

Abstract
All multisubunit DNA-dependent RNA polymerases (RNAP) are zinc metalloenzymes, and at least two zinc atoms are present per enzyme molecule. RNAP residues involved in zinc binding and the functional role of zinc ions in the transcription mechanism or RNAP structure are unknown. Here, we locate four cysteine residues in the Escherichia coli RNAP largest subunit, β′, that coordinate one of the two zinc ions tightly associated with the enzyme. In the absence of zinc, or when zinc binding is prevented by mutation, the in vitro-assembled RNAP retains the proper subunit stoichiometry but is not functional. We demonstrate that zinc acts as a molecular chaperone, converting denatured β′ into a compact conformation that productively associates with other RNAP subunits. The β′ residues coordinating zinc are conserved throughout eubacteria and chloroplasts, but are absent from homologs from eukaryotes and archaea. Thus, the involvement of zinc in the RNAP assembly may be a unique feature of eubacterial-type enzymes.
Keywords: Transcription, RNA polymerase, assembly mutations, zinc
DNA-dependent RNA polymerase (RNAP), alone or in complex with regulatory factors, is central to all steps of the transcription cycle, including promoter binding and melting, initiation of RNA synthesis, processive elongation, and release of RNA at terminators. Cellular RNAPs are large, multisubunit enzymes. A typical prokaryotic RNAP core contains 4–6 polypeptides with a total molecular mass of ∼400 kD. Core RNAP from eukaryotes and archaea contain 7–14 subunits with a total molecular mass in excess of 500 kD. Sequence alignments of RNAP subunits reveal extensive similarities (Allison et al. 1985; Sweetser et al. 1987). Each of the two largest RNAP subunits, which are the most evolutionarily conserved, contains eight to nine colinear segments with many invariant amino acids. Low-resolution (16–35 Å) three-dimensional models of Escherichia coli RNAP, as well as RNAPI and RNAPII from yeast, obtained by electron crystallography, also reveal significant similarities (Darst et al. 1989, 1991; Schultz et al. 1993; Polyakov et al. 1995).
All multisubunit RNAPs are zinc metalloenzymes, and at least two zinc atoms are present per enzyme molecule (Vallee and Auld 1990). It is currently believed that each of the largest subunits of eukaryotic enzymes coordinates one zinc ion. Sequence analysis supports this view: One putative zinc-binding motif was found close to the carboxy-terminal end of the second largest subunit from eukaryotes and archaea; another is close to the amino-terminal end of the largest subunit. Both motifs were shown to interact with radioactive zinc in blotting experiments involving denatured subunit fragments (Treich et al. 1991).
RNAP from E. coli is the best-studied enzyme of its class. It consists of a catalytic core of two α, one β′, and one β subunit (329, 1407, and 1342 amino acids, respectively). Binding of one of several specificity σ factors converts the core into the holoenzyme, capable of specific initiation at promoters. E. coli RNAP contains two zinc ions per molecule of holoenzyme (Scrutton et al. 1971). Experiments involving metal substitution and oxidative immobilization indicated that one zinc ion is associated with β′, whereas the other may be associated with either β′ or β (Wu et al. 1977). However, similar experiments with the highly homologous enzyme from Bacillus subtilis revealed that both zinc ions are bound to β′ (Halling et al. 1977).
Despite extensive research, the role of zinc atoms in RNAP structure and mechanism is not known. Addition of zinc markedly increases yields of active E. coli RNAP during in vitro reconstitution (Borukhov and Goldfarb 1993), and no RNAP activity can be recovered when both zinc atoms are removed (Giedroc and Coleman 1986). In contrast, removal of only one zinc ion does not prevent RNAP function (Solaiman and Wu 1985). Curiously, analysis of E. coli RNAP subunit sequences reveals only one recognizable zinc-binding motif in the β′ conserved segment A. This motif corresponds to a truncated version of the putative zinc-binding motif found in the largest subunits of eukaryotic RNAP (Treich et al. 1991).
Our working hypothesis is that zinc has primarily a structural role and promotes RNAP assembly by helping bring the large subunits together in a defined orientation. This hypothesis is in accord with available structural data, showing that protein loops stabilized by zinc ions are involved in the formation of quaternary structure in other multisubunit proteins (Vallee and Auld 1990). Here, we used in vivo metal substitution, localized hydroxyl radical cleavage, and site-directed mutagenesis to locate one of the two E. coli RNAP zinc ions in the largest subunit, β′. We also used in vitro RNAP reconstitution to demonstrate that β′ interacts with zinc in the absence of other RNAP subunits, and that this interaction is necessary for active RNAP assembly in vitro.
Results
Zinc suppresses the temperature-sensitive phenotype of rpoC assembly mutations
Recently, we mapped several rpoC(β′) mutations that affect RNAP assembly or result in temperature-sensitive RNAP activity (Nedea et al. 1999). Two mutations, R120 and XH56, occurred close to the evolutionarily conserved segment G of β′. Both R120 and XH56 changed amino acids that are strictly conserved within eubacterial RNAPs, but are absent from the β′ homologs from archaea and eukaryotes (Fig. 1A). Three additional amino acids in this region, Cys-888, Cys-895, and Cys-898, are also conserved in eubacteria (Fig. 1A). Groups of three or more cysteines can coordinate structural or catalytic zinc ions and ion clusters in proteins, and pairs of cysteines separated by two amino acids often serve as zinc-binding nuclei (Vallee and Auld 1990). We hypothesized that Cys-888, Cys-895, and Cys-898 may coordinate zinc, and that the R120 and XH56 mutations alter the zinc-binding site and thus interfere with RNAP assembly at the restrictive temperature (Nedea et al. 1999).
Figure 1.


The R120 and XH56 rpoC assembly mutations are suppressed by zinc. (A) Genetic context of the R120 and XH56 mutations. The bar at the top represents the 1407-amino-acid β′ subunit from E. coli. The lettered boxes indicate evolutionarily conserved segments. E. coli segment G is expanded underneath and is aligned to the corresponding segments from M. leprae (M.l.), T. maritima (T.m.), H. pylori (H.p.), and tobacco chloroplasts (T), as well as S. acidocaldarius (S.a.), and yeast RNAP I, II, and III (Yp1, Yp2, and Yp3, respectively). The XH56 and R120 mutations are indicated above the E. coli sequence; the three cysteines conserved within the eubacterial lineage are indicated below the tobacco sequence. (B) The overnight growth of the indicated E. coli strains on LB plates with or without 100 μm ZnCl2 at 30°C and 42°C.
Studies of the yeast GAL4 protein showed that mutations in the zinc-binding motif could be rescued by simple addition of excess zinc in the growth medium (Johnston 1987). Accordingly, we tested if the temperature-sensitive phenotype of the R120 and XH56 strains could be corrected by the addition of zinc to the medium. As controls, we used E. coli XL1-blue (rpoC+) as well as cells carrying rpoCTs4 (Nedea et al. 1999) and rpoA112 (Igarashi et al. 1990) temperature-sensitive mutations. The results are presented in Figure 1B. In the absence of zinc, only XL1-blue cells grew at the restrictive (42°C) temperature, as expected. In contrast, in the presence of 100 μm zinc in the growth medium, the temperature-sensitive phenotype of R120 and XH56 strains was corrected. Control cell carrying the Ts4 and rpoA112 mutations did not grow at 42°C even in the presence of zinc. Thus, this in vivo result supports the idea that R120 and XH56 affect a zinc-binding domain of β′.
Monitoring RNAP zinc-binding sites using localized radical cleavage
The Fe2+ ion under aerobic conditions generates reactive hydroxyl radicals, which can cleave nearby biopolymers. This process, known as the Fenton reaction, is often used for footprinting nucleic acid–protein complexes (Dixon et al. 1991). Zn2+ in zinc-finger proteins can be efficiently replaced by Fe2+ in vivo, and the resulting ‘iron fingers’ generate free radicals that cleave both nucleic acids and polypeptide chains close to the metal-binding site (Conte et al. 1996). Earlier studies demonstrated that both E. coli RNAP Zn2+ ions could be substituted for other metal ions without affecting RNAP structure and function. RNAP purified from cells grown on minimal medium supplemented with cobalt instead of zinc contained two molar equivalents of cobalt, and no zinc and was fully functional in vitro (Chatterji and Wu 1982). Similarly, in vitro reconstitution at controlled conditions allowed the preparation of functional RNAP containing Co, Cd, or Hg ions (Solaiman and Wu 1985).
We repeated the RNAP metal biosubstitution procedure of Chatterji and Wu (1982) and purified RNAP from cells grown in zinc-depleted, iron-enriched medium. The resultant RNAP was functional and absorbed light at 400–500 nm (data not shown). In contrast, the standard RNAP preparation does not absorb light in this range. We conclude that RNAP purified from zinc-depleted, iron-enriched medium contains tightly bound iron, and we refer to this preparation as RNAPFe.
Next we used RNAPFe in localized radical cleavage reaction. Open promoter complexes were formed on a DNA fragment containing the T7 A2 promoter with either RNAPFe or control RNAPZn immobilized on Ni2+–NTA agarose beads through His6-tagged β′. RNAP was then affinity labeled, using chimeric rifampicin–GTP compound and derivatized thymidine triphosphate, as described in Materials and Methods. Affinity labeling results in covalent attachment of Rif–GpCpT* to the conserved segment G of β′ (Severinov et al. 1996; the boldface type indicates radioactive phosphate; the asterisk indicates cross-linked nucleotide). The immobilized affinity-labeled complexes were washed from unincorporated reaction substrates and incubated with H2O2 and ascorbate, and the products were separated by SDS-PAGE and visualized by autoradiography (Fig. 2A).
Figure 2.


Localized radical cleavage of wild-type RNAP. (A) RNAPs containing zinc or iron were affinity labeled in β′ segment G using the T7 A2 promoter-containing DNA fragment as a template and treated with 0.03% H2O2 and 1 mm sodium ascorbate for times indicated. The reaction products were resolved on a 10% SDS-polyacrylamide gel and visualized by autoradiography. (Lanes 1,2) Marker lanes generated by treating the RNAP affinity labeled at β Lys-1065 (lane 1) or β′ conserved segment G (lane 2) with CNBr under single-hit conditions. The mobility of the recombinant β′821–1407 fragment is indicated by an arrow at the right of the gel. The bar at right represents E. coli β′. The affinity-labeling site in segment G is shown. The three catalytic Asp residues in segment D, the four evolutionarily conserved Cys residues in segment A, as well as the cysteines close to cleavage sites I and II are shown as shaded lines. (B) Genetic context of the β′ iron cleavage site II (see Fig. 1A legend for details). Cys-814 is highlighted by larger font size; an E. coli mutation S793F that results in streptolydigin-resistant RNAP (Severinov et al. 1995) is shown above the E. coli sequence. (↓) The position of natural split site in archaea.
Two cleavages were specific for RNAPFe (Fig. 2, lanes 7,8, sites I and II) and were thus due to the presence of tightly bound Fe2+. To map the iron-cleavage sites, the radio-tagged β or β′ subunits were cleaved at Met residues under the ‘single-hit’ conditions to yield families of nested, easily identifiable fragments (Fig. 2, lanes 1,2). From the relative migration of products, the major cleavage site (site I) was mapped at the amino-terminal side of β′ Met-932, which creates the smallest radioactive fragment produced under single-hit CNBr cleavage conditions. The second cleavage site (site II) was mapped close to Met-821–Met-822. We used the recombinant fragment of β′, β′821–1407 (Severinov et al. 1996) as a marker to ensure that the assignment of single-hit bands is correct (as can be seen in Fig. 2B, the band corresponding to site II cleavage migrates just above β′821–1407, which is indicated by an arrow). Thus, the site II cleavage occurred carboxy terminal to the β′ conserved segment F (Fig. 2B) involved in transcription elongation and termination (Waelbaecher et al. 1994), the binding of streptolydigin in bacterial RNAP (Severinov et al. 1995; Yang and Price 1995), and the binding of α-amanitin in eukaryotic RNAP (Bartolomei and Corden 1987). Similar to site I, there is a cysteine residue at or very close to cleavage site II, Cys-814, that is strictly conserved within the eubacterial lineage but is absent from archeal and eukaryotic β′ homologs.
Both RNAPZn and RNAPFe were cleaved with the same low efficiency near the β′ conserved segment D (Fig. 2A, lanes 4,7). It has been shown that three Asp residues in segment D that normally coordinate a loosely bound Mg2+, which is important for RNAP catalytic activity, can also coordinate a ferrous ion (Zaychikov et al. 1996). It should be noted that the experiment of Figure 2A was performed in the absence of Mg2+ ions. When the cleavage reaction was repeated in the presence of 10 mm Mg2+, the segment D cleavage was suppressed completely (data not shown). In contrast, cleavage specific for RNAPFe was not affected by 10 mm Mg2+ in the reaction (not shown). Thus, the observed segment D cleavage is likely explained by the presence of residual iron in the buffer.
Localized radical cleavage experiments were also performed with RNAPFe affinity labeled at the β subunit Lys-1065 (Grachev et al. 1987). No difference in radio-tagged β cleavage between RNAPFe and RNAPZn was observed (data not shown), suggesting that the second largest subunit does not participate in the binding of Zn2+.
Mutational analysis of the β′ zinc-binding site
The results of localized radical cleavage further support the idea that Cys-888, Cys-895, and Cys-898 are part of a zinc-binding site. Based on the identical kinetics of accumulation of the site I and site II cleavage products (data not shown), cleavage at both sites originates from the same ferrous ion, indicating that β′ residues Cys-888, Cys-895, Cys-898, and Cys-814 jointly form one of RNAP zinc-binding sites. We performed site-directed mutagenesis of site I and site II cysteines of the rpoC gene cloned on the expression plasmid pCYB2β′ (Nedea et al. 1999). Our goal was to use mutant enzymes (1) to confirm the role of sites I and II cysteines in the coordination of zinc ion, and (2) to study the structural and functional consequences of zinc ion removal. The first mutation, 3C3A, changed Cys-888, Cys-895, and Cys-898 of site I to Ala. The second mutation, C814A, changed the only site II cysteine to alanine. Plasmids pCYBβ′C814A and pCYBβ′3C3A expressing the mutant β′ subunits did not complement the chromosomal rpoC 39°C temperature-sensitive mutation at 42°C (Nedea et al. 1999). At 30°C these plasmids had no effect on cell growth. Thus, the mutant rpoC genes were either temperature-sensitive or recessive lethals.
The β′ subunit expressed from pCYBβ′His and its derivatives is His6 tagged at its carboxyl terminus, which enables the separation of RNAP containing plasmid-borne β′ from RNAP containing chromosome-encoded β′ by affinity chromatography on Ni2+ sorbent. Our repeated attempts to purify RNAPs harboring the C814A mutation from cells grown at 30°C using affinity chromatography failed (data not shown). We conclude that C814A completely abolishes the ability of β′ to enter E. coli RNAP core in vivo. In contrast, affinity-purified RNAP carrying the 3C3A mutation was active in vitro (Fig. 3). Because 3C3A was engineered near the XH56 and R120 mutations that make RNAP activity temperature sensitive (XH56) or interfere with RNAP assembly in vitro (R120) (Gross et al. 1977), we asked whether RNAP3C3A is active at high temperature of 42°C and whether active RNAP3C3A can be obtained after in vitro denaturation/renaturation. As can be seen, the 3C3A enzyme was functional at high temperature (Fig. 3A), but it did not assemble after denaturation in vitro (Fig. 3B) and thus resembled RNAPR120.
Figure 3.
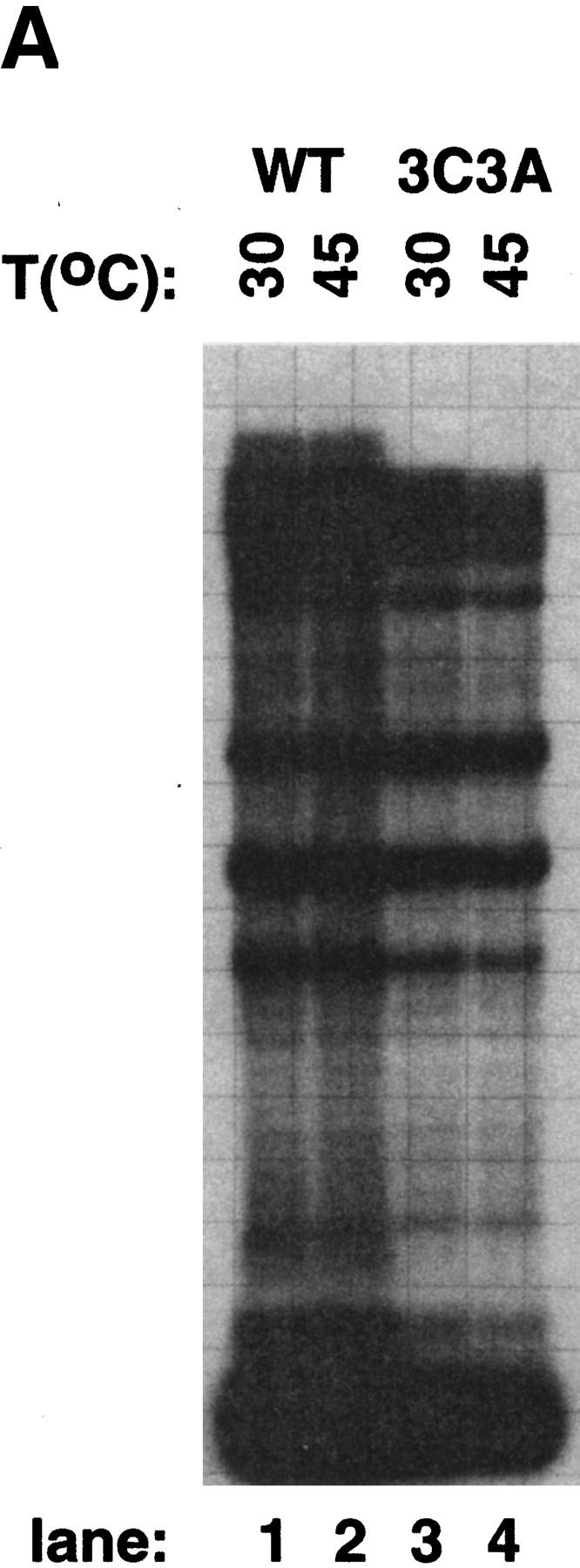
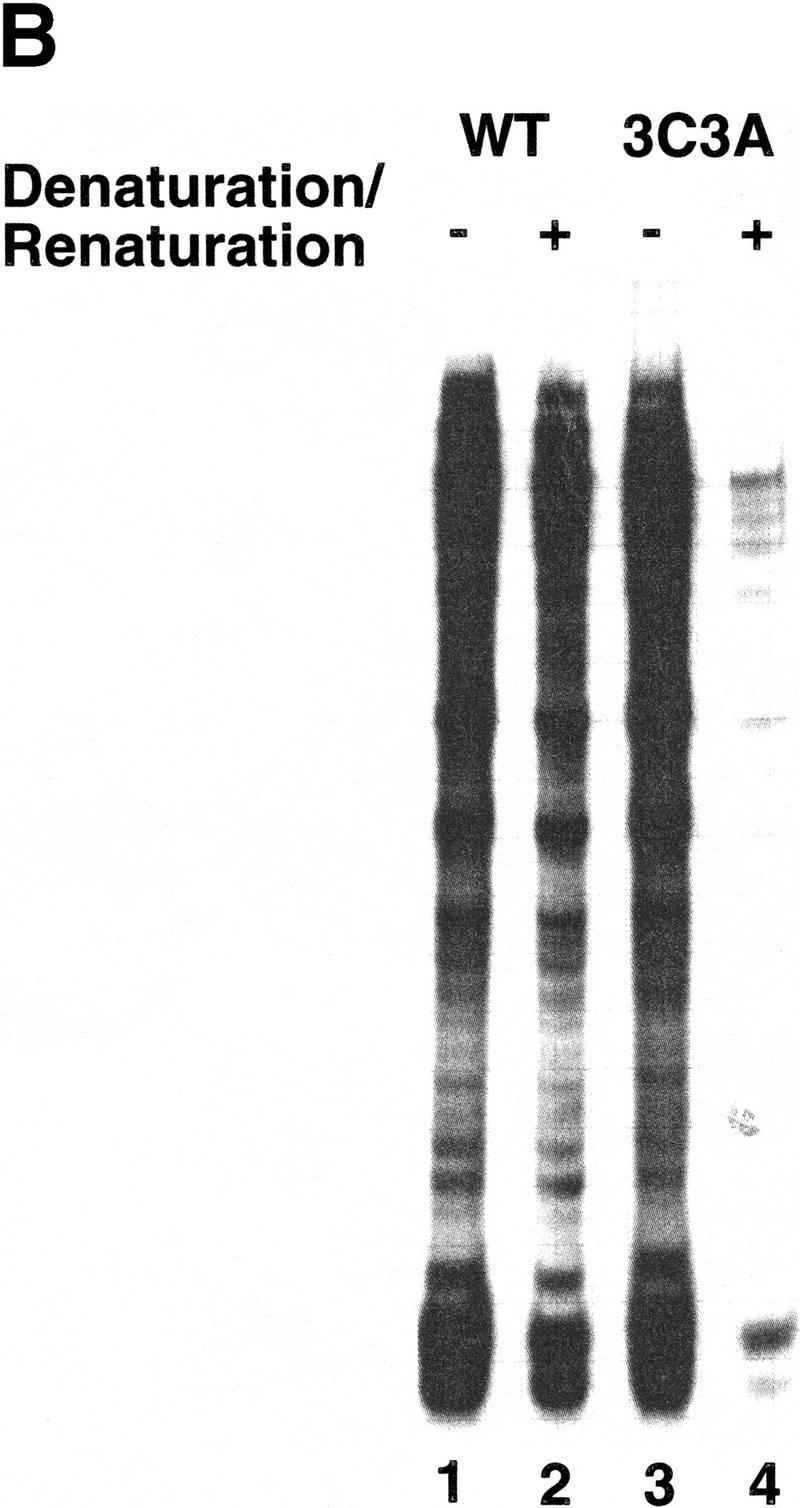

In vitro transcription by 3C3A RNAP. (A) RNAP3C3A is temperature resistant in vitro. RNAP3C3A was affinity purified from 397C cells harboring the pCYB2β′3C3A plasmid and used to transcribe the T7 A1 promoter-containing DNA fragment at the indicated temperatures. Reaction products were separated on a denaturing 20% polyacrylamide gel and revealed by autoradiography. (B) RNAP3C3A is assembly defective in vitro. RNAP3C3A and RNAPWT were subjected to a denaturation/renaturation cycle by the addition of 6 m guanidine-HCl, followed by dialysis into transcription buffer. The samples were then assayed in a steady-state transcription assay, using the T7 A1 promoter as a template. Reaction products were analyzed as in A. (C) Localized radical cleavage of iron-containing RNAP3C3A. RNAPs containing plasmid-borne, His6-tagged wild-type or 3C3A β′ were affinity purified from 397C E. coli cells grown in the presence of iron and in the absence of zinc. Affinity labeling, hydroxy-radical cleavage, and reaction-product analyses were performed as described in the legend to Fig. 2.
To rigorously test our interpretation that the 3C3A triple mutation disrupts an RNAP zinc-binding site we prepared iron-substituted 3C3A enzyme and performed localized radical cleavage (Fig. 3B). As expected, no cleavage at site I and site II was observed in 3C3A RNAPFe, further supporting the notion that both cleavages originate from the same iron atom. Interestingly, a major cleavage site close to one of the termini of β′ was present in the 3C3A enzyme (lane 3). A well-defined putative zinc-binding motif is present in the amino-terminal segment A (amino acids 85–117) of β′ (Borukhov et al. 1991). No such motif is present close to β′ carboxyl terminus. Therefore, we tentatively assigned this new cleavage to the amino terminus of β′, and we mapped it close to amino acid position 115 using recombinant amino-terminally truncated β′ fragments β′88–1407 and β′175–1407 as markers.
In vitro reconstitution of mutant enzymes in the presence of Zn2+ and RNAPWT reconstitution in the absence of Zn2+ leads to inactive RNAP complex
We investigated the assembly defect caused by the C814A and 3C3A mutations using in vitro RNAP reconstitution from recombinant subunits and a Ni2+-coimmobilization assay to test the ability of His6-tagged mutant β′ to interact with RNAP α, β, and σ70 subunits. In parallel, we studied RNAPWT assembly in the absence of zinc. Reconstitution mixtures were incubated with Ni2+–NTA agarose beads, the beads were washed and the bound material was eluted with 100 mm imidazole and analyzed by SDS-PAGE (Fig. 4). As can be seen, α, β, and σ70 were specifically coimmobilized with C814A and 3C3A β′ subunits in the expected stoichiometry. Similarly, wild-type β′ assembled into a holoenzyme-like complex both in the presence and in the absence of zinc. We conclude that C814A and 3C3A do not prevent the β′ interaction with other RNAP subunits and that this interaction can occur in the absence of added zinc or when zinc binding is prevented by mutation.
Figure 4.


Zn2+ is not important for interactions of β′ with other RNAP subunits. (A) Binding of α2β to immobilized His6–β‘ studied by the Ni2+-coimmobilization assay. The α subunit was renatured with β, σ, and His6–β′WT in the presence or in the absence of 10 μm ZnCl2 (lanes 1–5 and 6–10, respectively), or with His6–β′3C3A (lanes 11–15) or His6–β′C814A (lanes 16–20) in the presence of ZnCl2, loaded onto Ni2+–NTA–agarose beads (L), and the unbound protein (or flowthrough) removed (F). The beads were then washed with buffer containing 5 mm imidazole (W1) and 25 mm imidazole (W2), and the bound proteins were eluted with buffer containing 100 mm imidazole (E). The protein fractions were analyzed by SDS-PAGE. Control experiments indicated that when His6–β‘ was omitted from the reconstitution mixture, no nonspecific binding of β and α to the Ni2+–NTA–agarose beads occurred (data not shown; Wang et al. 1997). (B) Abnormal chromatographic behavior of RNAP assembled in the absence of zinc. Proteins eluted from Ni2+–NTA beads (lanes 5,10,15,20 in A) were loaded on a SEC-400 gel-filtration column.
The C814A, 3C3A, and the wild-type RNAP complexes assembled in the absence of zinc were transcriptionally inactive and did not interact with promoter DNA (data not shown). All three complexes exhibited abnormal and similar chromatographic behavior on SEC-400 size-exclusion HPLC column: They eluted early as broad peaks. In contrast, wild-type RNAP assembled in the presence of zinc eluted later as a sharp peak (Fig. 4). Limited proteolysis with trypsin also confirmed that structural organization of reconstituted C814A and 3C3A enzyme is different than the wild-type RNAP reconstituted in the presence of zinc and is similar (or identical) to that of RNAPWT reconstituted without zinc (data not shown). We conclude that in the absence of zinc, or when zinc binding is prevented by the C814A or 3C3A mutations, the specific interaction of β′ with α2β still occurs but leads to inactive complex, probably resembling the ‘immature’ state detected in the earlier studies (Zillig et al. 1977).
β′ becomes assembly competent in the presence of zinc
The results from the previous section suggest that (1) zinc is required for proper folding of β′, or (2) zinc acts at stages after β′ association with α2β to properly fold RNAP complex. To distinguish between these possibilities, we performed RNAP reconstitution from β′ or α2β renatured separately either in the presence or in the absence of zinc. The results are presented in Figure 5A. When β′ and α2β were renatured separately in the presence of zinc and combined together, active RNAP was assembled, as expected (lane 3). The yield of the assembled enzyme was about five times lower than that obtained by standard reconstitution procedure, when α, β, and β′ were renatured together in the presence of zinc (lane 1). As expected, no RNAP activity was recovered when α, β, and β′ were renatured in the absence of zinc, either together or separately (lane 2; data not shown). In contrast, when β′ was renatured in the presence of zinc, RNAP activity was recovered even when α2β was assembled in the absence of zinc (lane 5). Importantly, zinc had to be present during the renaturation of β′ from the fully denatured state, as addition of zinc to β′ renatured in its absence did not restore the activity (lanes 6,8).
Figure 5.

The β′ subunit becomes assembly competent in the presence of zinc ions. (A) The β′ subunit or a mixture of the α and β subunits (labeled α2β) were renatured separately either in the presence or in the absence of 10 μm ZnCl2. Proteins were then dialyzed in a buffer without zinc, combined as indicated, and incubated for 15 min at 30°C with the recombinant σ70 subunit to reconstitute RNAP either in the presence (lanes 6–8) or in the absence (lanes 1–5) of 10 μm ZnCl2. RNAP activity was then assayed using DNA fragment containing the T7 A1 promoter (see Fig. 3A legend). (Lanes 1,2) Controls, where standard RNAP reconstitution reactions containing α, β, and β′ were carried out either in the presence (lane 1) or in the absence (lane 2) of zinc. (B) Chromatographic analysis of the β′ subunits individually renatured in the presence or in the absence of zinc. The indicated β′ subunits were individually renatured and analyzed on an SEC-400 column attached to HPLC. Fractions were collected and analyzed by 8% SDS-PAGE (top). The gel at the bottom shows the results of in vitro transcription reaction using the indicated chromatographic fractions of wild-type β′ renatured in the presence of zinc. Fractions were supplemented with α2β and σ and assayed as in A.
When wild-type β′ was renatured individually in the absence of zinc and loaded on a gel-filtration column, it eluted as a diffuse, high-molecular-weight peak (Fig. 5B). In contrast, when β′ was renatured in the presence of zinc, an additional, narrow peak with higher retention time appeared. No such narrow peak was observed when the β′ subunits carrying the C814A and 3C3A mutations were renatured in the presence of zinc (Fig. 5B).
Both the diffuse peak and the narrow peak contained β′, as judged by SDS-PAGE analysis (Fig. 5B). Only the material from the narrow peak, however, was able to form active RNAP when combined with α2β and σ, as the gel shown on the bottom of Figure 5B demonstrates. From these experiments we conclude that (1) zinc is able to interact with β′ in the absence of other RNAP subunits; (2) zinc converts β′ into an assembly-competent, compact state; and (3) the C814A and 3C3A mutations prevent the interaction of zinc with β′ and thus interfere with the assembly of functional RNAP (Fig. 5B).
Discussion
The results of our work directly implicate four cysteine residues at positions 814, 888, 895, and 898 of the largest E. coli RNAP subunit, β′, in zinc binding and RNAP assembly. First, the temperature-sensitive phenotype of the R120 and XH56 rpoC assembly mutations that occurred very close to Cys-888, Cys-895, and Cys-898 can be suppressed by the simple addition of extra zinc in the medium. Second, using RNAP containing ferrous ions instead of zinc, we can demonstrate efficient cleavage close to Cys-888, Cys-895, and Cys-898 (site I) and Cys-814 (site II). Based on the identical kinetics of accumulation of cleavage products, both site I and site II cleavages likely originate from a single ferrous ion. Finally, both site I and site II cleavages are abolished by the 3C3A mutation, which is expected to destroy zinc binding.
The site I and site II cysteines are separated by >70 amino acids of β′ primary sequence and are located in different structural modules of the β′ subunit. The β′ modules can be physically separated from each other without preventing RNAP assembly in vitro (Severinov et al. 1996). The binding of zinc may bring the two modules of β′ together in correct orientation. Consistent with this interpretation, we were able to show that zinc converts β′ into an assembly-competent and compact conformation. In vitro, β′ interacts with other RNAP subunits even in the absence of zinc or when zinc binding is prevented by mutation. Hence, the primary determinants of β′ interaction with the α2β subassembly must lie outside of the zinc-binding domain defined in this work. Zinc had no effect on α2β formation, and no cleavage of the β subunit was detected. These results suggest that β is not involved in zinc binding, in agreement with earlier data obtained with RNAP from B. subtilis (Halling et al. 1977).
The 3C3A mutation behaves as either a recessive lethal or temperature sensitive in vivo and abolishes active RNAP assembly in vitro. RNAP3C3A purified from cells, however, is active, suggesting that binding of zinc in this site is not absolutely necessary for RNAP activity and assembly in vivo, at least at low temperature. Alternatively, it is also possible that other factors (e.g., chaperones) or mechanisms (e.g., cotranslational folding) may contribute to efficient RNAP assembly, even when zinc binding is prevented by mutation. The site I and site II cysteines are absent from the largest, β′-like subunits of eukaryotic and archeal RNAPs. Thus, RNAP assembly mechanisms in these organisms may differ from those used by prokaryotes. For example, small subunits of eukaryotic RNAPs interacting with the largest subunit can promote eukaryotic RNAP assembly in vivo (Gadal et al. 1999; Voutsina et al. 1999). Regardless of that fact, the β′ residues responsible for zinc coordination are strictly conserved in eubacteria and chloroplasts, suggesting that zinc plays an essential and ubiquitous role in RNAP assembly in these systems.
The C814A mutation appears to be more severe in vivo than 3C3A. This result is unexpected, as both C814A and 3C3A are expected to destroy the same zinc-binding site, and in vitro, recombinant β′ subunits carrying either of the two mutations appear to be equally defective in RNAP assembly. Therefore, the in vivo effect of C814A is evidently not a simple consequence of the disruption of the zinc-binding site. We speculate that C814A makes RNAP proteolitically labile, thus explaining our inability to prepare active RNAPC814A.
It has been shown previously that the two zinc ions bound to E. coli RNAP are not equal and that one, the so-called ‘loosely bound’ zinc ion can be removed or substituted for another metal ion under relatively mild reducing conditions without protein denaturation or loss of activity (Solaiman and Wu 1984). The 3C3A enzyme is functional, suggesting that the zinc-binding site defined in this work is ‘loose.’ We attempted to prove this conjecture experimentally by performing in vitro metal (zinc for iron) substitution experiments. Unfortunately, the conditions of in vitro metal exchange also promote Fenton cleavage, and no functional RNAP could be recovered (data not shown).
Localized radical cleavage results obtained with iron-substituted 3C3A enzyme are consistent with the idea that the universally conserved segment A also coordinates zinc. This zinc-binding site, however, appears to be inaccessible to Fenton oxidation in the wild-type enzyme. Disruption of the carboxy-terminal β′ zinc-binding site by the 3C3A mutation makes the segment A site accessible for cleavage, supporting the notion that segment A may indeed coordinate zinc. Thus, the two RNAP zinc-binding sites may interact with each other. Interestingly, genetic analysis of yeast RNAP I also suggested that segment A of the largest, β′-like subunit may interact with the putative zinc-binding motif located in the carboxyl terminus of the second largest, β-like subunit (McCusker et al. 1991). Our ongoing mutational analysis of β′ segment A should clarify its role in zinc binding, RNAP structure, and mechanism.
Materials and methods
Mutagenesis of the cloned rpoC gene
The pCYBβ′WT rpoC expression plasmid (Nedea et al. 1999) was used to generate rpoC mutants. The 3C3A mutation was generated by a single-step PCR using 3C3A, a 71-base DNA oligonucleotide as an upstream, mutagenic primer and a downstream primer that is complementary to rpoC codons 1400–1407 and contains an in-frame XhoI site. The 3C3A primer corresponds to rpoC codon positions 877–902 and introduces three Cys → Ala substitutions. In addition, it contains a SalI site, corresponding to the natural rpoC site at codon 877. The 2000-bp PCR product was cloned into pT7-blue blunt vector (Novagene), excised with SalI and XhoI, and cloned into appropriately treated pCYBβ′WT. After transformation in E. coli XL1-Blue, cells transformants were screened for the appearance of inducible β′ polypeptides on SDS–polyacrylamide gels, and the presence of mutation was confirmed by DNA sequencing.
To generate the C814A mutation, an 843-bp rpoC fragment was amplified with C814 DNA oligonucleotide as a downstream, mutagenic primer and an upstream primer corresponding to rpoC codons 541–548. The C814A oligonucleotide is complementary to rpoC codon positions 815–822 and introduces Cys-814 → Ala substitution. In addition, it contains a BspHI site, corresponding to the natural site at rpoC codon 819. The PCR product was cloned in pT7-blue, the 827-bp SphI–BspHI rpoC fragment was excised and subcloned into appropriately treated pACYC184. The resultant plasmid was treated with BspHI–HindIII and ligated with a 2500-bp BspHI–HindIII fragment of pCYBβ′WT containing the end of rpoC. Finally, a 2393-bp BsmI–XhoI fragment containing C814A mutation was recloned into the appropriately treated pCYBβ′WT. After transformation in E. coli XL1-Blue cells, transformants were screened for the appearance of inducible β′ polypeptides with increased mobility on SDS–polyacrylamide gels, and the presence of mutation was confirmed by DNA sequencing.
RNAP purification
The β′ subunits expressed from pCYBβ′WT or its derivatives contain a carboxy-terminal hexahistidine tag. pCYBβ′WT, pCYBβ′3C3A, and pCYBβ′C814A were transformed into 397C (Nedea et al. 1999) E. coli cells, and transformants were grown in 4 liters of Luria broth (LB) without addition of IPTG until late log phase. The cells were collected and disrupted by passing through a French press. The pellet, which contained most of the plasmid-borne β′ was used in RNAP reconstitution experiments (see below). The supernatant was used for RNAP purification by a combination of polyethyleneimine (Polymin-P) precipitation, metal ion affinity chromatography, and heparin–agarose affinity chromatography. RNAP was concentrated by filtration through a C-100 concentrator (Amicon, USA) to ∼1 mg/ml and stored in 50% (vol/vol) glycerol storage buffer at −20°C.
For RNAPFe purification, 397C cells transformed with pCYBβ′WT or pCYBβ′3C3A were grown in 2 liters of M9 minimal medium containing 50 μm FeCl3 and induced as described above. All components of the M9 medium, excluding CaCl2, Mg2SO4, and FeCl3, were treated with a Chelex-100 (Bio-Rad) ion exchanger (2 grams/liter) for 3 hr with agitation. CaCl2, Mg2SO4, and FeCl3 solutions were sterilized separately. The glassware was washed with nitric acid to remove traces of divalent ions.
Radical cleavage, affinity labeling, and transcription reactions
The standard 10-μl cross-linking reaction contained 0.5–1.0 μg of RNAP holoenzyme immobilized on ∼5 μl of Ni2+–NTA agarose (Qiagen, USA), 50 ng of the 106-bp DNA fragment containing the T7 A2 promoter (Severinov and Darst 1997), and 10 μm Rif6–GTP chimeric initiator molecule (Mustaev et al. 1994) in standard transcription buffer containing 40 mm Tris-HCl at pH 7.9, 40 mm KCl, and 10 mm MgCl2. Immobilized promoter complexes were incubated for 5 min at room temperature, and the excess of Rif-G was washed with excess of trancription buffer (Kashlev et al. 1996). Reaction was left in the final volume of 15 μl, followed by the addition of [α-32P]CTP (3000 Ci/mmole, DuPont-NEN) to a final concentration of 0.3 μm and 15-min incubation at 37°C. Unincorporated [α-32P]CTP was removed by washing the beads with a buffer containing 10 mm HEPES at pH 8.0, 50 mm NaCl, and 2 mm MnCl2. Reaction was left in 10 μl of the buffer, and 3′-deoxy- 3′-bromacetamidothymidine- 5′-triphosphate (Severinov et al. 1996) was added to the final concentration of 100 μm. Reaction proceeded for 30 min at 37°C, the beads were washed with 10 mm HEPES at pH 8.0, 50 mm NaCl, and 10 mm MgCl2, and affinity-labeled transcription complexes were immediately used for radical cleavage.
Radical cleavage was initiated by the addition of 0.03% H2O2 and 1 mm sodium ascorbate to the freshly prepared affinity-labeled transcription complexes. After incubation at 37°C for various times, reactions were terminated by the addition of thiourea to the final concentration of 100 mm. An equal volume of SDS-containing Laemmli loading buffer was added, and reaction products were immediately resolved by SDS-PAGE and revealed by autoradiography.
The standard steady-state transcription reaction contained in 15 μl of 50 ng of the T7 A1 promoter-containing DNA fragment, 500 mm CpA primer, 25 μm ATP, CTP, and GTP, and 0.3 μm [α-32P]UTP (3000 Ci/mmole, DuPont-NEN). Reaction proceeded for 15 min at room temperature, and the products were resolved by denaturing PAGE and revealed by autoradiography. In experiment shown in Figure 3B, 12 μl of transcription buffer containing 25 μg of RNAP was diluted to 50 μl with 8 m guanidine-HCl. The denaturing agent was then dialyzed away by an overnight microdialysis against 500 ml of reconstitution buffer (Borukhov and Goldfarb 1993). Transcription was performed directly in the reconstitution mixture by the addition of the T7 A1 template and transcription substrates as described above.
RNAP reconstitution
E. coli 397C cells (Christie et al. 1996) were used as a host for RNAP subunit expression to minimize contamination with wild-type β′. Standard RNAP reconstitution was performed essentially as described by Borukhov and Goldfarb (1993). In experiments that lacked Zn2+, all buffers were treated with Chelex-100 as described above, and 2 grams/liter Chelex was also present in the dialysis buffer during dialysis. Inclusion bodies containing overexpressed subunits were solubilized in denaturing buffer (Borukhov and Goldfarb 1993) and incubated overnight on ice in the presence of 10 mm EDTA prior to reconstitution. When RNAP subunits and subassemblies were renatured individually, β′, or an equimolar mixture of α and β were dissolved in the denaturing buffer to the final concentration of 0.25 mg/ml and dialyzed against two changes of 30-fold excess of the reconstitution buffer (with or without 10 μm of ZnCl2). Renatured β′ was mixed with α2β at molar ration 2:1, reactions were incubated for 30 min at 30°C with 1 molar equivalent of σ, followed by a steady state transcription assay performed as described above.
For chromatographic assays, individually renatured β′ was precipitated with ammonium sulfate, dissolved in 50 mm Tris-HCl at pH 7.9, 0.2 m KCl, 10% glycerol, 2 mm β-mercaptoethanol, and loaded on a Superose-6 column equilibrated in the same buffer and attached to Waters Biodiscovery 626 system. Chromatographic fractions were combined with α2β and assayed for transcription activity as described above.
Acknowledgments
This work was supported by a Career Award in Biomedical Sciences from the Borroughs Welcome Fund for Biomedical Research and National Institutes of Health (NIH) grant GM59295 to K.S. A.M. was supported by NIH grant GM30717 to Alex Goldfarb. D.M. is the recipient of a Charles and Johanna Busch postdoctoral fellowship. T.N. is in the graduate program of the Chemistry Department at Moscow State University, Russia.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
Recent crystallographic analysis of RNAP core from Thermus aquaticus established unequivocally that cysteine residues homologous to E. coli. β‘ Cys-814, -888, -895, and -898 coordinate zinc ion (G. Zhang, E. Campbell, L. Minakhin, C. Richter, K. Severinov, and S.A. Darst, in prep.)
Footnotes
E-MAIL severik@waksman.rutgers.edu; FAX (732) 445-5735.
References
- Allison LA, Moyle M, Shales M, Ingles CJ. Extensive homology among the largest subunits of eukaryotic and prokaryotic RNA polymerases. Cell. 1985;42:599–610. doi: 10.1016/0092-8674(85)90117-5. [DOI] [PubMed] [Google Scholar]
- Bartolomei MS, Corden JL. Localization of an alpha-amanitin resistance mutation in the gene encoding the largest subunit of mouse RNA polymerase II. Mol Cell Biol. 1987;7:586–594. doi: 10.1128/mcb.7.2.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borukhov S, Goldfarb A. Recombinant Escherichia coli RNA polymerase: Purification of individually overexpressed subunits and in vitro assembly. Protein Expr Purif. 1993;4:503–511. doi: 10.1006/prep.1993.1066. [DOI] [PubMed] [Google Scholar]
- Borukhov S, Severinov K, Kashlev M, Lebedev A, Bass I, Rowland GC, Lim PP, Glass RE, Nikiforov V, Goldfarb A. Mapping of trypsin cleavage and antibody-binding sites and delineation of a dispensable domain in the beta subunit of Escherichia coli RNA polymerase. J Biol Chem. 1991;266:23921–23926. [PubMed] [Google Scholar]
- Chatterji D, Wu FY-H. Selective substitution in vitro of an intrinsic zinc of Escherichia coli RNA polymerase with various divalent metals. Biochemistry. 1982;21:4651–4656. doi: 10.1021/bi00262a021. [DOI] [PubMed] [Google Scholar]
- Christie GE, Cale SB, Isaksson LA, Jin DJ, Xu M, Sauer B, Calendar R. Escherichia coli rpoC397 encodes a temperature-sensitive C-terminal frameshift in the beta′ subunit of RNA polymerase that blocks growth of bacteriophage P2. J Bacteriol. 1996;178:6991–6993. doi: 10.1128/jb.178.23.6991-6993.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte D, Narindrasorasak S, Sarkar B. In vivo and in vitro iron-replaced zinc finger generates free radicals and causes DNA damage. J Biol Chem. 1996;271:5125–5130. doi: 10.1074/jbc.271.9.5125. [DOI] [PubMed] [Google Scholar]
- Darst SA, Kubalek EW, Kornberg RD. Three-dimensional structure of Escherichia coli RNA polymerase holoenzyme determined by electron crystallography. Nature. 1989;340:730–732. doi: 10.1038/340730a0. [DOI] [PubMed] [Google Scholar]
- Darst SA, Edwards AM, Kubalek EW, Kornberg RD. Three-dimensional structure of yeast RNA polymerase II at 16 A resolution. Cell. 1991;66:121–128. doi: 10.1016/0092-8674(91)90144-n. [DOI] [PubMed] [Google Scholar]
- Dixon WJ, Hayes JJ, Levin JR, Weidner MF, Dombroski BA, Tullius TD. Hydroxyl radical footprinting. Methods Enzymol. 1991;208:380–413. doi: 10.1016/0076-6879(91)08021-9. [DOI] [PubMed] [Google Scholar]
- Gadal O, Shpakovski GV, Thuriaux P. Mutants in ABC10b, a conserved subunit shared by all three yeast RNA polymerases, specifically affect RNA polymerase I assembly. J Biol Chem. 1999;274:8421–8427. doi: 10.1074/jbc.274.13.8421. [DOI] [PubMed] [Google Scholar]
- Giedroc DP, Coleman JE. Structural and functional differences between the two intrinsic zinc ions of Escherichia coli RNA polymerase. Biochemistry. 1986;25:4969–4978. doi: 10.1021/bi00365a037. [DOI] [PubMed] [Google Scholar]
- Grachev MA, Kolocheva TI, Lukhtanov EA, Mustaev AA. Studies on the functional topography of Escherichia coli RNA polymerase. Highly selective affinity labeling by analogues of initiating substrates. Eur J Biochem. 1987;163:113–121. doi: 10.1111/j.1432-1033.1987.tb10743.x. [DOI] [PubMed] [Google Scholar]
- Gross GC, Fields DA, Bautz EK. Temperature-sensitive mutants of Escherichia coli with defects in the assembly of RNA polymerase in vitro. Eur J Biochem. 1977;81:333–338. doi: 10.1111/j.1432-1033.1977.tb11956.x. [DOI] [PubMed] [Google Scholar]
- Halling SM, Sanchez-Anzaldo FJ, Fukuda R, Doi RH, Meares CF. Zinc is associated with the β subunit of DNA-dependent RNA polymerase of Bacillus subtilis. Biochemistry. 1977;16:2880–2884. doi: 10.1021/bi00632a012. [DOI] [PubMed] [Google Scholar]
- Igarashi K, Fujita N, Ishihama A. Sequence analysis of two temperature-sensitive mutations in the alpha subunit gene (rpoA) of Escherichia coli RNA polymerase. Nucleic Acids Res. 1990;18:5945–5948. doi: 10.1093/nar/18.20.5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston M. Genetic evidence that zinc is an essential co-factor in the DNA binding domain of GAL4 protein. Nature. 1987;328:353–355. doi: 10.1038/328353a0. [DOI] [PubMed] [Google Scholar]
- Kashlev M, Nudler E, Severinov K, Borukhov S, Komissarova N, Goldfarb A. Histidine-tagged RNA polymerase of Escherichia coli and transcription in solid phase. Methods Enzymol. 1996;274:326–334. doi: 10.1016/s0076-6879(96)74028-4. [DOI] [PubMed] [Google Scholar]
- McCusker JH, Yamagishi M, Kolb JM, Nomura M. Suppressor analysis of temperature-sensitive RNA polymerase I mutations in Saccharomyces cerevisiae: Suppression of mutations in a zinc-binding motif by transposed mutant genes. Mol Cell Biol. 1991;11:746–753. doi: 10.1128/mcb.11.2.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustaev A, Zaychikov E, Severinov K, Kashlev M, Polyakov A, Nikiforov V, Goldfarb A. Topology of the RNA polymerase active center probed by chimeric rifampicin-nucleotide compounds. Proc Natl Acad Sci. 1994;91:12036–12040. doi: 10.1073/pnas.91.25.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedea EC, Markov D, Naryshkina T, Severinov K. Localization of E. coli rpoC mutations that affect RNA polymerase assembly and activity at high temperature. J Bacteriol. 1999;181:2663–2665. doi: 10.1128/jb.181.8.2663-2665.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyakov A, Severinova E, Darst SA. Three-dimensional structure of E. coli core RNA polymerase: Promoter binding and elongation conformations of the enzyme. Cell. 1995;83:365–373. doi: 10.1016/0092-8674(95)90114-0. [DOI] [PubMed] [Google Scholar]
- Schultz P, Celia H, Riva M, Sentenac A, Oudet P. Three-dimensional model of yeast RNA polymerase I determined by electron microscopy of two-dimensional crystals. EMBO J. 1993;12:2601–2607. doi: 10.1002/j.1460-2075.1993.tb05920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrutton MC, Wu C-W, Goldthwait DA. The presence and possible role of zinc in RNA polymerase obtained from Escheria coli. Proc Natl Acad Sci. 1971;68:2497–2501. doi: 10.1073/pnas.68.10.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinov K, Darst SA. A mutant RNA polymerase that forms unusual open promoter complexes. Proc Natl Acad Sci. 1997;94:13481–13486. doi: 10.1073/pnas.94.25.13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinov K, Markov D, Nikiforov V, Severinova E, Landick R, Darst SA, Goldfarb A. Streptolydigin-resistant mutants in an evolutionary conserved region of the β′ subunit of Escherichia coli RNA polymerase. J Biol Chem. 1995;270:23926–23929. doi: 10.1074/jbc.270.41.23926. [DOI] [PubMed] [Google Scholar]
- Severinov K, Mustaev A, Kukarin A, Muzzin O, Bass I, Darst SA, Goldfarb A. Structural modules of the large subunits of RNA polymerase. Introducing archaebacterial and chloroplast split sites in the beta and beta′ subunits of Escherichia coli RNA polymerase. J Biol Chem. 1996;271:27969–27974. doi: 10.1074/jbc.271.44.27969. [DOI] [PubMed] [Google Scholar]
- Solaiman D, Wu FY. Intrinsic zinc ion is essential for proper conformation of active Escherichia coli RNA polymerase. Biochemistry. 1984;23:6369–6377. doi: 10.1021/bi00321a013. [DOI] [PubMed] [Google Scholar]
- ————— Preparation and characterization of various Escherichia coli RNA polymerases containing one or two intrinsic metal ions. Biochemistry. 1985;24:5077–5083. doi: 10.1021/bi00340a018. [DOI] [PubMed] [Google Scholar]
- Sweetser D, Nonet M, Young RA. Prokaryotic and eukaryotic RNA polymerases have homologous core subunits. Proc Natl Acad Sci. 1987;84:1192–1196. doi: 10.1073/pnas.84.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Severinov K, Goldfarb A, Ebright R. Rapid RNA polymerase genetics: One-day, no-column preparation of reconstituted recombinant Escherichia coli RNA polymerase. Proc Natl Acad Sci. 1995;92:4902–4906. doi: 10.1073/pnas.92.11.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treich I, Riva M, Sentenac A. Zinc-binding subunits of yeast RNA polymerases. J Biol Chem. 1991;266:21971–21976. [PubMed] [Google Scholar]
- Vallee BL, Auld DS. Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry. 1990;29:5647–5659. doi: 10.1021/bi00476a001. [DOI] [PubMed] [Google Scholar]
- Voutsina A, Riva M, Carles C, Alexandraki D. Sequence divergence of the RNA polymerase shared subunit ABC14.5 (Rpb8) selectively affects RNA polymerase III assembly in Saccharomyces cerevisiae. Nucleic Acids Res. 1999;27:1047–1055. doi: 10.1093/nar/27.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Severinov K, Loizos N, Fenyö D, Heyduk E, Heyduk T, Chait BT, Darst SA. Determinants for Escherichia coli RNA polymerase assembly within the β subunit. J Mol Biol. 1997;270:648–662. doi: 10.1006/jmbi.1997.1139. [DOI] [PubMed] [Google Scholar]
- Weilbaecher R, Hebron C, Feng G, Landick R. Termination-altering amino acid substitutions in the β′ subunit of Escherichia coli RNA polymerase identify regions involved in RNA chain elongation. Genes & Dev. 1994;8:2913–2927. doi: 10.1101/gad.8.23.2913. [DOI] [PubMed] [Google Scholar]
- Wu C-W, Wu FY-H, Speckhard DC. Subunit location of the intrinsic divalent metal ions in RNA polymerase from Escherichia coli. Biochemistry. 1977;16:5449–5454. doi: 10.1021/bi00644a008. [DOI] [PubMed] [Google Scholar]
- Yang X, Price CW. Streptolydigin resistance can be conferred by alterations to either the beta or beta′ subunits of Bacillus subtilis RNA polymerase. J Biol Chem. 1995;270:23930–23933. doi: 10.1074/jbc.270.41.23930. [DOI] [PubMed] [Google Scholar]
- Zaychikov E, Martin E, Denissova L, Kozlov M, Markovtsov V, Kashlev M, Heumann H, Nikiforov V, Goldfarb A, Mustaev A. Mapping of catalytic residues in the RNA polymerase active center. Science. 1996;273:107–109. doi: 10.1126/science.273.5271.107. [DOI] [PubMed] [Google Scholar]
- Zillig W, Palm P, Heil A. Function and reassembly of subunits of DNA-dependent RNA polymerase. In: Losick R, Chamberlin M, editors. RNA polymerase. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1977. pp. 101–125. [Google Scholar]