

Abstract
Activation of the Cyclin B/Cdc2 kinase complex triggers entry into mitosis in all eukaryotic cells. Cyclin B1 localization changes dramatically during the cell cycle, precipitously transiting from the cytoplasm to the nucleus at the beginning of mitosis. Presumably, this relocalization promotes the phosphorylation of nuclear targets critical for chromatin condensation and nuclear envelope breakdown. We show here that the previously characterized cytoplasmic retention sequence of Cyclin B1, responsible for its interphase cytoplasmic localization, is actually an autonomous nuclear export sequence, capable of directing nuclear export of a heterologous protein, and able to bind specifically to the recently identified export mediator, CRM1. We propose that the observed cytoplasmic localization of Cyclin B1 during interphase reflects the equilibrium between ongoing nuclear import and rapid CRM1-mediated export. In support of this hypothesis, we found that treatment of cells with leptomycin B, which disrupted Cyclin B1–CRM1 interactions, led to a marked nuclear accumulation of Cyclin B1. In mitosis, Cyclin B1 undergoes phosphorylation at several sites, a subset of which have been proposed to play a role in Cyclin B1 accumulation in the nucleus. Both CRM1 binding and the ability to direct nuclear export were affected by mutation of these phosphorylation sites; thus, we propose that Cyclin B1 phosphorylation at the G2/M transition prevents its interaction with CRM1, thereby reducing nuclear export and facilitating nuclear accumulation.
Keywords: CRM1, CRS phosphorylation, cell cycle, Cyclin B1, cellular localization, nuclear export
Entry into mitosis in eukaryotic cells is controlled by activation of the Ser/Thr kinase Cdc2 bound to one of several B-type cyclins. Although Cyclin B proteins are synthesized and can interact with Cdc2 during interphase, Cdc2/Cyclin B complexes are held in an inactive state until the G2/M transition. This inactivation of Cdc2/Cyclin B during interphase is achieved through inhibitory phosphorylation of the Cdc2 subunit on Thr-14 and Tyr-15 by the Wee1 and Myt1 kinases, thus preventing premature initiation of mitosis (Norbury et al. 1991; Krek et al. 1992; Solomon et al. 1992; Atherton-Fessler et al. 1994; Kornbluth et al. 1994; Watanabe et al. 1994; McGowan et al. 1995; Mueller et al. 1995a,b). Dephosphorylation of Cdc2 by the dual specificity Cdc25 phosphatase then leads to activation of the Cdc2/Cyclin B kinase(s) and entry into mitosis (Dunphy and Kumagai 1991; Gautier et al. 1991; Strausfeld et al. 1991; Millar and Russell 1992).
Another aspect of mitotic regulation was revealed by the discovery that both the Cdc2/Cyclin complexes and their regulatory enzymes were concentrated in specific subcellular compartments. In HeLa cells, Cdc2/Cyclin B2 complexes are constitutively cytoplasmic, and primarily associated with the Golgi during interphase (Jackman et al. 1995). In contrast, Cdc2/Cyclin B1 complexes associate with microtubules during interphase, but transit precipitously into the nucleus at the G2/M transition, presumably to allow phosphorylation of nuclear substrates critical for mitosis. The Wee1 kinase, which negatively regulates Cdc2/Cyclin B through phosphorylation at Tyr-15 of Cdc2, is predominantly nuclear (Heald et al. 1993; Wu et al. 1996), whereas the Myt1 kinase, which can phosphorylate both Tyr-15 and Thr-14, is associated with the cytoplasmic face of the endoplasmic reticulum and Golgi (Liu et al. 1997). The localization of the Cdc25 phosphatase, which dephosphorylates these sites, has been somewhat controversial (Millar et al. 1991; Girard et al. 1992; Izumi et al. 1992; Seki et al. 1992; Heald et al. 1993), but it appears that different Cdc25 isoforms may be restricted to the nucleus or cytoplasm, perhaps in a cell-type-specific manner.
The finding that Cdc2/Cyclin B complexes and their regulators appear to be restricted in subcellular localization has important implications for the regulation of entry into mitosis, because particular regulators of Cdc2 may only have access to a subset of Cdc2/cyclin complexes. Furthermore, distinct Cdc2/cyclin complexes may be responsible for different aspects of mitosis because their accessibility to key substrates will depend on their localization. Coordination between the nuclear events of mitosis (e.g., nuclear envelope breakdown, chromosome condensation) and the cytoplasmic events of mitosis (e.g., Golgi disassembly, spindle formation) requires appropriately timed activation of Cdc2/Cyclin B kinases in both subcellular compartments. Thus, the mechanisms underlying the subcellular distribution of these key regulators are of great interest for understanding the regulated and concerted nature of the G2/M transition.
Recent work by a number of groups has revealed that nuclear trafficking of proteins is mediated by a family of saturable receptors that cooperate with soluble factors and nuclear pore proteins to move transport cargo in and out of the nucleus (for review, see Gorlich and Mattaj 1996; Corbett et al. 1997; Doye et al. 1997; Ullman et al. 1997). Recently, several such receptors have been discovered, including the importin α/β heterodimer, which mediates binding to classical nuclear localization sequences (NLSs) (Gorlich et al. 1994, 1995; Radu et al. 1995), transportin, which binds to a nuclear shuttling sequence (M9 confers bidirectional transport) (Pollard et al. 1996), and CRM1, a receptor for leucine-rich nuclear export sequences (NESs) (Fornerod et al. 1997; Fukuda et al. 1997; Neville et al. 1997; Ossareh-Nazari et al. 1997; Stade et al. 1997).
Analysis of chimeras between human Cyclin A (which is constitutively nuclear) and human Cyclin B1 in HeLa cells led to the discovery of a domain of Cyclin B1 (amino acids 109–151), which was necessary for the interphase localization of Cyclin B1 and sufficient to target Cyclin A to the cytoplasm (Pines et al. 1994). This domain was called the cytoplasmic retention sequence (CRS), based on the hypothesis that it acts as an anchoring site to tether Cyclin B1 to cytoplasmic components. In this report we show that rather than promoting cytoplasmic retention, the Cyclin B1 CRS is actually an NES that can autonomously direct export of a heterologous protein and confers binding to the nuclear export receptor, CRM1. Our results imply that the predominantly cytoplasmic accumulation of Cyclin B1 in interphase results from continuous nuclear import and rapid re-export rather than cytoplasmic immobilization.
In Xenopus, the CRS region of Cyclin B1, which is 61% identical to that of human Cyclin B1, contains four conserved serine phosphorylation sites. Mutation of these sites to alanine abolished the ability of the cyclin to promote oocyte maturation (Li et al. 1995, 1997). Biological activity was restored to this mutant when a nuclear localization sequence was fused to the amino terminus of the protein, suggesting that CRS phosphorylation might be important for nuclear accumulation of Cyclin B1 during oocyte maturation. We demonstrate here that binding of the CRS to CRM1 is inhibited by phosphorylation within the CRS, suggesting that cyclin phosphorylation can reduce its rate of nuclear export, promoting nuclear accumulation and entry into mitosis.
Results
Because Cyclin B1 phosphorylation site mutants have been phenotypically characterized in Xenopus oocytes and oocytes provide an experimentally tractable system in which to study nuclear transport, we wished to examine the regulation of Cyclin B1 localization in this system. Xenopus oocytes, which are physiologically arrested in G2, were manually separated into nuclear and cytosplasmic fractions. We found that Cyclin B1 was predominantly cytoplasmic (Fig. 1A), with a small fraction of the Cyclin B1 in the nucleus (germinal vesicle, GV). In theory, this cytoplasmic accumulation of Cyclin B1 could be the result of a lack of nuclear import (as would occur if the CRS bound to a cytoplasmic anchor) or to a steady state in which nuclear export of Cyclin B1 was more rapid than its import. To determine whether Cyclin B1 could serve as a nuclear export substrate, full-length Cyclin B1 was translated in vitro in reticulocyte lysates in the presence of [35S]methionine and injected into nuclei of stage VI oocytes. Immediately after injection or 4 hr after injection, the oocytes were dissected manually into nuclear and cytoplasmic fractions. Successfully injected oocytes were identified by the presence of pink nuclei, reflecting the presence of hemoglobin from the reticulocyte lysate. The nuclear and cytoplasmic fractions were analyzed by SDS-PAGE and autoradiography for the presence of radiolabeled protein. Only 12% of injected cyclin remained in the nuclei after 4 hr showing that Cyclin B1 can be actively exported (Fig. 1B).
Figure 1.

Characterization of Cyclin B1 nuclear export in Xenopus oocytes. (A) One, two, or five Xenopus stage VI oocytes were manually dissected into cytoplasmic and nuclear fractions and the distribution of endogenous Cyclin B1 was analyzed by SDS-PAGE followed by Western blotting with anti-Cyclin B1 sera. (T,C,N) Proteins extracted from total oocytes, cytoplasmic or nuclear fractions. Cdc25, which is cytoplasmic in oocytes, was used as a control. (B) Cyclin B1 was translated in reticulocyte lysates in the presence of [35S]methionine and injected into Xenopus oocyte nuclei along with 14C-labeled BSA. At 0 and 4 hr after injection, the oocytes were dissected and successfully injected oocytes were identified by the presence of pink coloring from the reticulocyte lysate. These oocytes were separated into cytoplasmic and nuclear fractions, extracted, and analyzed by SDS-PAGE followed by autoradiography.
In the several cases examined thus far, nuclear protein export requires the GTPase Ran/TC4. A Ran T24N mutant, locked in its GDP-bound form, blocks export of nuclear proteins, as does nuclear injection of RanGAP protein, which accelerates hydrolysis of GTP by Ran (Gorlich et al. 1997; Richards et al. 1997). To determine whether cyclin nuclear export was affected by Ran–GDP, we coinjected oocyte nuclei with 35S-labeled Cyclin B1 and purified T24N Ran protein or recombinant RanGap protein. Both of these treatments led to a significant diminution in the export of Cyclin B1 from the nucleus into the cytoplasm (Fig. 2; data not shown). This suggests that the GTP-bound form of Ran is likely to be required for nuclear export of Cyclin B1, as has been shown for other actively exported proteins (Gorlich et al. 1997; Richards et al. 1997).
Figure 2.
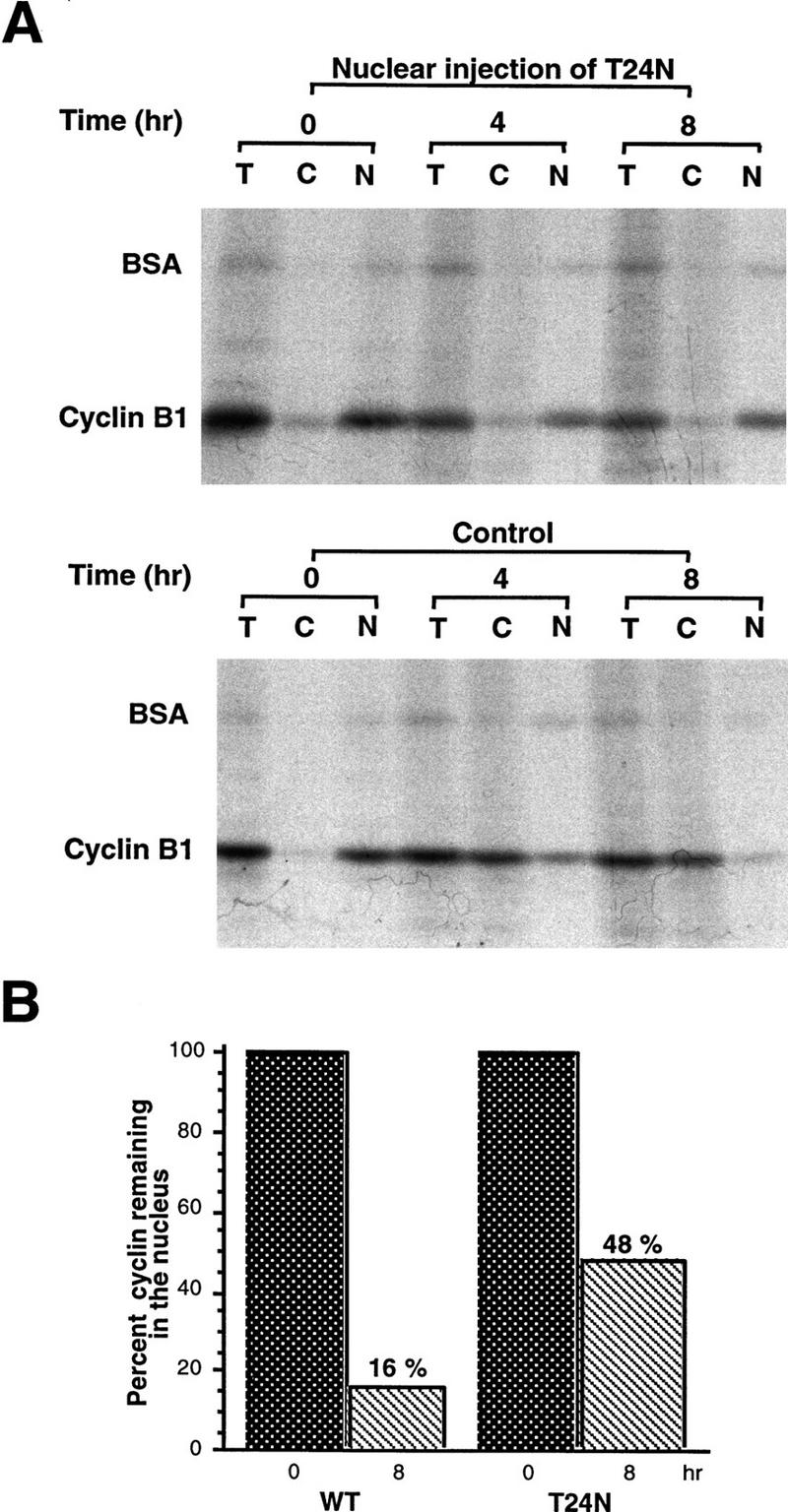
A GDP-bound form of Ran inhibits Cyclin B1 nuclear export. (A) A mixture of 35S-labeled Cyclin B1 and 14C-labeled BSA was coinjected into nuclei with recombinant T24N mutant Ran protein (GDP-bound form). Oocytes were dissected 4 hr and 8 hr later and proteins were extracted and analyzed by SDS-PAGE and autoradiography. (B) The bar graph represents a quantitation of the data in A, showing the percentage of Cyclin B1 remaining in nuclei after 0 and 8 hr.
Given the ability of Cyclin B1 to be exported from nuclei, we suspected that the CRS might actually mediate nuclear export of the cyclin, rather than blocking its nuclear import. To determine whether the CRS could promote nuclear export of a heterologous protein, we linked the isolated CRS from Xenopus Cyclin B1 (amino acids 76–125) to the carboxyl terminus of glutathione S-transferase (GST). GST–CRS or GST protein alone was then injected into nuclei (with dextran blue to identify oocytes with injected nuclei), and nuclear and cytosolic fractions were dissected at various times after injection and examined for the distribution of injected protein by immunoblotting with anti-GST sera. Although GST protein is small (25 kD), it is known to dimerize, and >90% of the injected GST protein was retained in the nucleus over a 7 hr time course (data not shown). Thus, GST protein is effectively too large to exit the nucleus by passive diffusion through pores, as has been noted by others (Wen et al. 1995; Meyer et al. 1996). In contrast, the injected GST–CRS protein was exported rapidly from the nuclei (Fig 3). These data demonstrate that the CRS can act as an autonomous NES.
Figure 3.


The CRS mediates nuclear export of Cyclin B1. (A) The isolated CRS from Xenopus Cyclin B1 (amino acids 76–125) was linked to the carboxyl terminus of GST. Either GST or GST–CRS protein was injected into oocyte nuclei along with 14C-labeled BSA. Oocytes were dissected at 0, 30, and 50 min after injection. Proteins were analyzed by SDS-PAGE followed by immunoblotting with GST antibody and detection with 125I-labeled protein A (quantitated by PhosphorImager). 14C-Labeled BSA was detected by autoradiography. (B) The graph represents a quantitation of the data in A, showing the percentage of injected proteins remaining in nuclei at 0 and 50 min.
Because the CRS could mediate nuclear export of Cyclin B1, we wished to determine whether the recently identified NES from protein kinase inhibitor (PKI) (Wen et al. 1995) could compete with Cyclin B1 for the nuclear export machinery. Human serum albumin was conjugated to multiple copies of the NES from PKI, or to an inactive scrambled mutant variant of the PKI NES (called PKI NOS) to serve as a control. These conjugates were then coinjected along with 35S-labeled Cyclin B1 into oocyte nuclei, and the location of the cyclin was examined 3 hr and 5 hr after injection as described above. Nuclear export of Cyclin B1 was delayed by the PKI–NES, but not by the NOS mutant (Fig. 4A,B). Additionally, microinjection of the PKI–NES conjugate into nuclei led to gradual nuclear accumulation of endogenous Cyclin B1, whereas injection of the PKI–NOS conjugate did not have this effect (Fig. 4C,D). This shows that Cyclin B1 can be imported into nuclei during interphase and suggests that its predominantly cytoplasmic location is promoted by nuclear export. Moreover, although the PKI peptide did not completely eliminate Cyclin B1 export, its ability to compete with Cyclin B1 for nuclear export suggests that Cyclin B1 and PKI utilize at least some of the same nuclear export machinery.
Figure 4.

The PKI NES reduces nuclear export and induces nuclear accumulation of Cyclin B1. (A) Human serum albumin was conjugated with the NES peptide from PKI (HSA–NES), or to an inactive scrambled mutant variant of the PKI NES as the unexportable control (HSA–NOS). The 35S-labeled Cyclin B1 and 14C-labeled BSA mixture was injected into oocyte nuclei either with 8 μg/μl HSA–NES or HSA–NOS. Oocytes were dissected at 0, 3, and 5 hr after injection, and proteins were analyzed by SDS-PAGE followed by autoradiography. (B) The data in A are graphed to show the change in ratio of nuclear/cytoplasmic injected Cyclin B1 over a 5-hr time course. (C) HSA–NES or HSA–NOS (16 μg/μl) was injected into oocyte nuclei with 10 μg/μl dextran blue 2000 as an injection marker. Oocytes were dissected at 0, 7, and 12 hr after injection and proteins were analyzed by SDS-PAGE followed by immunoblotting with Cyclin B1 antibody. (D) The amount of cyclin present in the nucleus at time 0 was quantitated; the increase in nuclear cyclin after PKI NES injection is plotted as a percentage of cyclin quantities initially found in the nucleus.
Recently, it has been reported that nuclear export of several substrates, including PKI, is mediated in part through NES binding of the importin β family member, CRM1 (Fornerod et al. 1997; Neville et al. 1997; Ossareh-Nazari et al. 1997; Stade et al. 1997). Because the CRS could mediate nuclear protein export, we suspected that the CRS also might promote binding to CRM1. To determine if this was the case, we coupled the GST–CRS fusion protein to a glutathione–Sepharose resin and used it as a bait to retrieve CRS-binding proteins from extracts of Xenopus oocytes. After incubation in Xenopus oocyte extracts, the beads were pelleted gently by centrifugation, washed extensively, and the bead-bound material was processed for immunoblotting with anti-CRM1 antisera. As shown in Figure 5A, the GST–CRS protein was able to bind to CRM1. No specific interaction between the CRS and other CRM1-related proteins implicated in protein export, including RanBP7 (Gorlich et al. 1997) and CAS (Scherf et al. 1996; Kutay et al. 1997) was detected in parallel experiments. (Fig. 5B). In addition, no bound CRM1 was detected with GST protein alone (Fig. 5A). Similarly, we found that recombinant GST–CRM1 protein linked to Sepharose beads could bind specifically to full-length, in vitro-translated Cyclin B1 (Fig. 5E). Thus, the nuclear export of Cyclin B1 may be mediated by CRM1.
Figure 5.





The nuclear export receptor, CRM1, mediates Cyclin B1 nuclear export through binding to CRS. (A) GST or GST–CRS fusion protein was coupled to glutathione–Sepharose beads. Twenty microliters of these resins were incubated in 100 μl of oocyte extract for 1 hr alone or in the presence of 200 nm leptomycin B. The beads were then pelleted and washed three times with oocyte extract (EB) buffer. The bead-bound proteins were analyzed by SDS-PAGE followed by Western blotting with anti-human Crm1 antibody. (B) The same samples as in A (with the exception of the leptomycin B-containing sample) were immunoblotted with antisera directed against Ranbp7 and Cas. (C) Oocytes were incubated with 200 nm leptomycin B in MB buffer for 2 hr before injection. A 35S-labeled Cyclin B1 and 14C-labeled BSA mixture was then injected into oocyte nuclei. Oocytes were dissected at 0, 3, and 7 hr after injection and proteins were analyzed by SDS-PAGE followed by autoradiography. (D) The graph represents a quantitation of the data in C, showing the percentage of injected proteins remaining in nuclei at 0 and 7 hr. (E) Human Crm1 was fused to the carboxyl terminus of GST protein. Either GST alone or GST–Crm1 fusion protein was coupled to glutathione–Sepharose beads. Twenty microliters of each resin was incubated with 2 μl of 35S-labeled Cyclin B1 in 100 μl of buffer with 0.3 μg/μl Ran G19V (a mutant form of Ran that lacks GTPase activity and hence remains constiutively GTP-bound) and 5 μm GTP at room temperature for 1 hr. The beads were then pelleted and washed three times with oocyte extract buffer. The beads-bound proteins were analyzed by SDS-PAGE followed by autoradiography.
Recently, it has been reported that the drug leptomycin B can specifically bind CRM1, thereby inhibiting CRM1-dependent nuclear protein export. Consistent with a role for CRM1 in export of Cyclin B1, we found that export of radiolabeled Cyclin B1 was effectively inhibited by treatment of oocytes with 200 nm leptomycin B (Fig. 5C,D). Moreover, leptomycin B treatment of egg extracts also prevented binding of CRM1 to the B1 CRS (Fig 5A).
Mutational analysis of a CRM1-dependent export substrate, HTLV I Rex protein, has identified a loose consensus NES (see Fig. 6A; Bogerd et al. 1996). By scanning the Cyclin B1 sequence, we noted a sequence conforming to the NES consensus within the CRS (amino acids 108–117) of Xenopus Cyclin B1 (Fig. 6A). We mutated one of the critical hydrophobic residues (F112) to an alanine to determine whether this would affect the NES function of the CRS. As shown in Figures 6, C and D, 35S-labeled F112A mutant Cyclin B1 injected into oocyte nuclei was not exported. In addition, an F112A mutant CRS variant no longer bound to CRM1 (Fig. 6E). Although this mutation disrupted the ability of Cyclin B1 to export from nuclei, it did not perturb its ability to interact with its binding partner, Cdc2 (Fig. 6B).
Figure 6.



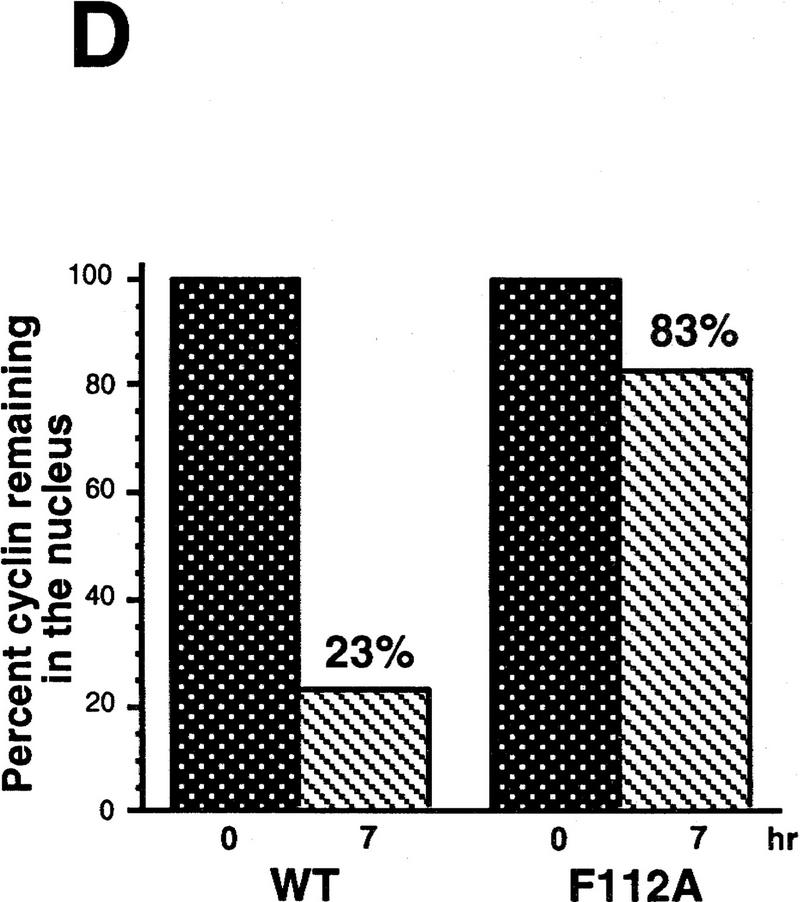

Cyclin B1 contains a Rev/Rex consensus NES within the CRS region. (A) The NESs of vertebrate Cyclin B1s were aligned and compared with the consensus sequence for a functional Rev/Rex NES. (B) A full-length Cyclin B1 mutant in which the Phe residue at position 112 was changed to Ala was translated in vitro and added to interphase extracts of Xenopus eggs (these contain Cdc2, but lack mitotic cyclins). The 35S-labeled F112A mutant cyclin and wild-type cyclin were similarly retrieved from extracts by p13 Sepharose (which can only bind Cyclin B1 indirectly through interactions with its binding partner, Cdc2), but not by control Sepharose. (C) A full-length Cyclin B1 mutant in which the conserved Phe-112 within CRS was mutated to Ala was translated in reticulocyte lysates in the presence of [35S]methionine and mixed with 14C-labeled BSA, a nonexportable injection control. The mixture was injected into Xenopus oocyte nuclei. After injection (0, 3, and 7 hr), the oocytes were dissected into cytoplasmic and nuclear fractions, proteins were extracted, and samples were then analyzed by SDS-PAGE, followed by PhosphorImager quantitation. (D) The graph represents a quantitation of the data in C, showing the percentage of injected proteins remaining in nuclei at the 0 and 7 hr time points. (E) Phe-112 of Cyclin B1 was mutated to Ala and the isolated CRS containing this mutation was fused to the carboxyl terminus of GST protein. GST alone, the wild-type GST–CRS protein, or F112A mutant GST–CRS protein were coupled to glutathione–Sepharose beads. Twenty microliters of each resin was incubated in oocyte extract for 60 min and the beads were then pelleted and washed three times with oocyte extract buffer. The bead-bound material was analyzed by SDS-PAGE followed by immunoblotting with anti-CRM1 antibody.
If cytoplasmic retention of Cyclin B1 is a consequence of CRM1-mediated export, then leptomycin B might be expected to induce nuclear accumulation of endogenous Cyclin B1. Treatment of oocytes with 200 nm leptomycin B induced gradual accumlation of nuclear Cyclin B1, consistent with the hypothesis that Cyclin B1 can shuttle through nuclei (Fig. 7A,B). We did not observe a concomitant decrease in the cytoplasmic level of Cyclin B1 because the cytoplasmic pool of Cyclin B1 in Xenopus oocytes is quite large and the cytoplasmic/nuclear ratio is >6:1. Injection of 35S-labeled Cyclin B1 into the cytoplasm of leptomycin B-treated oocytes or cytoplasmic injection of the F112A mutant protein also led to a slow nuclear accumulation of cyclin (data not shown). However, the nuclear accumulation of RanBP1, a shuttling component of the nuclear trafficking machinery, was considerably more striking, demonstrating that the nuclear transport machinery of the oocyte was fully operative (Fig. 7A,C). Therefore, we considered the possibility that slow nuclear import of Cyclin B might be a feature of the meiotically arrested G2 oocyte; progression to meiotic GV breakdown (at the G2/M transition) can take longer than 12 hr after treatment of oocytes with the meiotic inducer progesterone. To keep Cyclin B1 in the oocyte cytoplasm would require only that the rate of export exceeds the rather slow import rate.
Figure 7.

Leptomycin B treatment induces endogenous Cyclin B1 nuclear accumulation in Xenopus oocytes. (A) Oocytes were treated with 200 nm leptomycin B in MB buffer. At the indicated times after treatment, they were dissected into cytoplasmic and nuclear fractions. Proteins were extracted and analyzed by SDS-PAGE followed by immunoblotting with anti-Xenopus Cyclin B1 antibody. Although this antisera cross-reacts with other proteins from the extract, the indicated band is Cyclin B1; this has been confirmed by specific reactivity of the antisera against recombinant Cyclin B1 and not B2, and by observation of the periodic accumulation and destruction of this band in cycling extracts of Xenopus eggs. The samples blotted with anti-cyclin sera were also immunoblotted with anti-Ranbp1 sera, as a positive control protein for nuclear accumulation following leptomycin B treatment. (B) The graph represents a quantitation of the data in A, showing the fold increase of endogenous Cyclin B1 accumulated in nuclei at the indicated times after leptomycin B treatment. For every oocyte nuclear equivalent loaded on the gels, only one-eighth the amount of cytoplasm was loaded to facilitate observation of potential changes in cytoplasmic levels of protein. (C) The graph represents a quantitation of the data in A, showing the fold increase of endogenous RanBP1 in nuclei following leptomycin B treatment.
Given these data, we sought to examine the effects of leptomycin B treatment on nuclear Cyclin B1 accumulation in actively cycling somatic cells. Untreated HeLa cells or HeLa cells treated with 20 nm leptomycin B for varying times were lysed and separated into nuclear and cytoplasmic fractions by centrifugation at 1000g for 10 min. The pelleting of nuclei was confirmed by staining of the pellet with the fluorescent DNA intercalating dye Hoechst 33258. Nuclei were washed twice in PBS, and the nuclear and cytoplasmic fractions were resolved by SDS-PAGE. Immunoblotting of these fractions with a monoclonal antibody directed against human Cyclin B1 revealed that significant amounts of Cyclin B1 had accumulated in HeLa nuclei within 4 hr of leptomycin B treatment (Fig. 8A,B; compare this with the 38 hr time course in Xenopus oocytes). In addition, in HeLa cells, cytoplasmic depletion of Cyclin B1 was seen concomitant with nuclear accumulation. These results were confirmed by processing leptomycin B-treated HeLa cells for indirect immunofluorescence with the anti-Cyclin B1 monoclonal antibody. In untreated HeLa cells, Cyclin B1 is excluded from nuclei during interphase (Fig. 8C, top panel), as reported previously. The sharp boundary caused by the concentration of cyclin in perinuclear regions creates the appearance of a nuclear rim; however, this does not represent binding of Cyclin B1 to the nuclear envelope. In untreated cells, the occasional prophase cell could be seen staining with this antisera, consistent with the reported rapid entry of Cyclin B1 into nuclei just prior to entry into mitosis (data not shown; Pines and Hunter 1991). However, when HeLa cells were treated for 4 hr with leptomycin B, there was a dramatic nuclear accumulation of Cyclin B1 in a large proportion of interphase cells; a representative photomicrograph of such cells is shown in Figure 8C (bottom panel). To facilitate comparison, treated and untreated cells were photographed under the same conditions; this results in a somewhat overexposed nuclear signal in the leptomycin-treated cells in which cyclin is strongly concentrated in the nucleus. These results are entirely consistent with the idea that Cyclin B1 can cycle through the nucleus during interphase. In support of this hypothesis, Pines and colleagues, using time-lapse video microscopy, have observed cycling of GFP-labeled Cyclin B1 through nuclei during interphase; this cycling is abolished by leptomycin B treatment (J. Pines, pers. comm.).
Figure 8.

Leptomycin B treatment induces nuclear accumulation of Cyclin B1 in HeLa cells. (A) HeLa cells were treated with 20 nm leptomycin B. At the indicated times, cells were collected and separated into cytoplasmic and nuclear fractions. Sampled were analyzed by SDS-PAGE followed by Western blot with anti-human Cyclin B1 antibody (Santa Cruz). All of the nuclear or cytoplasmic fraction from 1.5 × 105 HeLa cells were loaded in each lane, as indicated. (B) The graph represents a quantitation of the data in A, showing the fold increase of endogenous Cyclin B1 in HeLa cell nuclei following leptomycin B treatment. Note the much shorter time course shown here than in the Xenopus experiment in 7B (4 hr vs. 40 hr). (C) Either untreated HeLa cells or HeLa cells treated for 4 hr with 20 nm leptomycin B were processed for indirect immunofluorescence with a monoclonal directed against human Cyclin B1, as described in Materials and Methods. (Left) Cyclin B1 immunofluorescence signals in representative interphase cells either treated or untreated with leptomycin B. (Right) The same cells stained with the DNA intercalating dye, Hoechst 33258. Fluorescence was observed and photographed with a 60× Planapochromat objective and CCD camera.
As mentioned above, phosphorylation of four sites (serines 94, 96, 101, and 113) within the CRS is thought to affect Cyclin B1 localization (Li et al. 1997). Given our findings, it seemed possible that phosphorylation might modulate the ability of the CRS to bind to CRM1 and thereby regulate its nuclear export rate. To test this model, we produced isolated GST–CRS fusion proteins in which the four serines in the CRS had been mutated to either Ala (nonphosphorylatable) or Glu (which often mimics constitutive phosphorylation). Beads coupled to each of these fusion proteins were incubated in oocyte extract and then examined for binding to CRM1, as described above. We found that compared with the wild-type CRS, the Ala mutant CRS bound more CRM1 from the extract, whereas the Glu mutant bound much less (Fig. 9A). Cyclin B1 containing the four Ala or four Glu mutations was then translated in vitro and injected into oocyte nuclei to monitor the effect of the mutations on cyclin export. The Ala mutant was exported very efficiently, with only 11% remaining in the nucleus after 4 hr (Fig. 9B,C). The Glu mutant was exported poorly, with 86% remaining in the nucleus after 4 hr, as compared with 51% of the wild-type protein (Fig. 9B,C). These data are consistent with the hypothesis that phosphoryation within the CRS impairs CRM1 binding and nuclear export.
Figure 9.

Phosphorylation within the CRS of Cyclin B1 impairs its ability to bind to Crm1 and inhibits Cyclin B1 nuclear export. (A) Mutant variants of the isolated CRS in which Ser-94, Ser-96, Ser-101, and Ser-113 had all been changed to either Ala or Glu were fused to the carboxyl terminus of GST protein. Equal amounts of these fusion proteins or the wild-type GST–CRS protein were coupled to glutathione–Sepharose beads. Twenty microliters of each resin was incubated in oocyte extract for 60 min and the beads were then pelleted and washed three times with oocyte extract buffer. The bead-bound material was analyzed by SDS-PAGE followed by immunoblotting with anti-CRM1 antibody. (B) Wild-type full-length Cyclin B1 or full-length mutant variants in which all four sites of Ser phosphorylation within the CRS had been mutated to either Ala or Glu was translated in the reticulocyte lysate in the presence of [35S]methionine and mixed with 14C-labeled BSA, a nonexportable injection control. The mixture was injected into Xenopus oocyte nuclei. At 0 and 4 hr after injection, the oocytes were dissected into cytoplasmic and nuclear fractions; proteins were extracted and samples were then analyzed by SDS-PAGE and quantitated with a PhosphorImager. (C) The bar graph represents a quantitation of the data in B, showing the percentage of each Cyclin B1 variant remaining in nuclei at 0 and 4 hr. Values were normalized to coinjected 14C-labeled BSA.
One complication in the interpretation of these results is that the localization of injected Cyclin B1 might, in principle, be the combined result of nuclear export and nuclear import rates, because Cyclin B1 can enter nuclei during interphase (e.g., Figs. 7 and 8). Although data from leptomycin B-treated oocytes suggests that the nuclear import rate of wild-type Cyclin B1 is quite slow during interphase, it remained formally possible that the Ala and Glu mutants grossly altered the efficiency of nuclear import of the exported Cyclin B1, rather than simply affecting export. To circumvent this problem, we analyzed the trafficking of the GST–CRS fusion proteins, because GST, which lacks a nuclear localization sequence, is not reimported into nuclei (Fig. 10A). Injection of the wild-type and mutant Glu GST–CRS fusion proteins into oocyte nuclei revealed that the GST–CRS Glu mutant was exported more slowly than the wild-type GST–CRS (Fig. 10B).
Figure 10.
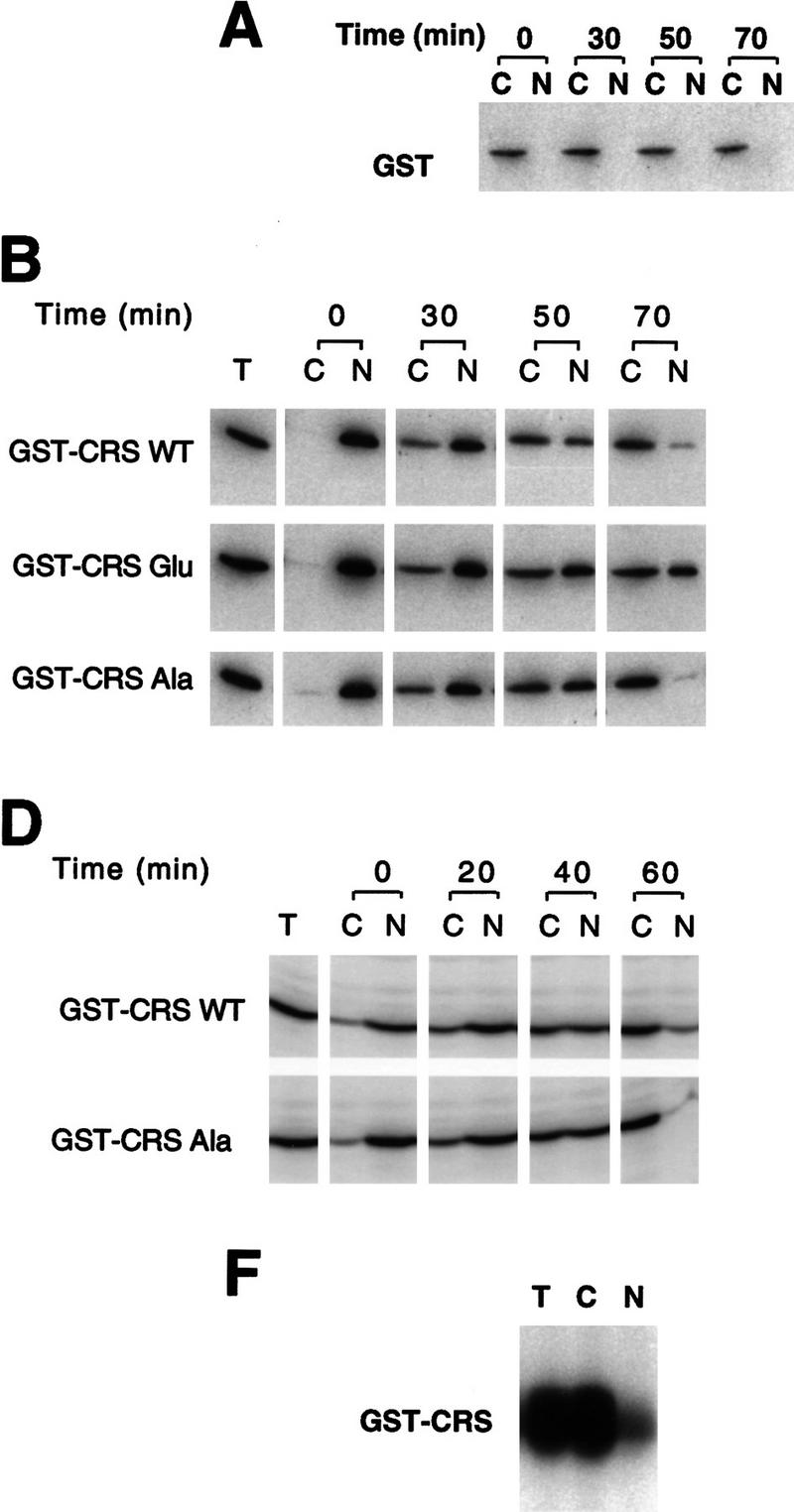
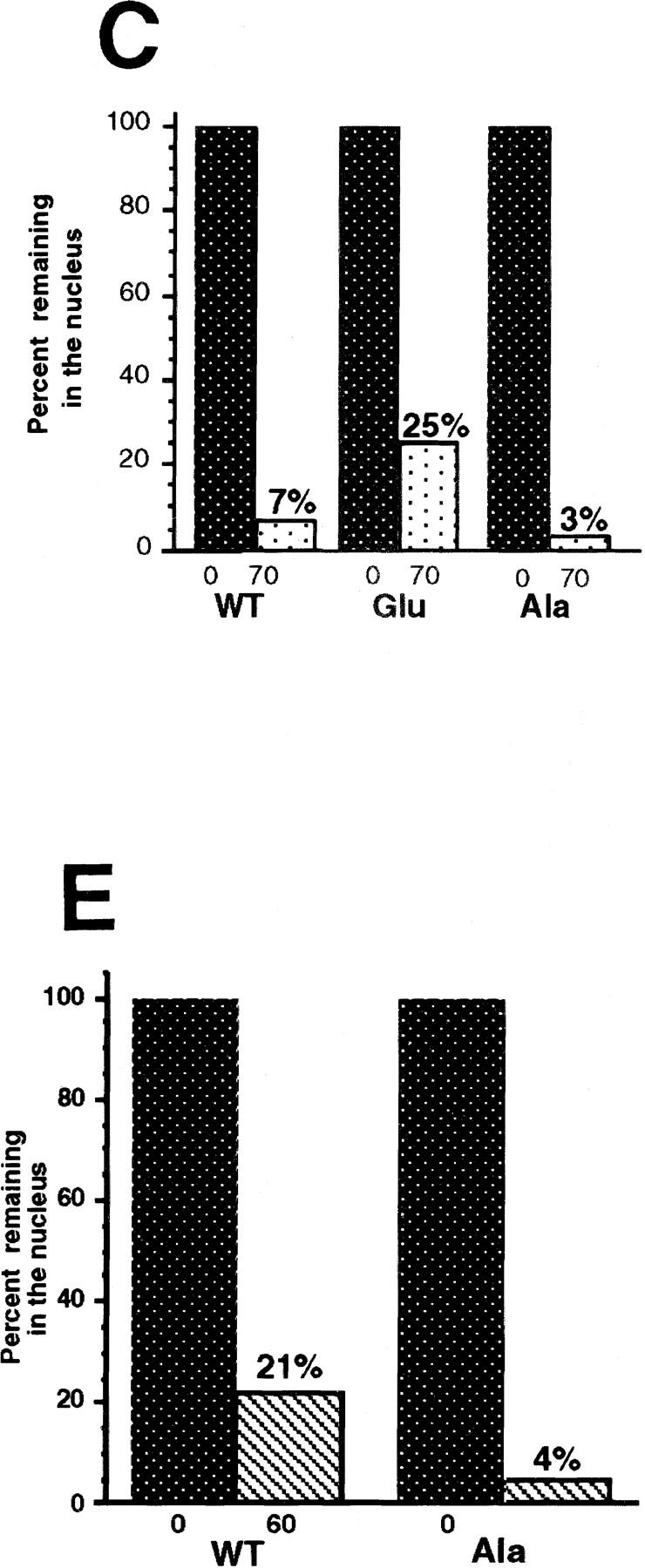
Phosphorylation of Cyclin B1 within the CRS directly modulates nuclear export. (A) GST protein was injected into the cytoplasm of oocytes and incubated for 30, 50, or 70 min. Oocytes were dissected into nuclear and cytoplasmic fractions, resolved by SDS-PAGE and immunoblotted with anti-GST sera. Injected GST remains cytoplasmic. (B) Fifty nanograms of GST protein fused to the wild-type, Ala, or Glu mutant CRS sequences was injected into oocyte nuclei along with 14C-labeled BSA. Oocytes were dissected at 0, 30, 50, and 70 min after injection and proteins were analyzed by SDS-PAGE followed by immunoblotting with GST antibody and 125I-labeled protein A or autoradiography to detect the 14C-labeled BSA. (C) Quantitation of the data in B. The 125I-labeled protein A signal was quantitated using a PhosphorImager. The bar graph shows the percent of each injected cyclin variant remaining in the nucleus immediately after and 70 min after injection. (D) GST fused to the wild-type or Ala mutant CRS sequences was injected into oocyte nuclei along with 14C-labeled BSA. Oocytes were dissected at 0, 20, 40, and 60 min after injection and proteins were analyzed by SDS-PAGE followed by Western blotting with GST antibody or autoradiography to detect the 14C-labeled BSA. Approximately 2.5-fold less protein was injected in this experiment than the experiment shown in B. (E) Quantitation of the data in D. The bar graph shows the percent of each injected cyclin variant remaining in the nucleus immediately after and 60 min after injection. (F) Oocytes were dissected into cytoplasmic and nuclear fractions and extracts were prepared from each fraction or from whole oocytes. Each extract was diluted to 1.45 μg/μl. Twenty microliters of GST–CRS wild-type beads was incubated in 45 μl of total (T), cytoplasmic (C), or nuclear (N) oocyte extract, 1 μm cold ATP and 15 μCi of [γ3P]ATP. After 20 min incubation at room temperature, beads were pelleted and washed with extract buffer three times. The bead-bound material was analyzed by SDS-PAGE, developed by PhosphorImager and quantitated.
In this experiment, the GST–CRS Ala mutant was exported from nuclei at a rate comparable with the wild-type GST–CRS (Fig. 10B,C), with 7% of the wild-type and 3% of the Ala mutant protein remaining in the nucleus after 70 min (as compared with 25% of the Glu mutant). This was somewhat surprising, because we had observed that CRM1 from oocyte extracts bound preferentially to the GST–CRS Ala mutant compared with the wild-type GST–CRS (Fig. 9A). One possible explanation for this discrepancy between CRM1-binding and nuclear export rates would be that the degree of phosphorylation of the wild-type GST–CRS was different in the CRM1- binding experiment and in this export experiment. In the CRM1-binding assay the GST–CRS was incubated in total oocyte extract, whereas in the export assay, the GST–CRS was injected directly into nuclei. As expected, in total oocyte extracts we found a robust kinase activity able to phosphorylate the wild-type CRS (Fig. 10F). However, when the GST–CRS fusion protein was incubated in equal amounts of total protein from separate nuclear and cytoplasmic extracts (equivalent to nuclei from 45 oocytes and cytoplasm from 3.6 oocytes), seven times more phosphate was incorporated into the CRS in the cytoplasmic extract than in the presence of GV extract (Fig. 10F). The low level of CRS phosphorylation observed in nuclear extracts suggested that the large bolus of GST–CRS protein that we injected into nuclei (Fig. 10B) may have overwhelmed the basal nuclear kinase activity, thereby minimizing differences between the (largely unphosphorylated) wild-type CRS protein and (unphosphorylatable) Ala proteins. To test this hypothesis we varied the amount of GST–CRS Ala and GST–CRS wild-type proteins injected; reducing the level of injected protein accentuated the difference in export rates of the Ala and wild-type GST–CRS proteins. One such experiment is shown in Figure 10D, in which 2.5-fold less protein was injected than in the experiments shown in Figure 10B. Under these circumstances, the GST-CRS wild-type protein was exported more slowly than the GST–CRS Ala mutant (Fig. 10D,E). Collectively, these data lead us to postulate that CRS phosphorylation occurs at high levels in the cytoplasm and at low levels in the nucleus during interphase.
Discussion
The CRS contains an NES
We have demonstrated that the CRS sequence of Cyclin B1 can serve as a nuclear export sequence that targets Cyclin B1 for CRM1-mediated nuclear export during interphase. This suggests that the predominantly cytoplasmic localization of Cyclin B1 at this time is the result of a dynamic traffic in which cyclin transits continually in and out of the nucleus.
Recent studies have shown that the leucine-rich NES of PKI directs nuclear export through association with the export mediator CRM1 (Fornerod et al. 1997; Neville et al. 1997; Ossareh-Nazari et al. 1997; Stade et al. 1997). We have shown that the Cyclin B1 CRS also binds to CRM1, and that cyclin export is delayed by an excess of the PKI NES. We also found that cyclin export was slowed by depletion of Ran–GTP, which was proposed to be a cofactor for nuclear protein export (Richards et al. 1997). Although the sequence requirements for NES function are only loosely defined, the CRS does contain a sequence LCQAFSDVLI (residues 108–117 of Xenopus Cyclin B1) that resembles the sequences critical for functioning of the HIV Rev, HTLV I Rex, and PKI NESs, at least in the spacing of hydrophobic amino acids (Fischer et al. 1995; Wen et al. 1995; Bogerd et al. 1996). We found that altering a critical hydrophobic residue in that sequence (F112) abolished both CRM1 binding and nuclear export of Cyclin B1. Collectively, our data strongly suggest that the Cyclin B1 CRS directs export from the nucleus via a CRM1-mediated pathway similar to that recently characterized for PKI.
Regulation of Cyclin B1 export through CRS phosphorylation
Previous studies established that Cyclin B1 undergoes phosphorylation at four serine residues within the CRS (Izumi and Maller 1991; Li et al. 1995). Data from several studies suggests that phosphorylation is a general feature of B-type cyclins from many species (Meijer et al. 1989; Gautier and Maller 1991; Izumi and Maller 1991), although a function for these phosphorylations has not been established. The data presented here indicate that CRS phosphorylation can have a significant impact on Cyclin B1 localization by reducing the affinity of Cyclin B1 for CRM1, thereby decreasing its nuclear export rate. A nonphosphorylatable mutant of Cyclin B1 maintained high export rates, and prevented nuclear accumulation of Cyclin B1, potentially explaining the failure of this mutant to promote oocyte maturation. In contrast, a mutant designed to mimic full phosphorylation of Cyclin B1 interacted much more weakly with CRM1 and was exported from the nucleus more slowly.
Whereas the cell cycle regulation of phosphorylation at sites within the CRS has not been examined specifically, Kobayashi et al. (1991) noted a Cyclin B1 modification (detected as a mobility shift of the protein in denaturing gels) in Xenopus oocytes that increased concomitant with germinal vesicle breakdown, perhaps indicating an increase in CRS phosphorylation. Given our findings, it is tempting to speculate that the rapid accumulation of Cyclin B1 in the nucleus at the G2/M transition could be governed by increasing phosphorylation of CRS residues, thereby reducing the affinity of Cyclin B1 for CRM1 and decreasing nuclear export.
In addition to the CRS phosphorylation-mediated control of nuclear export rates, it is possible that nuclear import rates are regulated and contribute to the relocalization of Cyclin B1 at the G2/M transition. Neither Cyclin B1 nor Cdc2 contains a classical NLS, and the mechanism by which the Cdc2/Cyclin B1 complex enters the nucleus is not yet clear. However, we have found that the nuclear import factor, importin β, can bind to Cdc2/Cyclin B1 complexes and experiments are in progress to determine whether this association reflects a role for importins in mediating nuclear import of CDK/Cyclin complexes (J.D. Moore and S. Kornbluth, unpubl.). Although the possibility that nuclear import can be regulated by changes in CRS phosphorylation clearly merits further investigation, it is interesting to note that Ala and Glu mutant Cyclin B1 proteins bind equally well to importin β (J.D. Moore and S. Kornbluth, unpubl.).
Localization of enzymes controlling Cyclin B1 CRS phosphorylation
The identity of the kinase(s) responsible for phosphorylating sites within the CRS remains unknown. On the basis of sequence comparisons with consensus substrate sites, Li et al. (1997) suggested that a MAP kinase or Cdc2/cyclin itself might phosphorylate Ser-94 or Ser-96 [as observed in vitro by Izumi and Maller (1991)], and that casein kinase II might phosphorylate Ser-101 and Ser-113 (Li et al. 1997). The latter site is adjacent to one of the conserved hydrophobic residues in the part of the CRS resembling the Rev and PKI NESs. It is unclear which of the phosphorylation sites regulate the CRS–CRM1 interaction, or whether multiple sites are involved. Further studies into the particular sites involved and the enzymes that control their phosphorylation are underway.
Even without detailed information regarding the identity of the CRS kinase(s) and phosphatase(s), our data suggest that a further layer of regulation may stem from differential localization of these enzymes. At high levels of injected protein, we found that the nuclear export rates of wild-type and nonphosphorylatable GST–CRS fusion proteins injected into the nucleus were very similar. Because phosphorylation of the wild-type CRS would have slowed its export, this result suggested that the CRS-directed kinase activities are relatively scarce in the interphase nucleus or that CRS phosphatases are very active in the nucleus, or both. Phosphorylation of the CRS was very low in nuclear extracts compared with that seen in cytoplasmic extracts, in which the CRS–CRM1 interaction was impaired relative to the nonphosphorylatable mutant.
When we injected lower levels of GST–CRS protein, the Ala and the wild-type proteins were exported at different rates, consistent with the results obtained with the full-length cyclins. These data suggested that nuclear kinases could phosphorylate the trace amounts of radiolabeled full-length cyclins injected, but that the higher amounts of injected GST–CRS protein exceeded the ability of the nuclear kinases to maintain a high stoichiometries of phosphorylation on the wild-type GST–CRS.
It is not clear why the GST–CRS fusion proteins are exported more rapidly than the full-length cyclin protein. The full-length cyclin has the capacity to be reimported into nuclei after export, whereas the GST–CRS fusion protein does not. However, the slow rate of nuclear accumulation of Cyclin B1 in leptomycin B-treated oocytes suggests that reimport may not have a significant effect on total nuclear efflux during a typical 8 hr nuclear export experiment. Therefore, we consider it possible that the context in which the CRS appears determines the nuclear export rate of the CRS-containing protein, or reimport of recently exported Cyclin B1 is more rapid than import of the overall pool, reflected by the slow accumulation of nonexportable cyclin. Although the full-length cyclins and the GST–CRS proteins were exported at different rates, the export rate of wild-type CRS-containing proteins consistently exceeded that of Glu CRS-containing proteins. This suggests that injected wild-type CRS-containing proteins were never fully phosphorylated in any of our experiments and that elevating phosphorylation of the CRS within the nucleus could provide an effective means of increasing nuclear accumulation of cyclin at the G2/M transition.
Functional implications of Cyclin B1 export
The accumulation of nuclear Cyclin B1 after leptomycin B treatment was less striking in Xenopus oocytes than in HeLa cells. Whether this represents a species difference or a difference in the import pathways operating in these two cell types bears further investigation. However, in both systems it appears that Cyclin B1 can be imported into nuclei during interphase, despite the fact that the steady state favors localization of Cyclin B1 to the cytoplasm.
It seems likely that the slow apparent import rate of Cyclin B1 we observed in the leptomycin B-treated oocytes reflects the nature of the G2-arrested oocyte system: Unprimed Xenopus oocytes can take >12 hr to accumulate sufficient active nuclear Cdc2/Cyclin B for germinal vesicle breakdown in response to progesterone treatment, presumably reflecting, at least in part, the slow accumulation of nuclear Cdc2/Cyclin B as oocytes transition from G2/M. The fact that the gradual accumulation of Cyclin B1 in oocytes, which we observed when nuclear export was blocked, may be physiologically significant is suggested by preliminary experiments in which mRNA encoding the F112A mutant Cyclin B1 produced more rapid progression through the meiotic G2/M transition than did similarly injected wild-type Cyclin B1 mRNA (J. Yang and S. Kornbluth, unpubl.).
In the somatic cell cycle, nuclear shuttling during interphase provides an appealing mechanism for coordinating nuclear and cytoplasmic aspects of entry into mitosis. The fact that a single Cdc2/cyclin complex can receive inputs from both nuclear and cytoplasmic regulators provides a simple way of integrating different signals from these compartments. For instance, entry into mitosis is predicated on the completion of DNA replication and repair of DNA damage (Murray et al. 1992). In the absence of these conditions, cell cycle checkpoints act to inhibit Cdc2/cyclin activation. Transit of Cdc2/Cyclin B1 complexes through the nucleus provides the opportunity for these complexes to continuously sample the nuclear environment and receive checkpoint information without invoking nucleocytoplasmic trafficking of checkpoint signals.
In addition to receiving inputs from different compartments, the shuttling of Cdc2/Cyclin B1 complexes between nucleus and cytoplasm may help to ensure that nuclear envelope breakdown, chromosome condensation, spindle formation, and other events of mitosis are appropriately timed and that the nucleus and cytoplasm enter mitosis simultaneously.
Materials and methods
In vitro translation
Xenopus Cyclin B1 wild-type, Ala, and Glu mutants were kindly provided by Dr. D. Donoghue (University of California, San Diego) in the vector SP64T, containing cyclin variants cloned downstream of an SP6 promoter. 35S-Labeled proteins were produced by use of the SP6-coupled TNT reticulocyte system (Promega) according to manufacturer’s instructions.
Oocyte microinjection and subfractionation
Stage VI oocytes of Xenopus laevis were prepared for microinjection, as described (Swenson et al. 1989). Eight nanoliters of protein sample was injected into oocyte nuclei along with 14C-labeled BSA. Either 10 mg/ml dextran blue 2000 or red reticulocyte lysate was coinjected to mark the injected nuclei. Successfully injected oocytes (those with blue or red nuclei) were separated into nuclear and cytoplasmic fractions by manual dissection under mineral oil (Lund et al. 1990). Fractions from 5 to 10 oocytes were homogenized in buffer (65 mm Tris at pH 7.6, 10 mm EGTA at pH 8.0, 1 mm PMSF, 5 ng/μl aprotinin/leupeptin) and spun for 5 min at 13,000g to remove insoluble material. The supernatants were precipitated with 3 volumes of acetone, pelleted, and dried. The pellets were resuspended in 10 μl of SDS-PAGE sample buffer per oocyte and analyzed by SDS-PAGE, followed by autoradiography or Western blotting. In the injection experiments, the absolute time course of Cyclin B export varies between batches of oocytes (i.e., between frogs). In all experiments in which we compare export rates in treated oocytes (e.g., T24N, leptomycin B, NES) with those in untreated oocytes, samples to be compared were done with the same batch of oocytes. The same is true for experiments comparing the nuclear export rates of different cyclin mutants. Multiple repetitions of each experiment confirmed that the relative rates of export of the mutants (or of wild-type cyclin in treated vs. untreated oocytes) were the same from experiment to experiment.
Site-directed mutagenesis
For construction of the Phe-112 → Ala mutant Cyclin B1, we used ExSite PCR-Based Site-Directed Mutagenesis Kit (Stratagene). Wild-type Xenopus Cyclin B1 cDNA in the pSP64T vector was used as the template. The primers were (1) 5′-GCTAGCGATGTCCTCATTCACGTTAAAGATGTTGATG, in which a silent mutation was used to generate an NheI site to facilitate mutant selection; and (2) 3′-AGCCTGGCAGAGCTCATCAGGGAGGCAACC.
Expression and purification of recombinant proteins
The isolated CRS from Xenopus Cyclin B1 (amino acids 76–125), or mutant variants in which the four phosphorylated Ser residues had been mutated to Glu or Ala were cloned into the vector pGexKG to generate GST–CRS fusion proteins. All constructs and the vector alone were expressed in Topp 3. Escherichia coli (Stratagene). The recombinant fusion proteins were expressed and purified as described (Solomon et al. 1992).
Preparation of total, cytoplasm, and nuclear oocyte extracts
Xenopus oocytes were prepared and manually dissected as described above. Either total, cytoplasm or nuclear fractions were homogenized in extract buffer (20 mm HEPES, 0.25 m sucrose, 0.1 m NaCl, 2.5 mm MgCl2 at pH 7.2) and centrifuged twice at 13,000g for 10 min. The clear supernatants were collected and diluted to 1.45 mg/ml for assays of GST–CRS phosphorylation.
Phosphorylation assay
Twenty microliters of glutathione–Sepharose beads coupled to GST or to GST–CRS wild-type, Ala, or Glu was incubated in 45 μl of extract with 1 μm ATP and 15 μCi [γ32P]ATP at room temperature for 20 min. The beads were then pelleted and washed with 1 ml of extract buffer three times. The bead-bound material was analyzed by SDS-PAGE followed by autoradiography.
Pull-down experiments
GST fusion proteins were coupled to glutathione–Sepharose beads and blocked with boiled fetal bovine serum to block nonspecific interactions of proteins with the sepharose beads. The beads were then incubated with either oocyte extract or in vitro-translated protein in boiled fetal bovine serum at 4°C for an hour. The beads were then washed with EB buffer and the binding proteins were resolved by SDS-PAGE followed by immunoblotting or autoradiography.
Fractionation of HeLa cell extracts
Two subconfluent 10-cm dishes of HeLa cells were treated with 20 nm leptomycin B. At various timepoints, cells were washed with PBS and scraped off the dish and harvested by centrifugation at 1 K for 10 min at 4°C. Cells were then resuspended in hypotonic buffer (20 mm Tris-HCl at pH 7.4, 10 mm KCl, 1 mm EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin) for 10 min on ice and lysed by dounce homogenization. Nuclei were pelleted by centrifugation at 1 K for 10 min and washed with PBS twice. Both the cytoplasmic and nuclear fractions were mixed with SDS sample buffer and analyzed by SDS-PAGE and immunoblotting. The nuclear and cytoplasmic fractions were normalized to contain equal numbers of cell equivalents (∼1.5 × 105 cells/lane).
Immunofluorescence
For immunolocalization of Cyclin B1, HeLa cells were grown to subconfluency on coverslips in DMEM with 10% fetal bovine serum. Where indicated, cells were incubated for 4 hr with the same medium containing 20 ng/ml leptomycin B. Leptomycin-treated or control coverslips were fixed at room temperature with 4% paraformaldehyde and then permeabilized with cold 2% Triton X-100 in PBS. Nonspecific sites were blocked by incubation with 2% BSA, 0.2% Triton X-100 in PBS, followed by incubation with a monoclonal antibody to human Cyclin B1 (GNS1, 1/200; Santa Cruz Biotechnology). Bound antibody was detected with RITC-conjugated antibody to mouse IgG1. During the final wash, cells were stained with Hoechst 33258 to visualize the DNA, after which they were mounted on slides with gelvatol.
Acknowledgments
We are grateful to Dr. Jonathan Pines for sharing his results with us prior to publication, and for suggesting the F112A mutant. We thank Dr. Minoru Yoshida for generously providing leptomycin B. We thank Ashley Smith for her assistance with the immunofluorescence. Dr. Katherine Swenson made these experiments possible by providing helpful advice and a rapid and excellent tutorial in oocyte injection, for which we are grateful. We thank Dr. D. Forbes for her generous support and for providing for the synthesis of the PKI NES and NOS peptides and conjugates. We thank Dr. G. Grosveld for generously providing anti-CRM1 antisera, Dr. J. Maller for anti-Cyclin B1 sera, Dr. Mary Dasso for anti-RanBP1, and Drs. U. Brinkmann and D. Gorlich for kindly providing anti-CAS and anti-RanBP7 sera. We thank Dr. F. Melchior for her gift of purified RanGAP and Dr. D. Donoghue for the Ala and Glu mutant Cyclin B1 clones. We thank Dr. K. Ullman for protocols and advice. We thank Dr. D. Lew for critical reading and editing of the manuscript. This work was supported by a grant from the American Cancer Society to S.K. and a fellowship from the Leukemia Research Foundation to J.D.M.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL kornb001@mc.duke.edu; FAX (919) 613-8642.
References
- Atherton-Fessler S, Liu F, Gabrielli B, Lee MS, Peng C-Y, Piwnica-Worms H. Cell cycle regulation of the p34cdc2 Inhibitory kinases. Mol Biol Cell. 1994;5:989–1001. doi: 10.1091/mbc.5.9.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Fridell RA, Benson RE, Hua J, Cullen BR. Protein sequence requirements for function of the human T-cell leukemia virus type 1 Rex nuclear export signal delineated by a novel in vivo randomization-selection assay. Mol Cell Biol. 1996;16:4207–4214. doi: 10.1128/mcb.16.8.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett AH, Silver PA. Nucleocytoplasmic transport of macromolecules. Microbiol Mol Biol Rev. 1997;61:193–211. doi: 10.1128/mmbr.61.2.193-211.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doye V, Hurt E. From nucleoporins to nuclear pore complexes. Curr Opin Cell Biol. 1997;9:401–411. doi: 10.1016/s0955-0674(97)80014-2. [DOI] [PubMed] [Google Scholar]
- Dunphy WG, Kumagai A. The cdc25 protein contains an intinsic phosphatase activity. Cell. 1991;67:189–196. doi: 10.1016/0092-8674(91)90582-j. [DOI] [PubMed] [Google Scholar]
- Fischer U, Huber J, Boelens WC, Mattaj IW, Lührmann R. The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell. 1995;82:475–483. doi: 10.1016/0092-8674(95)90436-0. [DOI] [PubMed] [Google Scholar]
- Fornerod M, Ohno M, Yoshida M, Mattaj IW. Crm1 is an export receptor for Leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- Gautier J, Maller JL. Cyclin B in Xenopus oocytes: Implications for the mechanism of pre-MPF activation. EMBO J. 1991;10:177–182. doi: 10.1002/j.1460-2075.1991.tb07934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier J, Solomon MJ, Booher RN, Bazan JF, Kirschner MW. Cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell. 1991;67:197. doi: 10.1016/0092-8674(91)90583-k. [DOI] [PubMed] [Google Scholar]
- Girard F, Strausfeld U, Cavadore JC, Russell P, Fernandez A, Lamb NJ. Cdc25 is a nuclear protein expressed constitutively throughout the cell cycle in nontransformed mammalian cells. J Cell Biol. 1992;118:785–794. doi: 10.1083/jcb.118.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D, Dabrowski M, Bischoff FR, Kutay U, Bork P, Hartmann E, Prehn S, Izaurralde E. A novel class of RanGTP binding proteins. J Cell Biol. 1997;138:65–80. doi: 10.1083/jcb.138.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D, Mattaj IW. Nucleocytoplasmic transport. Science. 1996;271:1513–1518. doi: 10.1126/science.271.5255.1513. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Vogel F, Mills AD, Hartmann E, Laskey RA. Distinct functions for the two importin subunits in nuclear protein import. Nature. 1995;377:246–248. doi: 10.1038/377246a0. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Prehn S, Laskey RA, Hartmann E. Isolation of a protein that is essential for the first step of nuclear protein import. Cell. 1994;79:767–778. doi: 10.1016/0092-8674(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Heald R, McLoughlin M, McKeon F. Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated cdc2 kinase. Cell. 1993;74:463–474. doi: 10.1016/0092-8674(93)80048-j. [DOI] [PubMed] [Google Scholar]
- Izumi T, Maller JL. Phosphorylation of Xenopus cyclins B1 and B2 is not required for cell cycle transitions. Mol Cell Biol. 1991;11:3860–3867. doi: 10.1128/mcb.11.8.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T, Walker DH, Maller JL. Periodic changes in phosphorylation of the Xenopus cdc25 phosphatase regulate its activity. Mol Biol Cell. 1992;3:927–939. doi: 10.1091/mbc.3.8.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman M, Firth M, Pines J. Human cyclins B1 and B2 are localized to strikingly different structures: B1 to microtubules, B2 primarily to the Golgi apparatus. EMBO J. 1995;14:1646–1654. doi: 10.1002/j.1460-2075.1995.tb07153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Minshull J, Ford C, Golsteyn R, Poon R, Hunt T. On the synthesis and destruction of A- and B- type cyclins during oogenesis and miotic maturation in Xenopus laevis. J Cell Biol. 1991;114:755–765. doi: 10.1083/jcb.114.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornbluth S, Sebastian B, Hunter T, Newport J. Membrane localization of the kinase which phosphorylates p34cdc2 on Threonine 14. Mol Biol Cell. 1994;5:273–282. doi: 10.1091/mbc.5.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek W, Marks J, Schmitz N, Nigg EA, Simanis V. Vertebrate p34cdc2 phosphorylation site mutants: Effects upon cell cycle progression in the fission yeast Schizosaccharomyes pombe. J Cell Sci. 1992;102:43–53. doi: 10.1242/jcs.102.1.43. [DOI] [PubMed] [Google Scholar]
- Kutay UB, Bischoff FR, Kostka S, Kraft R, Gorlich D. Export of Importin α from the nucleus is mediated by a specific nuclear transport factor. Cell. 1997;90:1061–1071. doi: 10.1016/s0092-8674(00)80372-4. [DOI] [PubMed] [Google Scholar]
- Li J, Meyer AN, Donoghue DJ. Requirement for phosphorylation of cyclin B1 for Xenopus oocyte maturation. Mol Biol Cell. 1995;6:1111–1124. doi: 10.1091/mbc.6.9.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Nuclear localization of cyclin B1 mediates its biological activity and is regulated by phosphorylation. Proc Natl Acad Sci. 1997;94:502–507. doi: 10.1073/pnas.94.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Stanton JJ, Wu Z, Piwnica-Worms H. The human Myt1 kinase preferentially phosphorylates cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi. Mol Cell Biol. 1997;17:571–583. doi: 10.1128/mcb.17.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Paine PL. Non-aqueous isolation of transcriptionally active nuclei from Xenopus oocytes. Meth Enzymol. 1990;181:36–43. doi: 10.1016/0076-6879(90)81110-g. [DOI] [PubMed] [Google Scholar]
- McGowan CH, Russell P. Cell cycle regulation of human WEEl. EMBO J. 1995;14:2166–2175. doi: 10.1002/j.1460-2075.1995.tb07210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L, Arion D, Golsteyn R, Pines J, Brizuela L, Hunt T, Beach D. Cyclin is a component of the sea urchin M-phase specific histone H1 kinase. EMBO J. 1989;8:2275–2282. doi: 10.1002/j.1460-2075.1989.tb08353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer BE, Meinkoth JL, Malim MH. Nuclear transport of human immunodeficiency virus type 1, visna virus, and equine infectious anemia Rev proteins: Identification of a family of transferrable export signals. J Virol. 1996;70:2350–2359. doi: 10.1128/jvi.70.4.2350-2359.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JBA, Russell P. The cdc25 M-Phase inducer: An unconventional protein phosphatase. Cell. 1992;68:407–410. doi: 10.1016/0092-8674(92)90177-e. [DOI] [PubMed] [Google Scholar]
- Millar JBA, Blevitt J, Gerace L, Sadhu K, Featherstone C, Russell P. p55-cdc25 is a nuclear protein required for the intitiation of mitosis in human cells. Proc Natl Acad Sci. 1991;88:10500–10504. doi: 10.1073/pnas.88.23.10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Dunphy WG. Cell cycle regulation of a Xenopus Wee1-like kinase. Mol Biol Cell. 1995a;6:119–134. doi: 10.1091/mbc.6.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Kumagai A, Dunphy WG. Myt1: A membrane-associated inhibitory kinase that phosphorylates cdc2 on both Threonine-14 and Tyrosine-15. Science. 1995b;270:86–90. doi: 10.1126/science.270.5233.86. [DOI] [PubMed] [Google Scholar]
- Murray AW. Creative blocks: Cell-cycle checkpoints and feedback controls. Nature. 1992;359:599–604. doi: 10.1038/359599a0. [DOI] [PubMed] [Google Scholar]
- Neville MS, Stutz F, Lee L, Davis LI, Rosbash M. The importin-beta family member Crm1p bridges the interaction between Rev and the nuclear pore complex during nuclear export. Curr Biol. 1997;7:767–775. doi: 10.1016/s0960-9822(06)00335-6. [DOI] [PubMed] [Google Scholar]
- Norbury C, Blow J, Nurse P. Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J. 1991;10:3321–3329. doi: 10.1002/j.1460-2075.1991.tb04896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of Crm1 in signal-mediated nuclear protein export. Science. 1997;278:141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- Pines J, Hunter T. Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport. J Cell Biol. 1991;115:1–17. doi: 10.1083/jcb.115.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J, Hunter T. The differential localization of human cyclins A and B is due to a cytoplasmic retention signal in cyclin B. EMBO J. 1994;13:3772–3781. doi: 10.1002/j.1460-2075.1994.tb06688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard VW, Michael WM, Nakielny S, Siomi MC, Wang F, Dreyfuss G. A novel receptor-mediated nuclear protein import pathway. Cell. 1996;86:985–994. doi: 10.1016/s0092-8674(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Radu A, Blobel G, Moore MS. Identification of a protein complex that is required for nuclear protein import and mediates docking of import substrate to distinct nucleoporins. Proc NatI Acad Sci. 1995;92:1769–1773. doi: 10.1073/pnas.92.5.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards SA, Carey KL, Macara IG. Requirement for guanosine triphosphate-bound Ran for signal-mediated nuclear protein export. Science. 1997;276:1842–1844. doi: 10.1126/science.276.5320.1842. [DOI] [PubMed] [Google Scholar]
- Scherf R, Pastan I, Willingham NC, Brinkmann U. The human Cas protein which is homologous to CSE1 yeast chromosome segregation gene product is associated with microtubules and mitotic spindle. Proc Natl Acad Sci. 1996;93:2670–2674. doi: 10.1073/pnas.93.7.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T, Yamashita K, Nishitani H, Takagi T, Russell P, Nishimoto T. Chromosome condensation caused by loss of RCC1 function requires the cdc25C protein that is located in the cytoplasm. Mol Biol Cell. 1992;3:1373–1388. doi: 10.1091/mbc.3.12.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon MJ, Lee T, Kirschner MW. Role of phosphorylation in p34cdc2 activation: Identification of an activating kinase. Mol Biol Cell. 1992;3:13–27. doi: 10.1091/mbc.3.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stade K, Ford CS, Guthrie C, Weiss K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90:1041–1050. doi: 10.1016/s0092-8674(00)80370-0. [DOI] [PubMed] [Google Scholar]
- Strausfeld U, Labbe J-C, Fesquet D, Cavadore J-C, Picard A, Sadhu K, Russell P, Doree M. Dephosphorylation and activation of a p34cdc2/cyclin B complex in vitro by human CDC25 protein. Nature. 1991;351:242–245. doi: 10.1038/351242a0. [DOI] [PubMed] [Google Scholar]
- Swenson KI, Jordan JR, Beyer EC, Paul DL. Formation of gap junctions by expression of connexins in Xenopus oocyte pairs. Cell. 1989;57:145–155. doi: 10.1016/0092-8674(89)90180-3. [DOI] [PubMed] [Google Scholar]
- Ullman KS, Powers MA, Forbes DJ. Nuclear export receptors: From Importin to Exportin. Cell. 1997;90:967–970. doi: 10.1016/s0092-8674(00)80361-x. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Broome M, Hunter T. Regulation of the human Wee1Hu CDK tyrosine-15 kinase during the cell cycle. EMBO J. 1994;14:1878–1891. doi: 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- Wu L, Shiozaki K, Aligue H, Russell P. Spatial organization of the Nim1-wee1-cdc2 mitotic control network in Schizosaccharomyces pombe. Mol Biol Cell. 1996;7:1749–1758. doi: 10.1091/mbc.7.11.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]