

Abstract
Signal transduction by the TGF-β family involves sets of receptor serine/threonine kinases, Smad proteins that act as receptor substrates, and Smad-associated transcription factors that target specific genes. We have identified discrete structural elements that dictate the selective interactions between receptors and Smads and between Smads and transcription factors in the TGF-β and BMP pathways. A cluster of four residues in the L45 loop of the type I receptor kinase domain, and a matching set of two residues in the L3 loop of the Smad carboxy-terminal domain establish the specificity of receptor–Smad interactions. A cluster of residues in the highly exposed α-helix 2 of the Smad carboxy-terminal domain specify the interaction with the DNA-binding factor Fast1 and, as a result, the gene responses mediated by the pathway. By establishing specific interactions, these determinants keep the TGF-β and BMP pathways segregated from each other.
Keywords: TGF-β, signal transduction, Smad proteins, BMP pathway
The transforming growth factor β (TGF-β) family of polypeptide growth factors regulate cell division, differentiation, motility, adhesion, and death in virtually all metazoan tissues (Massagué 1990; Roberts and Sporn 1990; Kingsley 1994; Gaddy-Kurten et al. 1995; Hogan 1996; Mehler et al. 1997). Members of this family include the TGF-βs, the activins, the bone morphogenetic proteins (BMPs), and other related factors. Signal transduction by these factors involves three classes of molecules: a family of membrane receptor serine/threonine kinases, a family of cytoplasmic proteins (the Smad family) that serves as substrates for these receptors, and nuclear DNA-binding factors that associate with Smads forming transcriptional complexes (Heldin et al. 1997; Massagué 1998). Signaling is initiated by binding the growth factor to a specific pair of receptor kinases, an event that induces the phosphorylation and activation of one kinase, known as the type I receptor, by the other kinase or type II receptor (Wrana et al. 1994). The activated type I receptor phosphorylates a subset of Smads, known as receptor-regulated Smads (R-Smads), which then move into the nucleus (Heldin et al. 1997; Massagué 1998). On their way to the nucleus, R-Smads associate with the related protein Smad4 (Lagna et al. 1996), a tumor-suppressor gene product (Hahn et al. 1996). In the nucleus, this complex may associate with specific DNA-binding proteins that direct it to the regulatory region of target genes. The first identified Smad-associated DNA-binding factor is the forkhead family member Fast1, which mediates activation of Mix.2 in response to activin-type signals during Xenopus embryogenesis (X. Chen et al. 1996). The integrity of this signaling network is essential for normal development and tissue homeostasis, and its disruption by mutation underlies several human inherited disorders and cancer (Heldin et al. 1997; Massagué 1998).
Because of the diversity of processes controlled by different TGF-β family members, there is an intense interest in elucidating the basis for the specificity of their signal transduction pathways. The TGF-β and activin type I receptors, which have nearly identical kinase domains (Cárcamo et al. 1994; ten Dijke et al. 1994), interact with and phosphorylate Smad2 (or the closely related Smad3) (Baker and Harland 1996; Graff et al. 1996; Macias-Silva et al. 1996; Zhang et al. 1996; Nakao et al. 1997), which then interacts with DNA-binding factors such as Fast1 (X. Chen et al. 1996, 1997; Liu et al. 1997). The BMP receptors interact with Smad1 (or the closely related Smads 5, 8, or, in Drosophila, Mad) (Sekelsky et al. 1995; Graff et al. 1996; Hoodless et al. 1996; Liu et al. 1996; Yingling et al. 1996; Y. Chen et al. 1997), which do not recognize Fast1 (X. Chen et al. 1996). Although the TGF-β and BMP pathways are well segregated from each other, their receptors and R-Smads are structurally very similar. Therefore, the specificity of the receptor and Smad interactions in each pathway may be dictated by discrete structural elements. Here we describe the identification of such elements in the type I receptors and the R-Smads, and their role in specifying receptor–Smad interactions and Smad interactions with transcription factors.
Results
Determinants of specificity in the type I receptor
We searched the cytoplasmic domain of TGF-β family type I receptors for regions that might determine the specificity of their interactions with R-Smads. One candidate was the GS domain, a 30-amino-acid region located just upstream of the kinase domain in all type I receptors (Wieser et al. 1995). The GS domain contains sites whose phosphorylation by the type II receptor activate the type I receptor kinase (Wrana et al. 1994). Phosphorylation sites in receptor tyrosine kinases function as docking sites for signal transduction molecules (Pawson and Scott 1997). However, replacing the GS domain in the TGF-β type I receptor (TβR-I) with the GS domain from one of the most divergent member of the TβR-I family in vertebrates, ALK2, did not alter the signaling specificity of TβR-I (Wieser et al. 1995; data not shown). This result argued against a role of the GS domain in determining the specificity of receptor–Smad interactions.
A nine-amino-acid segment in the receptor kinase domain, known as the L45 loop, was also of interest (Fig. 1A). It has been shown that replacement of all but the L45 loop in the kinase domain of TβR-I with the corresponding regions from ALK2 yields a construct that still mediates TGF-β responses (Feng and Derynck 1997). As predicted from the conserved structure of protein kinases, the L45 loop links β-strands 4 and 5, and is not part of the catalytic center (Taylor et al. 1992). The L45 loop differs between type I receptors of different signaling specificity, such as the TGF-β receptors and the BMP receptors, but is highly conserved between receptors of similar signaling specificity such as TβR-I and the activin receptor ActR-IB, or the BMP receptors from human (BMPR-IA and BMPR-IB) and Drosophila (Thick veins) (Fig. 1A).
Figure 1.



(A) L45 loop sequences of the TGF-β type I receptor family. Conserved amino acids are boxed. Three groups of functionally related receptors have each a characteristic L45 loop sequence. ALK1 is also known as TSR-1, and ALK2 as ActR-I or Tsk7L. (B) R-Smad association with Smad4. (Scheme) a TGF-β signal transduction pathway with a type II receptor (II), a type I receptor (I), R-Smad phosphorylation (P), Smad4 (4), and a DNA-binding factor (F). COS1 cells were transfected with Flag-tagged Smad1 or Smad2, HA-tagged Smad4, the indicated wild-type (WT) or mutant type I receptors, and the corresponding type II receptors TβR-II or BMPR-II. R-Smad binding to Smad4 was determined after incubation with TGF-β or BMP2. (C) Nuclear translocation of R-Smads induced by wild-type and L45 mutant type I receptors. HepG2 cells were transfected with Flag-tagged Smad1 (solid bars) or Smad2 (hatched bars), the indicated type I receptors, and their corresponding type II receptors. Cells were incubated with TGF-β1 or BMP2 for 1 hr and subjected to anti-Flag immunofluorescence.
To investigate the role of the L45 loop we concentrated our efforts on TβR-I and BMPR-IB. The L45 loops of these two receptors differ by three nonconservative amino acid substitutions (Fig. 1A). We made constructs encoding these receptors with their L45 loops swapped by introducing N267I, D269G, N270T, and T272S mutations in TβR-I, and the reciprocal mutations in BMPR-IB. These constructs showed a complete switch in their ability to activate Smad1 and Smad2. Compared to the wild-type receptors, TβR-I with the BMPR-I L45 loop [TβR-I(LB) construct] lost the ability to induce the formation of a Smad2–Smad4 complex and gained the ability to induce the formation of a Smad1–Smad4 complex (Fig. 1B). The reciprocal pattern was observed with BMPR-IB containing the TβR-I L45 loop [BMPR-IB(LT) construct] (Fig. 1B). These mutations also switched the ability of the receptors to induce translocation of Smad1 and Smad2 into the nucleus (Fig. 1C).
The L45 exchange mutations switched the signaling specificity of the receptors. BMPR-IB(LT) gained the ability to mediate TGF-β- and activin-like responses including activation of the 3TP-lux reporter construct, which contains a TGF-β response element from plasminogen activator inhibitor-1 and three AP-1-binding sites (Wrana et al. 1992) (Fig. 2A), and a reporter construct (A3-CAT) that contains activin- and TGF-β-responsive Fast1-binding sites from the Mix.2 promoter (Huang et al. 1995) (Fig. 2B). TβR-I(LB) lost the ability to mediate these responses (Fig. 2A,B) but gained the ability to mediate a BMP-like response, namely activation of the Vent.2 promoter from Xenopus (Candia et al. 1997) in P19 mouse embryonal carcinoma cells (Fig. 2C). Valine mutations of two conserved threonines (T272 and T274) at or near the TβR-I L45 loop did not impair 3TP–lux activation by TβR-I (data not shown). Further evidence for a switch in signaling specificity was obtained using Xenopus embryo ectoderm explants. In these explants, TGF-β/activin signaling induces dorsal mesoderm and, indirectly, neural tissue via Smad2 (Baker and Harland 1996; Graff et al. 1996), whereas BMP signaling induces ventral mesoderm via Smad1 (Graff et al. 1996; Liu et al. 1996; Thomsen 1996). These effects can be observed using activated mutant forms of the corresponding type I receptors (Suzuki et al. 1997; Hata et al. 1998; see Fig. 2D). However, an activated BMPR-IB receptor containing the L45 loop from TβR-I [BMPR-IB(QD)(LT) construct] lost the ability to induce expression of the ventral mesoderm marker globin and gained the ability to induce the dorsal mesoderm marker muscle actin and the pan-neural marker NRP-1 (Fig. 2D). The reciprocal construct TβR-I(TD)(LB) showed an incomplete switch in signaling specificity in this assay system, losing the capacity to induce muscle actin without a gain of globin induction or a loss of NRP-1 induction (Fig. 2D).
Figure 2.




Exchanging the L45 loops switches the signaling specificity of TβR-I and BMPR-IB. (A) Activation of the TGFβ-responsive reporter 3TP-luciferase in TβR-I-defective R1B/L17 cells transfected with wild-type or mutant receptors. Cells were incubated with TGF-β (T) or BMP2 (B), and luciferase activity was determined in triplicate samples. (Inset) HA-tagged receptors immunoprecipitated from metabolically labeled cells as controls. (B) Activation of the A3–CAT reporter containing activin- and TGF-β-responsive Mix.2 elements. R1B/L17 cells were transfected with Fast1 and receptor constructs. TβR-I transfectants were incubated with TGF-β and BMPR-IB transfectants with BMP2, and CAT activity was determined. (C) Activation of the BMP-responsive reporter Vent.2–luciferase in P19 cells transfected with TβR-II and wild-type or mutant TβR-I. Cells were incubated with BMP2 (B) or TGF-β (T), and luciferase activity was determined. (D) Induction of markers of dorsal mesoderm (muscle actin), ventral mesoderm (globin), and neural tissue (NRP-1) in Xenopus embryos. RNAs encoding the indicated constitutively active receptor forms were injected into the animal pole of two-cell embryos. Expression of muscle actin, globin, NRP-1, and EF-1α (as control) in animal caps from these embryos was determined. Animal caps from uninjected embryos (control), whole embryos (embryo), and a sample without reverse transcription (RT−) were included.
The switch in the signaling specificity of TβR-I(LB) and BMPR-IB(LT) correlated with a switch in their ability to recognize and phosphorylate Smads 1 and 2. The interaction between TGF-β family receptors and R-Smads is transient but can be visualized using mutant Smads lacking the receptor phosphorylation region (Lo et al. 1998). As shown by coprecipitation of affinity-labeled receptors with phosphorylation-defective Smads, TβR-I(LB) gained affinity for Smad1 and lost affinity for Smad2 compared to the wild-type receptors, whereas BMPR-IB(LT) lost affinity for Smad1 and gained affinity for Smad2 (Fig. 3A). This switch extended to the pattern of receptor-dependent Smad phosphorylation. TβR-I and BMPR-I mediate carboxy-terminal phosphorylation of Smad2 (Macias-Silva et al. 1996) and Smad1 (Kretzschmar et al. 1997b), respectively (Fig. 3B); basal phosphorylation (Fig. 3B) is a result of MAP kinase action on inhibitory sites located in the central region of Smads (Kretzschmar et al. 1997a). In contrast to the effects of the wild-type receptors, transfection of TβR-I(LB) elevated the phosphorylation of Smad1, whereas transfection of BMPR-IB(LT) elevated the phosphorylation of Smad2 (Fig. 3B). Interestingly, the increases in Smad phosphorylation caused by transfection of the L45 mutant receptors were ligand independent. Indeed, TβR-I(LB) and BMPR-IB(LT) were hyperactive compared to the wild-type receptors in in vitro kinase assays (data not shown). The phenotype of a TβR-I allele containing a mutation (G261E) three residues upstream of the L45 loop had suggested previously that this region is involved in receptor activation (Weis-Garcia and Massagué 1996). However, despite their elevated kinase activity, the L45 mutant receptors had a clear switch in substrate specificity as TβR-I(LB) did not elevate Smad2 phosphorylation and BMPR-IB(LT) did not elevate Smad1 phosphorylation (Fig. 3B). We conclude that the subtype-specific residues in the receptor L45 loop determine the specificity of Smad recognition, phosphorylation, and activation.
Figure 3.


(A) Receptor–Smad association in COS-1 cells transfected with the indicated type I receptors, the corresponding type II receptors, and Flag-tagged Smad1(1–454) or Smad2(1–456). Receptors were cross-linked to 125I-labeled TGF-β1 (left) or 125I-labeled BMP2 (right). Smad-bound receptors were visualized by anti-Flag immunoprecipitation, SDS-PAGE, and autoradiography (top). Total cell lysates were analyzed to control for receptor expression (middle). Smad expression was controlled by immunoprecipitation from metabolically labeled cells (bottom). (B) Smad phosphorylation was determined in L17 cells transfected with Flag-tagged Smads, the indicated type I receptors, and the corresponding type II receptors. Cells were labeled with [32P]phosphate, incubated with TGF-β1 or BMP2, and immunoprecipitated with anti-Flag.
Matching determinants of specificity in R-Smads
The conserved carboxy-terminal domain of R-Smad proteins, which is known as the “Mad homology-2” (MH2) domain, interacts with specific TGF-β family receptors and has specific effector functions. When expressed on its own in tissue culture cells or Xenopus embryos, the Smad2 MH2 domain is able to interact with the TGF-β receptor (Lo et al. 1998), associate with Fast1 (Liu et al. 1997), and generate TGF-β and activin-like effects (Baker and Harland 1996; Hata et al. 1997). These observations suggested that the receptor and DNA-binding protein interactions of R-Smads are specified by determinants in the MH2 domain.
To search for such determinants, we investigated 21-amino-acid residues of the MH2 domain that are not conserved between Smad1 and Smad2, but are highly conserved in Smads 1, 5, 8, and Mad, or in Smads 2 and 3 (Fig. 4A). The location of these residues in the three-dimensional structure of the protein can be inferred from the crystal structure of the Smad4 MH2 domain (Shi et al. 1997). The Smad4 MH2 monomer contains two β-sheets capped on one side by three α-helices (H3, H4, and H5) forming a bundle and, on the other side, by two large loops (L1 and L2) and an α-helix (H1). Smads form homo-oligomers in the cell (Lagna et al. 1996; Wu et al. 1997) and in solution (Shi et al. 1997). In the crystal structure, the Smad4 MH2 domain forms a disc-shaped trimer, with the loop/helix region of one monomer forming an interface with the three-helix bundle of the next monomer (Fig. 4B, inset). Mutations in tumor-derived, inactive alleles of Smad2 and Smad4 often map to this interface (Shi et al. 1997). At the amino acid sequence level, most of the structural elements of the Smad4 MH2 domain are conserved in the R-Smads (Fig. 4A), which suggests that this three-dimensional structure is also conserved in R-Smads.
Figure 4.
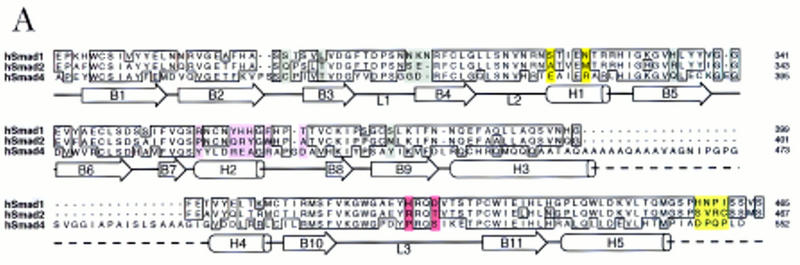
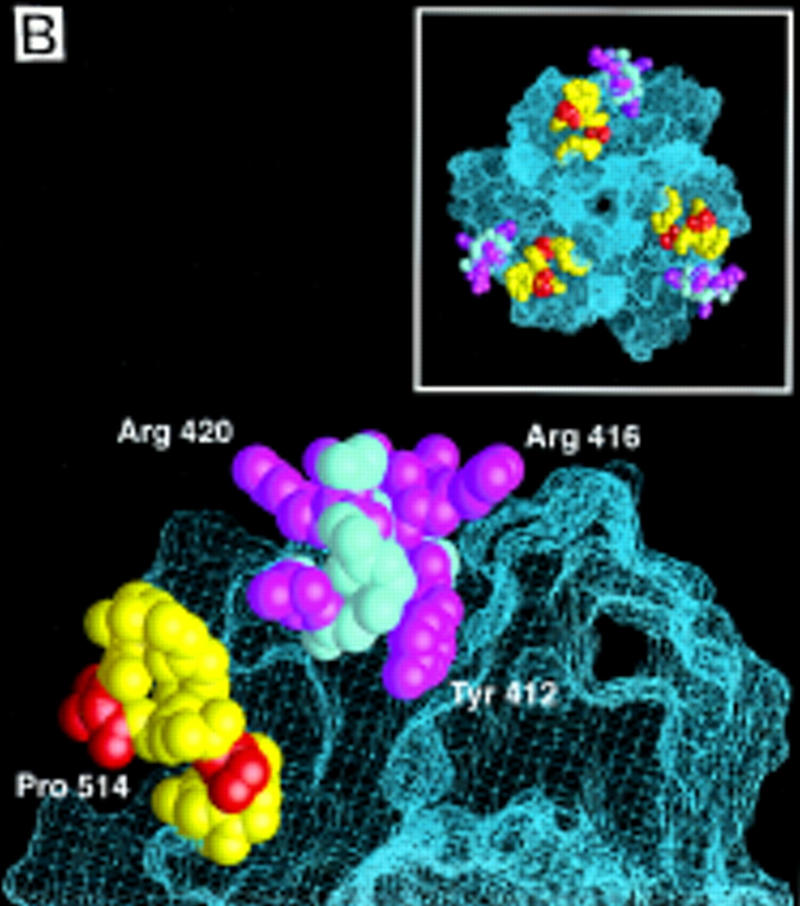
(A) Sequence alignment of the MH2 domains of Smad1, 2, and 4, with the Smad4 MH2 domain secondary structure elements indicated below. Identical residues are boxed. Subtype-specific residues map to α-helix 1 (yellow), α-helix 2 and its vicinity (purple), the L3 loop (red), and immediately upstream of the carboxy-terminal receptor phosphorylation motif SS(V/M)S (green). The remaining subtype-specific residues (gray) are scattered in the primary sequence but clustered in the crystal structure near the point of connection to the amino-terminal half of the molecule (Shi et al. 1997). (B) A close-up, lateral view of the Smad4 MH2 crystal structure showing the L3 loop (yellow) with subtype specific residues (red) and the α-helix 2 (cyan) with subtype-specific residues (magenta). (Inset) Frontal view of the location of the L3 loop and helix 2 of each MH2 monomer in the crystallographic trimer.
Seven of the 21 subtype-specific amino acid residues (gray in Fig. 4A) are clustered on the amino-terminal side of the disc, near the point of connection to the amino-terminal half of the Smad molecule; these residues are exposed only partially to solvent (Shi et al. 1997). Two subtype-specific residues (yellow in Fig. 4A) are located in α-helix 1, and six other (purple in Fig. 4A) are at or near α-helix 2, which is highly exposed on the edge of the disc (Fig. 4B). Of the remaining subtype-specific residues, two (red in Fig. 4A) are located in the L3 loop, a structure protruding from each monomer on the carboxy-terminal side of the disc (see Fig. 4B), and the last four (green in Fig. 4A) are located immediately upstream of the carboxy-terminal receptor phosphorylation motif SS(V/M)S. Neither these four amino acids nor the phosphorylation motif itself is required for association with the TGF-β receptor (Macias-Silva et al. 1996; Lo et al. 1998).
Mutational analysis has shown that the L3 loop of Smad4 is essential for interaction with R-Smads (Shi et al. 1997), whereas the L3 loop of R-Smads is essential for interaction with TGF-β receptors (Lo et al. 1998). Furthermore, the two subtype-specific amino acids in this loop determine the specificity of the Smad-receptor interactions (Lo et al. 1998). To determine whether the specificity of a R-Smad L3 loop matches the specificity of the receptor L45 loop, we investigated whether a Smad2 construct containing the Smad1 L3 loop sequence [Smad2(L1) construct] and the mutant TβR-I(LB) receptor construct would complement each other in the rescue of a TGF-β response. The association of Smad2 with Fast1 in response to agonist was used as a readout in these experiments. Formation of this complex recapitulates various additional signaling events (see Fig. 1B). The Smad2(L1) construct bound Fast1 in response to BMP but not in response to TGF-β (Fig. 5A), which is consistent with the ability of Smad2(L1) to recognize BMPR-IB but not TβR-I (Lo et al. 1998). TβR-I(LB) failed to mediate Smad2 association with Fast1. However, TβR-I(LB) mediated Smad2(L1) association with Fast1 (Fig. 5B). Furthermore, the combination of TβR-I(LB) and Smad2(L1) rescued, partially at least, the ability to activate a Mix.2 reporter construct in response to TGF-β (Fig. 5C). Therefore, the specificity of TGF-β receptor–Smad interaction is determined by the L45 loop of the type I receptor and a complementary L3 loop in Smad2.
Figure 5.



Matching receptor L45 loops and R-Smad L3 loops. (A) The L3 loop determines Smad activation by a specific receptor but not Smad interaction with Fast1. COS1 cells were transfected with Flag-tagged Smad constructs, myc-tagged Fast1, and TGF-β receptors or BMP receptors. Cells were incubated with the corresponding receptor ligands, TGF-β1 or BMP4, and Smad association with Fast1 was determined. [Ig(H)] immunoglobulin heavy chain. (B,C) TβR-I(LB) rescues the ability of TGF-β to induce Smad2(L1) association with Fast1 (B) and activation of the A3–luciferase Mix.2 reporter (C). R1B/L17 cells transfected with various constructs, as indicated, were incubated with 0.5 nm TGF-β for 20 hr, and luciferase activity was measured.
Determinants of Smad interaction with a DNA-binding partner
How a specific gene is targeted for activation by Smads has been delineated in the case of Mix.2. Activation of Mix.2 by activin or TGF-β requires the formation of a Smad2–Smad4–Fast1 complex that binds to a specific promoter sequence known as the activin response element (ARE) (X. Chen et al. 1996, 1997; Liu et al. 1997). In this complex, the DNA-binding domain of Fast1 mediates specific binding to the ARE (X. Chen et al. 1996), whereas the Smads act as transcriptional activators and enhancers of DNA binding (Liu et al. 1997). The interaction between Smad2 and Fast1 is direct, as determined by their ability to interact as recombinant proteins in solution or in a yeast two-hybrid assay (X. Chen et al. 1997, unpubl.).
To identify a structural element that might specify the interaction of Smad2 with Fast1, we investigated whether candidate Smad2 sequences introduced into Smad1 would allow it to recognize Fast1 and activate a Mix.2 ARE reporter in response to BMP. The presence of six subtype-specific residues in the helix 2 of the MH2 domain (see Fig. 4A), and the prominent exposure of helix 2 on the edge of the MH2 trimer (Fig. 4B) made this region a good candidate for this interaction. Exchanging the six subtype-specific helix 2 residues of Smad1 and Smad2 did not alter the specificity of their receptor interactions. Smad1 containing the helix 2 sequence of Smad2 [Smad1(H2) construct] bound Smad4 in response to BMP, and the reciprocal construct, Smad2(H1), bound Smad4 in response to TGF-β (Fig. 6A, top). However, these helix 2 mutations switched the pattern of interactions with Fast1. Smad1(H2) gained the ability to associate with Fast1 in response to BMP, whereas Smad2(H1) failed to do so in response to TGF-β (Fig. 6A, bottom). Correlating with this switch, Smad1(H2) was able to mediate activation of a Mix.2 reporter in response to BMP, whereas Smad2(H1) was unable to mediate activation of this reporter (Fig. 6B). The Fast1 interaction specified by the Smad2 helix 2 was independent of the target promoter as Smad1(H2) was also able to activate a GAL4 reporter construct in cooperation with a Fast1–GAL4 DNA-binding domain fusion (Fig. 6C). These results suggest that α-helix 2 of Smad2 is primarily responsible for the specificity for Fast1 and, as a result, the gene responses activated by the pathway. Extending these observations to the BMP pathway, Smad2(H1) gained the ability to mediate activation of a Vent.2 reporter in response to TGF-β (Fig. 6D).
Figure 6.




The α-helix 2 of Smad2 specifies the interaction with the DNA-binding factor Fast1. (A) Interaction of wild-type R-Smads and helix 2 exchange mutants with Smad4 and Fast1. HA-tagged Smad4 or myc-tagged Fast1 constructs were cotransfected into COS1 cells with the indicated Flag-tagged forms of Smad1 or Smad2. Transfectants were incubated with TGF-β (T) or BMP2 (B) and the associations of R-Smads with Smad4 (top) and with Fast1 (bottom) were determined. The helix 2 exchange mutants bound Smad4 in response to their agonists, but Smad2(H1) lost the ability to associate with Fast1 whereas Smad1(H2) gained the ability to bind Fast1 in response to BMP. (B) Activation of a Mix.2 reporter by wild-type R-Smads and helix 2 exchange mutants. L17 cells were cotransfected with the indicated forms of Smad1 or Smad2, Fast1, the A3-luciferase construct, and TGF-β receptors or BMP receptors. Cells were incubated with the corresponding receptor ligands, and luciferase activity was determined. Smad2(H1) lost the ability to activate the reporter, whereas Smad1(H2) gained the ability to do so in response to BMP. (C) Fast1-dependent activation of a GAL4 reporter by Smad1(H2). L17 cells were cotransfected with the indicated forms of Smad1, a Fast1 fusion with the DNA-binding domain from yeast GAL4, a GAL luciferase reporter, and BMP receptors. Cells were incubated with or without BMP2, and luciferase activity was determined. (D) Activation of the Vent.2–luciferase reporter in P19 cells cotransfected with TβR-I, TβR-II, and the indicated Smad2 constructs. Cells were incubated with or without TGF-β, and luciferase activity was determined in triplicate samples.
Discussion
We have identified key determinants of specificity at three levels in the TGF-β and BMP signaling pathways. These determinants are encoded by specific amino acid residues in the L45 loop of the kinase domain in the type I receptors, and in the L3 loop and the α-helix 2 of the MH2 domain in R-Smads. In each case, the residues involved are few and highly conserved in receptors or R-Smads that have similar signaling specificity. The interaction between these proteins may involve additional surface contacts, but our results suggest that pathway specificity is largely determined by these residues. Exchanging these residues at any of the three levels between TGF-β and BMP pathway components switches the signaling specificity of these pathways.
The L45 loop of type I receptor kinases had drawn attention previously because replacing the entire kinase domain except this loop in TβR-I with the corresponding regions from the functionally divergent receptor kinase ALK2 still allows mediation of TGF-β responses (Feng and Derynck 1997). The L3 loop of Smads has drawn attention as a target of inactivating mutations in Drosophila and Caenorhabditis elegans Smad family members (Sekelsky et al. 1995; Savage et al. 1996). As inferred from the effect of similar mutations in vertebrate Smads, the L3 loop participates in different interactions that are essential for signaling. In Smad4 the L3 loop is required for interaction with activated R-Smads (Shi et al. 1997), whereas in R-Smads the L3 loop is required for interaction with the receptors and, furthermore, it specifies these interactions (Lo et al. 1998). The present results show that matching combinations of L45 loops and L3 loops determine the specificity of the receptor–Smad interaction. Exchanging the subtype-specific residues in either the L45 loop or the L3 loop causes a switch in the specificity of this interaction, with an attendant switch in the signaling specificity of the pathway. As evidence of a functional match between a receptor L45 loop and a R-Smad L3 loop, the switch in the signaling specificity of a TGF-β receptor construct containing the BMP receptor L45 loop can be reversed by a Smad2 construct containing the matching L3 loop sequence from Smad1.
Our results suggest that the interaction supported by the L45 and L3 loops achieves signal transduction by increasing selectively the affinity of a particular receptor kinase for a particular subtype of R-Smads. The docking interaction between receptors and R-Smads is independent of their catalytic interaction. The carboxy-terminal SSXS phosphorylation motif of R-Smads and the adjacent upstream sequence are neither required for association with the receptors in vivo nor for the specificity of this interaction (Lo et al. 1998). However, effective R-Smad phosphorylation in vivo requires this docking interaction. Mutations that disrupt receptor docking strongly inhibit Smad phosphorylation and signal transduction. Of note, no stable interaction has been observed between the recombinant receptor kinase domains and Smads 1 or 2 in solution. Under these conditions, the TβR-I and BMPR-IB kinases can phosphorylate both Smad1 and Smad2, and mutations in the L45 loop do not inhibit these reactions (Y.G. Chen and J. Massagué, unpubl.). Therefore, the interaction supported by the L45 and L3 loops might be cooperative, requiring the correct assembly of multivalent receptor complexes and R-Smad complexes in the cell.
The present work also provides evidence that the choice of DNA-binding partner and, consequently, the choice of target genes are determined by helix 2 in the MH2 domain of R-Smads. In the crystal structure of the Smad4 MH2 domain, helix 2 protrudes from the edge of the Smad trimer with several highly exposed residues. The sequence of helix 2 is divergent between R-Smads that mediate TGF-β (or activin) responses and those that mediate BMP responses, but is highly conserved within each subgroup of R-Smads. Using as models the Mix.2 gene response to TGF-β and the Vent.2 gene response to BMP, we show that the helix 2 of Smad2 and Smad1, respectively, determine the ability to mediate these responses. We further show that helix 2 from Smad2 specifies the selective interaction of Smads with the ARE-binding factor Fast1. Factors that mediate other Smad2- or Smad1-dependent gene responses remain to be identified. The ability of helix 2 to determine these interactions may provide ways to identify such factors. The role of helix 2 in Smad4 is also not known, although a mutation (R420H) in this region has been reported in lung carcinoma (Nagatake et al. 1996).
The identification of determinants of specificity at three levels in TGF-β signal transduction suggests a general model for the organization of the selective protein–protein interactions that configure this signaling network (Fig. 7). The determinants of specificity identified here segregate the TGF-β and BMP pathways from each other. Still, each pathway can generate different responses in different cell types. Specificity at that level may depend on the repertoire of gene-targeting factors that the Smad complex encounters in the nucleus of a given cell.
Figure 7.

Determinants of specificity in TGF-β signal transduction. In the TGF-β or BMP receptor complexes, the type I receptor recognizes and phosphorylates a specific R-Smad, such as Smad2 in the TGF-β pathway or Smad1 in the BMP pathway (Heldin et al. 1997; Massagué 1998). The R-Smad then associates with Smad4 (not shown) and moves into the nucleus. Specific association with the DNA-binding factor Fast1 in the nucleus takes the Smad2–Smad4 complex to specific target genes such as Mix.2, activating their transcription (X. Chen et al. 1996, 1997; Liu et al. 1997). Selection of a R-Smad by a receptor is specified by the type I receptor L45 loop and the R-Smad L3 loop, whereas selection of a DNA-binding factor (such as Fast1 in the case of Smad2) is specified by the α-helix 2 of the R-Smad. Exchanging any of these three elements between the TGF-β and BMP receptors or between Smad1 and Smad2 causes a switch in the signaling specificity of these two pathways. Specific activation of other target genes by Smad1 or Smad2 complexes is presumed to involve different DNA-binding partners.
Materials and methods
Cell culture
R1B/L17 and COS-1 cells were maintained as described previously (Y.G. Chen et al. 1997). HepG2 cells were maintained in minimal essential medium (MEM; GIBCO-BRL) supplemented with 10% fetal bovine serum (FBS), nonessential amino acids, and 2 mm sodium pyruvate. Mouse embryonal carcinoma P19 cells were cultured in DMEM medium supplemented with 10% FBS.
Protein interaction, phosphorylation, and immunofluorescence assays
Mutant receptor and Smad constructs were generated by PCR using appropriate oligonucleotides. Helix 2 exchange mutants were generated by exchanging the 6 residues highlighted in the helix 2 region in Figure 4A. Mutations were verified by DNA sequencing. Wild-type and mutant receptors were carboxy-terminally tagged with a hemagglutinin (HA) epitope and were subcloned into the mammalian expression vector pCMV5. Cells were transfected transiently with the indicated constructs or empty vector by the DEAE–dextran method, as described (Y.G. Chen et al. 1997). Phosphorylation of Smad1 and Smad2 was tested in R-1B/L17 cells by cotransfecting Flag-tagged Smad constructs and the indicated receptor constructs, labeling the cells with [32P]orthophosphate for 2 hr, followed by incubation with 1 nm TGF-β1 or 5 nm BMP2 for 30 min, and anti-Flag immunoprecipitation (Lo et al. 1998). Expression levels of transfected proteins was determined by immunoprecipitation from [35S]methionine/cysteine-labeled cells. Flag-tagged R-Smad interaction with HA-tagged Smad4 or myc-tagged Fast 1 was determined in COS-1 cells by anti-Flag immunoprecipitation and anti-HA or anti-myc Western immunoblotting (Lagna et al. 1996; Liu et al. 1997). For Smad immunofluorescence assays, HepG2 cells were transfected overnight with DNA constructs as indicated, using the standard calcium–phosphate–DNA precipitation method. Twenty-four hours after transfection, cells were transferred onto chamber slides (Nunc, Inc.). Two days later, cells were stimulated with 5 nm BMP2 or 1 nm TGF-β1 for 1 hr and processed for anti-Flag immunofluorescence as described previously (Lo et al. 1998). The percentage of cells showing nuclear staining was determined by counting 200–300 positive cells.
Reporter assays
Activation of the p3TP-luciferase reporter construct (Cárcamo et al. 1995) was analyzed in R1B/L17 cells as described previously (Y.G. Chen et al. 1997). To measure the activity of a Xvent2–luciferase reporter (Candia et al. 1997), P19 cells were transfected with this construct, TβR-I, and TβR-II. The next day, cells were incubated with 0.5 nm TGF-β1 or 1 nm BMP2, and luciferase activity was measured 20 hr later. To measure the activity of a Mix.2 ARE reporters (A3-CAT or A3-luciferase) (Huang et al. 1995), R1B/L17 cells were transfected with these reporters, Fast1, and the indicated receptor constructs. The next day, cells were treated with 0.5 nm TGF-β1 or 1 nm BMP2 for 20 hr and the reporter gene activity was determined as described previously (Liu et al. 1997). A GAL4 DNA-binding domain fusion with Fast1 was created by subcloning Fast1 into pGAD424 (Clontech). GAL4–Fast1 activation was determined in R-1B/L17 cells by cotransfection with the indicated constructs, and incubation with BMP2 for 14 hr on the following day.
Xenopus injections and animal cap assay
Receptor RNA (10 nl, 2 ng) was injected into the animal pole of two-cell embryos. Animal caps were explanted at the blastula stage and incubated to the tailbud stage (stage 28). RT–PCR of the indicated markers was performed as described previously (Lagna et al. 1996).
Receptor assays
TGF-β1 and BMP2 were labeled with sodium 125I as described previously (Cheifetz et al. 1990). To detect receptor–Smad interactions, COS-1 cells were transfected transiently with constructs that encode Smad1 and Smad2 lacking the last 11 amino acids [Smad1(1–454) and Smad2(1–456) constructs] and the indicated receptor constructs. After 40–48 hr, cells were labeled by cross-linking to receptor-bound [125I]TGF-β1 or [125I]BMP2, as described previously (Lo et al. 1998).
Acknowledgments
We thank G. Lagna and A. Hemmati-Brivanlou for animal cap assays and discussions, M. Whitman, C. Niehrs, and M. Kretzschmar for reporter constructs, W. Mark for P19 cells, and C. Pouponnot for helpful discussions. National Institutes of Health grants to J.M. and Memorial Sloan-Kettering Cancer Center supported this work. Y.G.C. and A.H. are Research Associates and J.M. an Investigator of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL j-massague@ski.mskcc.org; FAX (212) 717-3298.
References
- Baker J, Harland RM. A novel mesoderm inducer, mMadr-2, functions in the activin signal transduction pathway. Genes & Dev. 1996;10:1880–1889. doi: 10.1101/gad.10.15.1880. [DOI] [PubMed] [Google Scholar]
- Candia AF, Watabe T, Hawley SH, Onichtchouck D, Zhang Y, Derynck R, Niehrs C, Cho KW. Cellular interpretation of multiple TGF-β signals: Intracellular antagonism between activin/BVg1 and BMP-2/4 signaling mediated by Smads. Development. 1997;124:4467–4480. doi: 10.1242/dev.124.22.4467. [DOI] [PubMed] [Google Scholar]
- Cárcamo J, Weis FMB, Ventura F, Wieser R, Wrana JL, Attisano L, Massagué J. Type I receptors specify growth inhibitory and transcriptional responses to TGF-β and activin. Mol Cell Biol. 1994;14:3810–3821. doi: 10.1128/mcb.14.6.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárcamo J, Zentella A, Massagué J. Disruption of TGF-β signaling by a mutation that prevents transphosphorylation within the receptor complex. Mol Cell Biol. 1995;15:1573–1581. doi: 10.1128/mcb.15.3.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheifetz S, Hernandez H, Laiho M, ten Dijke P, Iwata KK, Massagué J. Distinct transforming growth factor-β receptor subsets as determinants of cellular responsiveness to three TGF-β isoforms. J Biol Chem. 1990;265:20533–20538. [PubMed] [Google Scholar]
- Chen X, Rubock MJ, Whitman M. A transcriptional partner of MAD proteins in TGF-β signalling. Nature. 1996;383:691–696. doi: 10.1038/383691a0. [DOI] [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- Chen Y, Takeshita A, Ozaki K, Kitano S, Hanazawa S. Transcriptional regulation by transforming growth factor β of the expression of retinoic acid and retinoid X receptor genes in osteoblastic cells is mediated through AP-l. J Biol Chem. 1996;271:31602–31606. doi: 10.1074/jbc.271.49.31602. [DOI] [PubMed] [Google Scholar]
- Chen Y, Bhushan A, Vale W. Smad8 mediates the signaling of the receptor serine kinase. Proc Natl Acad Sci. 1997;94:12938–12943. doi: 10.1073/pnas.94.24.12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Liu F, Massagué J. Mechanism of TGFβ receptor inhibition by FKBP12. EMBO J. 1997;16:3866–3876. doi: 10.1093/emboj/16.13.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Derynck R. A kinase subdomain of transforming growth factor-β (TGF-β) type I receptor determines the TGF-β intracellular signaling activity. EMBO J. 1997;16:3912–3922. doi: 10.1093/emboj/16.13.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaddy-Kurten D, Tsichida K, Vale W. Activins and the receptor serine kinase superfamily. Recent Prog Horm Res. 1995;50:109–129. doi: 10.1016/b978-0-12-571150-0.50010-x. [DOI] [PubMed] [Google Scholar]
- Graff JM, Bansal A, Melton DA. Xenopus Mad proteins transduce distinct subsets of signals for the TGFβ superfamily. Cell. 1996;85:479–487. doi: 10.1016/s0092-8674(00)81249-0. [DOI] [PubMed] [Google Scholar]
- Hahn SA, Schutte M, Hoque ATMS, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- Hata A, Lo R, Wotton D, Lagna M, Massagué J. Mutations increasing autoinhibition inactivate the tumour suppressors Smad2 and Smad4. Nature. 1997;388:82–86. doi: 10.1038/40424. [DOI] [PubMed] [Google Scholar]
- Hata A, Lagna G, Massagué J, Hemmati-Brivanlou A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes & Dev. 1998;12:186–197. doi: 10.1101/gad.12.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin C-H, Miyazono K, ten Dijke P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Hogan BLM. Bone morphogenetic proteins: Multifunctional regulators of vertebrate development. Genes & Dev. 1996;10:1580–1594. doi: 10.1101/gad.10.13.1580. [DOI] [PubMed] [Google Scholar]
- Hoodless PA, Haerry T, Abdollah S, Stapleton M, O’Connor MB, Attisano L, Wrana JL. MADR1, a MAD-related protein that functions in BMP2 signalling pathways. Cell. 1996;85:489–500. doi: 10.1016/s0092-8674(00)81250-7. [DOI] [PubMed] [Google Scholar]
- Huang H-C, Murtaugh LC, Vize PD, Whitman M. Identification of a potential regulator of early transcriptional responses to mesoderm inducers in the frog embryo. EMBO J. 1995;14:5965–5973. doi: 10.1002/j.1460-2075.1995.tb00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley DM. The TGF-β superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes & Dev. 1994;10:16–21. doi: 10.1101/gad.8.2.133. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Massagué J. Opposing BMP and EGF signalling pathway converge on the TGFβ family mediator Smad1. Nature. 1997a;389:618–622. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Liu F, Hata A, Doody J, Massagué J. The TGF-β mediator Smad1 is directly phosphorylated and functionally activated by the BMP receptor kinase. Genes & Dev. 1997b;11:984–995. doi: 10.1101/gad.11.8.984. [DOI] [PubMed] [Google Scholar]
- Lagna G, Hata A, Hemmati-Brivanlou A, Massagué J. Partnership between DPC4 and SMAD proteins in TGFβ signalling pathways. Nature. 1996;383:832–836. doi: 10.1038/383832a0. [DOI] [PubMed] [Google Scholar]
- Liu F, Hata A, Baker J, Doody J, Cárcamo J, Harland R, Massagué J. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature. 1996;381:620–623. doi: 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- Liu F, Pouponnot C, Massagué J. Dual role of the Smad4/DPC4 tumor suppressor in TGFβ-inducible transcriptional responses. Genes & Dev. 1997;11:3157–3167. doi: 10.1101/gad.11.23.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo RS, Chen YG, Shi YG, Pavletich N, Massagué J. The L3 loop: A structural motif determining specific interactions between SMAD proteins and TGF-β receptors. EMBO J. 1998;17:996–1005. doi: 10.1093/emboj/17.4.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias-Silva M, Abdollah S, Hoodless PA, Pirone R, Attisano L, Wrana JL. MADR2 is a substrate of the TGFβ receptor and phosphorylation is required for nuclear accumulation and signaling. Cell. 1996;87:1215–1224. doi: 10.1016/s0092-8674(00)81817-6. [DOI] [PubMed] [Google Scholar]
- Massagué J. The transforming growth factor-β family. Ann Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- ————— TGFβ signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Mehler MF, Mabie PC, Zhang D, Kessler JA. Bone morphogenetic proteins in the nervous system. Trends Neurosci. 1997;20:309–317. doi: 10.1016/s0166-2236(96)01046-6. [DOI] [PubMed] [Google Scholar]
- Nagatake M, Takagi Y, Osada H, Uchida K, Mitsudomi T, Saji S, Shimokata K, Takahashi T, Takahashi T. Somatic in vivo alterations of the DPC4 gene at 18q21 in human lung cancers. Cancer Res. 1996;56:2718–2720. [PubMed] [Google Scholar]
- Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J, Heldin CH, Miyazono K, ten Dijke P. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Signaling through scaffold, anchoring and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB. The transforming growth factor-βs. In: Sporn MB, Roberts AB, editors. Peptide growth factors and their receptors. Heidelberg, Germany: Springer-Verlag; 1990. pp. 419–472. [Google Scholar]
- Savage C, Das P, Finelli A, Townsend SR, Sun C-Y, Baird SE, Padgett RW. Caenorhabditis elegans genes sma-2, sma-3 and sma-4 define a conserved family of transforming growth factor β pathway components. Proc Natl Acad Sci. 1996;93:790–794. doi: 10.1073/pnas.93.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekelsky JJ, Newfeld SJ, Raftery LA, Chartoff EH, Gelbart WM. Genetic characterization and cloning of Mothers against dpp, a gene required for decapentaplegic function in Drosophila melanogaster. Genetics. 1995;139:1347–1358. doi: 10.1093/genetics/139.3.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Hata A, Lo RS, Massagué J, Pavletich NP. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature. 1997;388:87–93. doi: 10.1038/40431. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Chang C, Yingling JM, Wang XF, Hemmati-Brivanlou A. Smad5 induces ventral fates in Xenopus embryos. Dev Biol. 1997;184:402–405. doi: 10.1006/dbio.1997.8548. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Knighton DR, Zheng J, Ten Eyck LF, Sowadski JM. Structural framework for the protein kinase family. Annu Rev Cell Biol. 1992;8:429–462. doi: 10.1146/annurev.cb.08.110192.002241. [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Yamashita H, Ichijo H, Franzén P, Laiho M, Miyazono K, Heldin C-H. Characterization of type I receptors for transforming growth factor-β and activin. Science. 1994;264:101–104. doi: 10.1126/science.8140412. [DOI] [PubMed] [Google Scholar]
- Thomsen G. Xenopus mothers against decapentaplegic is an embryonic ventralizing agent that acts downstream of the BMP-2/4 receptor. Development. 1996;122:2359–2366. doi: 10.1242/dev.122.8.2359. [DOI] [PubMed] [Google Scholar]
- Weis-Garcia F, Massagué J. Complementation between kinase-defective and activation-defective TGF-β receptors reveals a novel form of receptor cooperativity essential for signaling. EMBO J. 1996;15:276–289. [PMC free article] [PubMed] [Google Scholar]
- Wieser R, Wrana JL, Massagué J. GS domain mutations that constitutively activate TβR-I, the downstream signaling component in the TGF-β receptor complex. EMBO J. 1995;14:2199–2208. doi: 10.1002/j.1460-2075.1995.tb07214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Cárcamo J, Zentella A, Doody J, Laiho M, Wang X-F, Massagué J. TGF-β signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J. Mechanism of activation of the TGF-β receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- Wu R-Y, Zhang Y, Feng X-H, Derynck R. Heteromeric and homomeric interactions correlate with signaling activity and functional cooperativity of Smad3 and Smad4/DPC4. Mol Cell Biol. 1997;17:2521–2528. doi: 10.1128/mcb.17.5.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling JM, Das P, Savage C, Zhang C, Padgett RW, Wang X-F. Mammalian Dwarfins are phosphorylated in response to TGF-β and are implicated in control of cell growth. Proc Natl Acad Sci. 1996;93:8940–8944. doi: 10.1073/pnas.93.17.8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Feng X-H, Wu R-Y, Derynck R. Receptor-associated Mad homologues synergize as efectors of the TGFβ response. Nature. 1996;383:168–172. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]