐opioid receptor stimulation on Ca2+ homeostasis in rat ventricular myocytes subjected to ischemic insults. J Pharmacol Exp Ther. 2004; 310: 606–613. [DOI] [PubMed] [Google Scholar]
- 36. Xiao CY, Hara A, Yuhki K, Fujino T, Ma H, Okada Y, Takahata O, Yamada T, Murata T, Narumiya S, et al Roles of prostaglandin I(2) and thromboxane A(2) in cardiac ischemia‐reperfusion injury: a study using mice lacking their respective receptors. Circulation. 2001; 104: 2210–2215. [DOI] [PubMed] [Google Scholar]
- 37. Xiao CY, Yuhki K, Hara A, Fujino T, Kuriyama S, Yamada T, Takayama K, Takahata O, Karibe H, Taniguchi T, et al Prostaglandin E2 protects the heart from ischemia‐reperfusion injury via its receptor subtype EP4. Circulation. 2004; 109: 2462–2468. [DOI] [PubMed] [Google Scholar]
- 38. Hendrickson SC, St Louis JD, Lowe JE, Abdel‐aleem S. Free fatty acid metabolism during myocardial ischemia and reperfusion. Mol Cell Biochem 1997; 166: 85–94. [DOI] [PubMed] [Google Scholar]
- 39. Gross GJ, Hsu A, Falck JR, Nithipatikom K. Mechanisms by which epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J Mol Cell Cardiol. 2007; 42: 687–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Granville DJ, Gottlieb RA. Having a heart attack? Avoid the “HETE”! Am J Physiol Heart Circ Physiol. 2006; 291: H485–H487. [DOI] [PubMed] [Google Scholar]
- 41. Ghosh M, Stewart A, Tucker DE, Bonventre JV, Murphy RC, Leslie CC. Role of cytosolic phospholipase A(2) in prostaglandin E(2) production by lung fibroblasts. Am J Respir Cell Mol Biol. 2004; 30: 91–100. [DOI] [PubMed] [Google Scholar]
- 42. Yue TL, Wang C, Gu JL, Ma XL, Kumar S, Lee JC, Feuerstein GZ, Thomas H, Maleeff B, Ohlstein EH. Inhibition of extracellular signal‐regulated kinase enhances Ischemia/Reoxygenation‐induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ Res. 2000; 86: 692–699. [DOI] [PubMed] [Google Scholar]
- 43. Lips DJ, Bueno OF, Wilkins BJ, Purcell NH, Kaiser RA, Lorenz JN, Voisin L, Saba‐El‐Leil MK, Meloche S, Pouyssegur J, et al MEK1‐ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation. 2004; 109: 1938–1941. [DOI] [PubMed] [Google Scholar]
- 44. Darling CE, Jiang R, Maynard M, Whittaker P, Vinten‐Johansen J, Przyklenk K. Postconditioning via stuttering reperfusion limits myocardial infarct size in rabbit hearts: role of ERK1/2. Am J Physiol Heart Circ Physiol. 2005; 289: H1618–H1626. [DOI] [PubMed] [Google Scholar]
- 45. Frias MA, Rebsamen MC, Gerber‐Wicht C, Lang U. Prostaglandin E2 activates Stat3 in neonatal rat ventricular cardiomyocytes: a role in cardiac hypertrophy. Cardiovasc Res. 2007; 73: 57–65. [DOI] [PubMed] [Google Scholar]
- 46. Frias MA, Somers S, Gerber‐Wicht C, Opie LH, Lecour S, Lang U. The PGE2‐Stat3 interaction in doxorubicin‐induced myocardial apoptosis. Cardiovasc Res. 2008; 80: 69–77. [DOI] [PubMed] [Google Scholar]
- 47. Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007; 282: 11613–11617. [DOI] [PubMed] [Google Scholar]
- 48. Han C, Demetris AJ, Stolz DB, Xu L, Lim K, Wu T. Modulation of Stat3 activation by the cytosolic phospholipase A2alpha and cyclooxygenase‐2‐controlled prostaglandin E2 signaling pathway. J Biol Chem. 2006; 281: 24831–24846. [DOI] [PubMed] [Google Scholar]
- 49. Schaub MC, Hefti MA. The PGE2‐Stat3 connection in cardiac hypertrophy. Cardiovasc Res. 2007; 73: 3–5. [DOI] [PubMed] [Google Scholar]
- 50. Qian JY, Harding P, Liu Y, Shesely E, Yang XP, LaPointe MC. Reduced cardiac remodeling and function in cardiac‐specific EP4 receptor knockout mice with myocardial infarction. Hypertension. 2008; 51: 560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]