

Abstract
Despite a strong correlation between telomerase activity and malignancy, the outcome of telomerase inhibition in human tumor cells has not been examined. Here, we have addressed the role of telomerase activity in the proliferation of human tumor and immortal cells by inhibiting TERT function. Inducible dominant-negative mutants of hTERT dramatically reduced the level of endogenous telomerase activity in tumor cell lines. Clones with short telomeres continued to divide, then exhibited an increase in abnormal mitoses followed by massive apoptosis leading to the loss of the entire population. This cell death was telomere-length dependent, as cells with long telomeres were viable but exhibited telomere shortening at a rate similar to that of mortal cells. It appears that telomerase inhibition in cells with short telomeres lead to chromosomal damage, which in turn trigger apoptotic cell death. These results provide the first direct evidence that telomerase is required for the maintenance of human tumor and immortal cell viability, and suggest that tumors with short telomeres may be effectively and rapidly killed following telomerase inhibition.
Keywords: TERT, telomere, dominant negative, proliferation, cancer
The termini of most eukaryotic chromosomes are composed of terminal repeats called telomeres. These repeats serve to protect chromosome integrity and ensure complete replication of essential genes. Telomeric repeats are produced de novo by the ribonucleoprotein enzyme telomerase, a large molecular weight complex containing both an RNA subunit and multiple protein components (for review, see Greider 1996, 1998; Harley and Sherwood 1997).
Genes encoding telomerase protein components have been identified in single-celled eukaryotes such as yeast (Lundblad and Szostak 1989; Lendvay et al. 1996; Counter et al. 1997; Lingner et al. 1997; Nakamura et al. 1997) and ciliates (Collins et al. 1995; Lingner et al. 1997; Bryan et al. 1998; Collins and Gandhi 1998). These telomerase components include Tetrahymena p80 and p95, Saccharomyces cerevisiae telomerase reverse transcriptase EST2 and its homolog in fission yeast Schizosaccharomyces pombe, ciliate telomerase reverse transcriptase p123 and p133, and Oxytricha telomerase reverse transcriptase.
The cloning of telomerase genes in lower eukaryotes has facilitated the identification of homologs in mammals. A homolog to the tetrahymena P80 protein, TEP1 (TP1/TLP1) has been identified in humans, rats, and mice (Harrington et al. 1997a; Nakayama et al. 1997). TEP1 binds the telomerase RNA and associates with telomerase activity in tumor cell lines (Harrington et al. 1997a). Mammalian homologs to the telomerase reverse transcriptases (TERTs) cloned in yeast and ciliates have also been identified (Harrington et al. 1997b; Kilian et al. 1997; Meyerson et al. 1997; Nakamura et al. 1997; Greenberg et al. 1998). The mammalian protein TERT (hTRT/hEST2/TP2/TCS1) contains consensus reverse transcriptase motifs and is associated with telomerase activity. Mutational analysis of both yeast and human TERT, in vitro reconstitution of telomerase activity with hTERT and the RNA subunit, and the high phylogenic conservation of TERT, suggest that TERT and the telomerase RNA form the core catalytic function of the multicomponent telomerase holoenzyme (Harrington et al. 1997b; Lingner et al. 1997; Weinrich et al. 1997; Beattie et al. 1998; Nakayama et al. 1998).
A number of experiments suggest that telomerase activity is important for the long-term replicative capability of normal cells and unicellular organisms. Primary human fibroblasts do not express telomerase activity and exhibit a limited ability to replicate in culture. Such cells cease to divide after 30–50 doublings and become senescent. This, and the observation that telomere length correlates with replicative capacity (Hayflick 1961; Counter et al. 1992) has led to the telomere hypothesis, which postulates that telomere length specifies the number of divisions a cell can undergo prior to senescence (Harley 1991; Harley et al. 1990). This hypothesis has gained recent support with the demonstration that overexpression of the TERT subunit can lead to telomere lengthening and an extension of replicative lifespan in telomerase-negative fibroblasts in culture (Bodnar et al. 1998; Vaziri and Benchimol 1998). Additionally, disruption or mutation of telomerase genes in lower organisms has shown that telomerase activity in these organisms is largely responsible for telomere maintenance and subsequent long-term survival (Lundblad and Szostak 1989; Lendvay et al. 1996; Lingner et al. 1997; Nakamura et al. 1997). Mice and mouse ES cells lacking the telomerase RNA gene (mTR) both have shown telomere shortening and can eventually impair long-term viability (Lee et al. 1998; Niida et al. 1998).
Unlike normal human fibroblasts, most tumor cell populations possess infinite replicative capacity. Additionally, many studies have demonstrated a strong correlation between telomerase activity and malignancy (for review, see Shay and Bacchetti 1997). In agreement, the catalytic subunit of human telomerase hTERT is upregulated in tumor and immortalized cells (Meyerson et al. 1997; Nakamura et al. 1997) suggesting that TERT expression and telomerase activity play a role in the immortality of tumor cells. However, some exceptions to this model exist. Human tumors and cell lines that lack telomerase activity have been reported. Mechanisms for alternate lengthening of telomeres have been postulated on the basis of mechanisms discovered in yeast (Bryan and Reddel 1997; Lundblad 1997). It is not known to what extent these or other mechanisms may be functioning in telomerase-positive tumor cells. Additionally, mice lacking the gene for the mTR possess no telomerase activity yet are viable for up to six generations (Blasco et al. 1997). Cell lines have been derived from these mice, which are competent to form tumors in nude mice, suggesting that telomerase activity per se is not required for tumor formation (Blasco et al. 1997). Late generation mice doubly null for mTR and INK4a tumor suppressor genes showed reduction in tumor formation and oncogenic potential (Greenberg et al 1999), indicating that telomerase activity is important in tumor formation in cells with critically short telomeres. In contrast, combination of lack of telomerase activity and p53 deficiency in mTR/p53 null mice also cooperated to initiate tumor formation in aging mTR null mice (Chin et al. 1999). However, the effect of telomerase inhibition on tumor cells has not been examined directly.
To better understand mechanisms underlying telomere maintenance, and to provide a model to predict the outcome of pharmacologic inhibition of telomerase in human tumors, we set out to specifically inhibit telomerase activity in human tumor and immortal cell lines. Previously, antisense oligonucleotides against human telomerase RNA have been used to inhibit telomerase activity in human malignant glioma cells (S. Kondo et al. 1998a,b; Y. Kondo et al. 1998). These tumor cells demonstrated increased sensitivity to DNA damaging agents and suppressed tumor growth due to apoptosis (Kondo et al. 1998). The molecular cloning of hTERT subunit has provided a tool to directly examine the role of telomerase activity in human tumor cell proliferation. Here we demonstrate that inhibition of telomerase activity via dominant-negative mutants of hTERT in human immortal cells can lead to rapid and dramatic apoptosis.
Results
TERT aspartic acid mutants act as dominant-negative inhibitors
We and others have identified previously several point mutants in the reverse transcriptase (RT) motifs of the telomerase catalytic subunit hTERT, which exhibit dramatically reduced telomerase activity (Harrington et al. 1997; Weinrich et al. 1997b; Nakamura et al. 1997). To determine whether these mutants would act in a dominant-negative fashion, four different mutants and the wild-type hTERT were each placed under the control of a CMV promoter followed by an IRES element and a reporter gene encoding an enhanced green fluorescent protein (EGFP). The resulting constructs express both the hTERT protein and the GFP reporter from the same transcript. These constructs were transfected into telomerase-positive human 293 cells. Each set of transfectents was sorted into three groups on the basis of the GFP expression level as follows: nontransfected (non), low expressors (low), and high expressors (high) by FACS. Endongenous TERT protein was undetectable because of its low expression level. Cells were then evaluated for telomerase activity (Fig. 1A) and hTERT protein levels (Fig. 1B). Cells expressing low or high levels of mutant hTERT exhibited dramatically reduced telomerase activity relative to the nontransfected cells from each population. In contrast, cells expressing either the wild-type hTERT protein or cells transfected with the GFP vector alone showed normal levels of telomerase activity. Each of the mutants could be coimmunoprecipitated with both TEP1 and the telomerase RNA (data not shown), further supporting the notion that each could interact with the telomerase complex and act in a dominant-negative manner to block the activity of endogenous TERT protein.
Figure 1.


TERT reverse transcriptase motif mutants function as dominant-negative mutants. (A) Telomerase activity of 293 cell extracts transiently transfected with GFP-selectable TERT proteins and assayed by the TRAP protocol. Each transfected cell was sorted into three populations as follows: not transfected (Non); low expression of GFP (Low); high expression of GFP (High). The transgenes used for transfection are TERT 3-1 mutant (3.1); TERT 5-1 mutant (5.1); TERT 5-2 mutant (5.2); 5.2, TERT 5-1,2 mutant (5.1); wild-type TERT (w.t. TERT); plasmid containing GFP only (GFP vector). Each transfected lysate (0.5 μg) was used in the TRAP assay. The seven lanes on the right represent TRAP activity of buffer alone (−) and serial dilution of nontransfected 293 lysate, the numbers indicate micrograms of total 293 lysate assayed. (Arrowhead) The internal control band of the PCR reaction used in TRAP. (B) Western analysis of the same lysates (15 μg/lane) as in A with an anti-hTERT peptide antibody (See Material and Methods). The endogenous TERT proteins present in nontransfected cells cannot be detected by this antibody.
To study the effects of long-term expression of the dominant-negative mutants on tumor cells, stable cell lines were generated in two telomerase-positive human cell lines. The A431 line is an epidermoid tumor cell line (derived from an 85-year-old patient), whereas 293 is an embryonic kidney cell line that was originally immortalized in culture with SV40 T antigen. Both cell lines exhibit a transformed phenotype and both cell lines efficiently form tumors in nude mice (data not shown). To provide control over telomerase inhibition, these cells were first transfected with a Drosophila ecdysone receptor, then with the mutant or wild-type hTERTs that were placed under the control of the ecdysone-inducible promoter element such that expression of the hTERT transgenes could be regulated with the ecdysone analog muristerone.
We first examined the telomere length of these stable cell lines prior to induction by measuring the length of the telomere restriction fragment (TRF) (Fig. 2A). All the A431 stable cell lines showed a homogeneous pattern of unusually short-telomere length (1–4 kb) (Fig 2A). In contrast, 293 stable cell lines showed a great deal of clone-to-clone variation in telomere length. Clonal stable cell lines exhibited a range of average TRF length from ∼12 to 3 kb, although none of the cell lines exhibit TRFs as short as those observed in the majority of the A431 stable clones.
Figure 2.



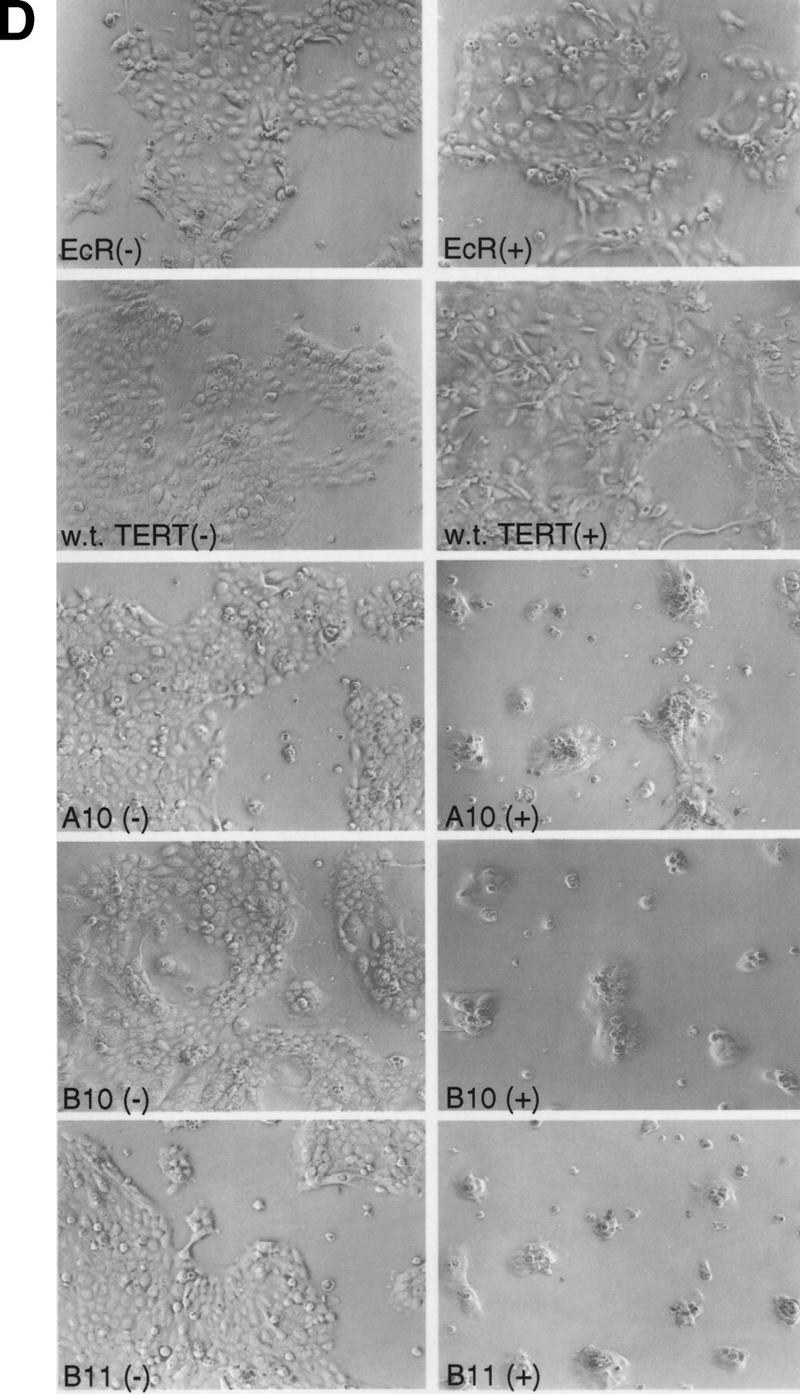
Effects of dominant-negative TERT mutants on A431 cells. (A) Telomere restriction fragment length of A431 and 293 stable clones (see Material and Methods). The sizes (kb) of molecular mass markers are labeled. The three sets of samples were run on different percentages of agarose gel, thus showing the different running patterns; A clones, TERT 3-1 mutant; B clones, TERT 5-1 mutants; C clones, TERT 5-1,2 mutants; D clones, wild-type TERT, and E clones, TERT 5-2 mutants. (B) Muristerone induces the expression of dominant-negative TERT mutants. Cell extracts (15 μg/lane) from A431 stable clones with or without the induction of muristerone were detected by Western blotting. The hTERTs are labeled. The same antibody as in Fig. 1 was used. (C) Expression of dominant-negative TERT mutants inhibits telomerase activity in A431 cells. TRAP assay of the same lysate (1 μg) as in B was performed. (Lanes 17–20) The activity of serial dilution of parental A431 cell lysate. The internal control for PCR reaction is shown as labeled. (D) Morphology of A431 cell lines expressing control vector (EcR), wild-type TERT (D2), and mutant TERTs (A10, B10, B11).
Telomerase inhibition in A431 cells leads to rapid cell death
We then investigated whether expression of dominant-negative hTERT mutants would have any effect on A431 cells, the cell line exhibiting the shortest average telomere length. Six isolated mutant clones and a wild-type hTERT clone were all highly inducible as assayed by muristerone-dependent protein expression (Fig. 2B). The induction of dominant-negative mutants by muristerone dramatically inhibited the telomerase activity in these cells, whereas the induction of the wild-type hTERT protein resulted in slightly increased telomerase activity (Fig. 2C). Each clone was split into two populations and grown in the presence or absence of exogenously added muristerone. Several days after induction, all of the mutant cell lines exhibited morphological changes accompanied by the apparent death of the cells over a 5–10 day period (Fig. 2D). The entire population rounded up off the plastic, exhibiting cytoplasmic blebbing and chromatin condensation, features of apoptosis (Fig 2D). In contrast, muristerone induction of clones expressing the wild-type hTERT protein, which differ from the mutants by only a single amino acid, exhibited normal morphology, although their telomeres were as short as those of mutant clones. The parental cell line expressing the ecdysone receptor also appeared normal in the presence of muristerone. We continuously maintained the mutant cells in the presence of muristerone. By day 32, all of the originally plated cells had died and a few individual surviving clones were proliferating. Western analysis showed that all of the surviving clones had lost the expression of the dominant-negative protein and exhibited wild-type levels of telomerase activity (data not shown).
A431 cell death is apoptotic
To understand the mechanistic outcome of telomerase inhibition on tumor viability, we next studied the kinetics of protein accumulation, growth, proliferation, and apoptosis in the A431 cell lines on the induction of mutant TERT. A431 EcR, wild-type, and mutant TERT cell lines were grown over an 8-day period in the presence or absence of muristerone. Both wild-type and mutant TERT protein was observed by 6 hr postinduction, reaching steady-state levels by 24 hr (Fig. 3A). The growth curves of these cells were examined as shown in Figure 3B. The EcR, wild-type, and mutant cell lines had comparable growth rate when uninduced (Fig. 3A). In contrast, growth curves from cells expressing the dominant-negative mutants showed little-to-no increase in cell number in comparison with EcR and wild-type cells (Fig. 3B), suggesting that telomerase inhibition exerted a specific effect on tumor cell viability. We also observed a telomerase-independent effect of muristerone on EcR-expressing parental cells, which resulted in a reduction in the growth rate of the EcR and wild-type cells in the presence of muristerone (Fig. 3B). The effect appeared to be caused by a decrease in the percentage of S-phase cells; no increase in the apoptotic rate was observed (see below).
Figure 3.


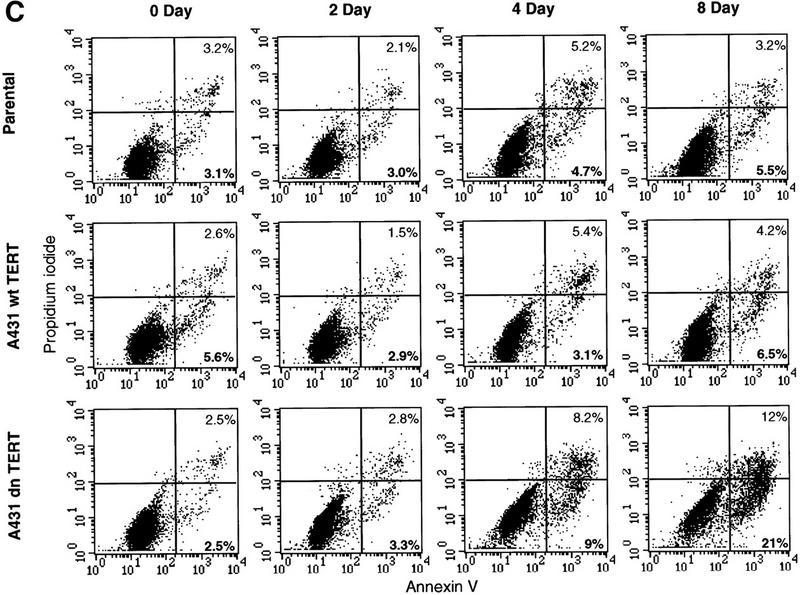

Kinetics of apoptosis induced by mutant TERT in A431 cells. Each A431 cell line (EcR, wild-type clone D2, and mutant clone B11) was plated into multiple sets of 60-mm tissue culture dishes at a density of 3 × 105 cells/dish and allowed to attach overnight and recover from trypsinization. Cells were then induced by changing to medium containing 1 μm muristerone. Sets of cells from each line were used for Western blotting, apoptosis analysis, and cell cycle analysis at indicated days. (A) Kinetics of exogenous TERT expression. One set of cells per indicated day was lysed, and the total cell lysate (5 μg/lane) was run on SDS-PAGE and Western blotted with the anti-TERT peptide antibody as in Fig. 1. (Arrow) The position of hTERT. (B) Growth curve of A431 cells. Equal number of cells (1.2 × 105) from each cell line were plated into six-well plates in triplicate in the presence or absence of muristerone (1 μm) and allowed to grow for 7 days with medium being changed on day 4. Cell numbers were counted each day with a hemocytometer. Data shown are mean of the triplicate from one of three experiments all showing similar results. (□) EcR (Left) Uninduced; (⋄) w.t. TERT; (●) B11. (Right) Induced; (□) ECR; (▵) w.t. TERT; (●) B11. (C) Flow cytometry analysis of cells double labeled with annexin V and PI. One set of cells from each cell line was harvested at indicated days. Cells were then stained and analyzed as described in Materials and Methods. (D) Anaphase chromosome bridges induced by mutant TERT. EcR, wild-type, and mutant A431 cells were seeded in two-well chamber slides. Cells were induced for 3 days and then fixed and stained by propidium iodide. Shown are four typical anaphase cells with bridging chromosomes in mutant A431 cells.
Cytological observation of the telomerase-inhibited cells at later time points suggested an appearance consistent with programmed cell death. To determine whether the cells were undergoing apoptosis, and to examine the time course of cell death, we performed flow cytometry analysis of living cells double labeled with annexin V and propidium iodide (PI) over the 8-day induction period (Fig. 3C). An early feature of apoptotic cells is the loss of plasma membrane asymmetry reflected by the exposure of phophatidylserine (PS) from the inner leaflet to the outer leaflet (Fadok et al. 1992). Thus the high-affinity PS-binding protein Annexin V has been used widely to detect early stage apoptotic cells (Vermes et al. 1995). When combined with PI, which stains DNA of cells with permeable membranes, this approach allows a further distinction of early apoptotic (annexin V+/PI−) and late apoptotic/necrotic (annexin V+/PI+) cells. EcR and wild-type A431 cells showed an average of 3.4% of annexin V+/PI+ cells and 4.3% of annexin V+/PI− cells. No increase of apoptotic cell death in these two cell lines could be detected by annexin V and PI. Nor was an increase in apoptosis observed in the mutant A431 following 2 days of induction. However, by day 4, a significant increase of both early (9%) and late apoptotic cells (8.2%) were detected in mutant A431 cells (Fig. 3C). By day 8, mutant A431 had undergone massive apoptosis. Annexin V and PI staining detected 21% annexin V+/PI− cells and 12% annexin V+/PI+ cells and these cells exhibited the same morphology as shown in Figure 2D. The increased apoptosis of A431 cells induced by mutant TERT expression were also detected by a different approach, TUNEL (TDT-mediated dUTP nick end labeling) analysis of the DNA stand breaks (data not shown). Parallel cell cycle analysis of the same set of cells by 5-bromo-2′-deoxyuridine (BrdU) incorporation and PI staining showed that EcR, wild-type, and mutant A431 cells all continued DNA synthesis and cell divisions throughout the induction period (data not shown). The three cell lines also showed similar cell cycle profiles (data not shown). In addition, the apoptosis assays and the cell cycle analysis revealed the nature of the muristerone-induced cell growth inhibition observed in all three cell lines. The percentage of cells in S phase was reduced in all three cell lines following the induction of muristerone, whereas the apoptotic rate appeared to be unaltered (Fig. 3C; data not shown), suggesting that the nonspecific muristerone growth inhibition was due to an increase in cell doubling time.
Toward the elucidation of a mechanism for the cell death observed in the telomerase-inhibited cells, mitoses from EcR, wild-type, and mutant A431 cells were examined for evidence of chromosomal alterations. A significant increase in the occurrence of anaphase chromosomal bridges, the result of chromosome end fusion, was observed in mutant A431 cells after induction by muristerone (Fig. 3D; Table 1), whereas no change in the frequency of abnormal mitoses was observed in the parental cell line or cells expressing wild-type TERT (Table 1). Previous experiments have also drawn a correlation between loss of telomerase activity, telomere shortening, and an increase in chromosome fusion events (Blasco et al. 1997). Taken together, it appears that expression of dominant-negative TERT protein in A431 cells inhibits telomerase activity while permitting continued cell division. This presumably results in further telomere shortening, which leads to chromosome damage and the subsequent activation of cellular apoptotic pathways.
Table 1.
Induction of aberrant anaphase mitoses events by mutant TERT proteins
| Cell line
|
Induction
|
No. of cells examined
|
Fraction with aberrant eventsa (%)
|
|---|---|---|---|
| A431-EcR | − | 50 | 16 |
| A431-EcR | + | 50 | 16 |
| A431-wt TERT | − | 50 | 15 |
| A431-wt TERT | + | 50 | 16 |
| A431-dn TERT | − | 50 | 14 |
| A431-dn TERT | + | 60 | 58 |
Fraction of aberrant anaphase mitoses were determined by calculating the ratio of mitosis events showing either chromosome bridges or lagging chromosomes to the total mitosis events observed.
Apoptosis is telomere-length dependent
We then asked whether the induction of apoptosis in A431 cells by inhibition of telomerase activity is dependent on the telomere length. We were unable to observe a change in telomere length in the A431 cells because of the rapid loss of the population. Nor were we able to isolate any A431 clones with long telomeres. Therefore, we examined the growth effect of dominant-negative hTERT mutants on 293 clones with long telomeres. Figure 4A shows the level and extent of TERT protein expression and inducibility for two mutant (A1, A2) and one wild-type TERT (D1)-expressing cell lines. Although the expression of TERT proteins was induced to a higher level in A2 and wild-type lines, all three lines exhibited a constitutive expression of TERT transgenes. The constitutive expression of dominant-negative TERT in A2 was sufficient to reduce endogenous telomerase activity in the uninduced cells (Fig. 4B). It is important to point out that endogenous TERT protein is present at very low levels (undetectable by Western blotting; data not shown) such that even modest amounts of expressed mutant protein may act as a dominant negative. Each 293 cell line was divided into two populations, and one was grown in the presence of muristerone. Telomere maintenance in these cell lines was determined by measuring the TRF length (Fig. 4C). Initial TRFs for A1 and A2 and wild-type lines were relatively long at 10 and 12 kb, respectively (Fig. 4C, lanes 1,5). Induced and uninduced populations from A1 and A2 that expressed the dominant-negative protein and exhibited reduced telomerase activity demonstrated pronounced telomere shortening. The rate of shortening was determined to be ∼60 nucleotides per population doubling, similar to the rate reported in telomerase negative fibroblasts (Counter et al. 1992). In contrast, the wild-type TERT-expressing cell line exhibited no changes in telomere length (Fig. 4C). No difference in growth rate between induced, uninduced, or wild-type TERT-expressing cell populations was observed (Fig. 4D). Telomere shortening, inhibition of telomerase activity, and presence of the dominant-negative protein persisted for >109 population doublings, until the average TRF length approached 4 kb. Subsequent passages in A1 and A2 revealed the loss of the dominant-negative protein, with the subsequent reactivation of telomerase activity and a stabilization of telomere length (data not shown). In contrast, the wild-type TERT-expressing cell line maintained expression of the hTERT protein over the same number of population doublings (data not shown). This suggested that the dominant-negative transgene was subject to telomere length-dependent negative selection, perhaps due to reduced viability of cells with critically short telomeres.
Figure 4.

Effects of dominant-negative TERT mutants on 293 cells with long telomeres. (A) Western analysis of 293 wild-type clones (D1) and mutant clones A1 and A2 by the same method as described in Fig. 2. The expression of TERTs are indicated by arrowhead. (B) TRAP assay of the wild-type, A1, and A2 clones. (Lanes 7–10) The serial dilution of nontransfected 293 lysate. One microgram of each lysate was assayed. (C) TRF of wild-type, A1, and A2 clones over a period of 109 population doublings of growth in the presence of 2 μm muristerone. The same method as described in Fig. 2 was used. (D) Comparison of long-term growth of wild-type and mutant 293 clones . Cells were grown in the presence or absence of muristerone and passed every 3 days as described in Materials and Methods and counted per passage. Data represent population doublings of these clones in each passage over a 40-passage period. (□) 293WT; (♦); top, 293 A1; bottom, 293A2.
Telomerase inhibition in short TRF 293 cells leads to loss of viability followed by telomerase reactivation
To determine whether telomerase inhibition affected the viability of 293 cells with relatively shorter telomeres, 293 mutant TERT lines with short TRF length were identified. Dominant-negative lines B12 and C4 were both highly inducible (Fig. 5A) and exhibited TRF lengths of ∼4 and 3 kb, respectively (Fig. 2A). A wild-type TERT-expressing cell line (D11) had similar TRF length (Fig. 2A) and was used as a control. Each cell line was divided into two populations grown in the presence or absence of muristerone and passed every 3 days. On induction, both B12 and C4 exhibited a significant change in growth properties (Fig. 5C). The induced cells exhibited variable altered morphology and failed to reach confluence in the first few passages (Fig. 5C). Colony formation assays of the cells from passage 1 were also performed, demonstrating a decrease in the number of viable cells (Fig. 5D; Table 2). Uninduced B12 and C4, and both induced and uninduced wild-type populations appeared normal and did not show any decrease in population doublings per passage (Fig. 5C; data not shown). At passage 4, the inhibited cell growth rates in B12 and C4 began to recover and by passage 8, both grew at control rates (Fig. 5C). Examination of telomerase activity at each passage revealed a reactivation of telomerase activity at passage 4 despite the continued presence of epitope-tagged dominant-negative protein (Fig. 5A,B). We were unable to observe any significant change in the size of the TRF over this relatively short duration of telomerase inhibition (data not shown). TEP1 protein levels and telomerase RNA levels remained the same in all passages. No change in endogenous hTERT mRNA was observed as assayed by quantitative RT–PCR with primers in the 3′ untranslated region (data not shown).
Figure 5.




Effects of dominant-negative TERT mutants on 293 cells with short telomeres. (A) Western analysis of 293 mutant clones B12 and C4. Cells were grown in the presence or absence of muristerone and passed every 3 days for a total of nine passages. (arrowhead) Expression of mutant TERTs. Mouse Anti-FLAG monoclonal antibody was used. (B) TRAP assay of B12 and C4 at each passage as in A. (Lanes 1–5) The serial dilution of 293 lysate. One microgram from each lysate was assayed. (C) Comparison of growth of B12 and C4 in the presence and absence of muristerone. Cells were grown, passed, and counted as in A. Data represent population doublings in each passage. (Left) (□) 293B12 (−); (♦) 293B12 (+). (Right) (□) 293C4 (−); (♦) 293C4 (+). (D) Colony formation assay of EcR-293, clone B12, and clone C4 as described in Materials and Methods. (−) Uninduced; (+) induced.
Table 2.
Colony formation of 293 clones
| Cell type
|
Colonies ≥1 mm
|
|||
|---|---|---|---|---|
| experiment 1
|
experiment 2
|
|||
| −muristerone
|
+muristerone
|
−muristerone
|
+muristerone
|
|
| 293–EcR | N.D. | N.D. | 72 | 25 |
| 293–D1 | 49 | 41 | N.D. | N.D. |
| 293–B12 | 25 | 3 | 37 | 0 |
| 293–C4 | 15 | 0 | 45 | 1 |
(N.D.) Not determined.
Discussion
We have generated and expressed dominant-negative telomerase catalytic subunits to determine the outcome of telomerase inhibition of human tumor and immortalized cells. Surprisingly, we observed rapid loss of viability in telomerase-inhibited cells carrying short TRFs. The effect appears to be telomere-length dependent as clones with long telomeres are unaffected by telomerase inhibition. Rather, telomerase inhibition shortened the telomere length of these cells at a rate of ∼60 nucleotides per population doubling, the same rate of telomere shortening as that of telomere-negative cells. The effect appears to be specific to telomerase inhibition because overexpression of the wild-type TERT subunit, which differs from the mutants by a single amino acid, does not negatively affect viability or telomere shortening. Because the exogenously expressed proteins are present at levels many fold higher than the endogenous TERT protein, it is formally possible that the dominant-negative subunit is acting nonspecifically to activate apoptotic pathways, perhaps by titration of some factor. Such a mechanism is unlikely, as expression of the wild-type TERT would be expected to act in a similar manner. Furthermore, the time course and kinetics of the apoptotic response suggest that further cell divisions are required prior to activation of apoptosis. Dominant-negative protein accumulates within 24 hr, whereas increased rates of apoptosis are not observed until 4 days after induction.
Telomerase activity is a dominant mechanism for telomere maintenance in telomerase-positive cells
These results suggest that telomerase activity is the dominant mechanism providing telomere maintenance and long-term viability to human immortalized cells. The ability of cells with long telomeres to proliferate in the absence of telomerase demonstrates that telomerase activity is not required for basic replicative functions of these cells. Instead, telomerase activity appears to be required to maintain a minimum telomere length. Alternate pathways for telomere maintenance in human immortalized cells have been suggested by the identification of telomerase-negative human cells that exhibit long telomeres (Bryan and Reddel 1997). This alternate lengthening of telomeres (ALT) has been postulated to function via recombination on the basis of the discovery of such a mechanism in yeast (Lundblad 1997). It might be expected that this mechanism may function effectively in telomerase-inhibited tumor populations; however, it was not observed in these experiments. Instead, cells exhibited three fates following telomerase inhibition, cell death, loss of the inhibitory transgene, or reactivation of telomerase activity despite the presence of the dominant-negative TERT.
Rapid apoptosis in telomerase-inhibited cells
Others have speculated that telomerase inhibition may lead to a phenotypic lag in which cells would continue to divide until the point at which the telomeres became critically short. We observe here that cells with short telomeres appear to require telomerase activity even for short-term viability. Therefore, we postulate that these cells are replicating with critically short telomeres, and as such may require telomerase activity at nearly every cell division. Inhibition of telomerase activity in these cells does not cause senescence, but loss of viability through activation of apoptotic pathways. Evidence of chromosomal end joining was observed in these telomerase-inhibited cells, suggesting that telomerase normally functions to protect the chromosome ends from fusion events. The signal to activate apoptosis is not known, although the presence of chromosome breakage events suggests that the signal may involve sensors of DNA damage. It is important to note that one such sensor of DNA damage, p53, is functionally absent from both A431 and 293 cell lines (these 293 cells harbor the SV40 T Antigen). Chromosome bridges were also observed in human immortal cells that expressed dominant-negative TRF2 (Karlseder et al. 1999). However, unlike our studies, the anaphase bridges were induced in TRF2 dominant-negative lines despite the presence of long telomeres. In addition, the apoptosis observed in TRF2-inhibited cells is p53 dependent (Karlseder et al. 1999), whereas in our experiments, p53 is apparently not required for the apoptotic response to telomerase inhibition. This system defines another apoptotic signal, independent of p53, which is activated between day 2 and day 4 following induction of the dominant-negative TERT protein. The ability to pinpoint the induction of apoptosis in these cells may provide a powerful tool to investigate the molecular nature of the apoptotic response to telomerase inhibition.
Telomerase reactivation in 293 cells
Following telomerase inhibition, a population of cells from the inhibited lines regained viability and returned to normal growth. This return correlated with a reactivation of telomerase activity despite the continued presence of the dominant-negative protein. This suggests that there exists a strong selection for telomerase activity in maintaining viability in these cells. The molecular mechanism for this reactivation could be attributed to at least two possible responses. First, it is possible that another telomerase activity is being activated that does not rely on the catalytic subunit. Although formally possible, there is no biochemical evidence to support this mechanism. It is more likely that the reactivation of telomerase activity in this line is due to abrogation of the ability of the dominant-negative protein to function through alteration or upregulation of other telomerase-associated components. Although no changes in expression were observed among the known components, alteration of unknown components may contribute to the acquisition of resistance to the dominant-negative protein. Because such alterations would be expected to evade the effects of the dominant negative, these changes should not affect agents that directly inhibit the catalytic activity of the endogenous protein.
Inhibition of telomerase activity has been proposed as a potential approach for the treatment of human malignancy. This work suggests that telomerase inhibition may serve as an effective tool for eliminating tumor cells that have short telomeres. Such tumors may provide reasonable targets for agents that inhibit telomerase. These experiments await the development of specific inhibitors of the components of the telomerase complex.
Materials and methods
Generation of stable clones expressing inducible hTERT and its dominant-negative mutants
The human TERT cDNAs containing point mutations on the reverse transcriptase motifs were constructed as described (Harrington et al. 1997b). Four resulting mutant cDNAs are TERT 3-1 (D712/A712), TERT 5-1(D868/A868), TERT 5-2 (D869/A869), and TERT 5-1,2 (D868, D869/A868, A869). In addition, the 3′ end of TERT 5-1, TERT 5-2, and TERT-5-1,2 was tagged by Flag epitope (DYKDDDDK), whereas the TERT 3-1 was not tagged.
For transient transfection and FACS sorting, the wild-type and mutant TERT ORFs were subcloned into pIRES–EGFP expression vector (CLONTECH), which contains a CMV promoter, a multiple cloning site, and an internal ribosome entry site, (IRES) element followed by the gene for the reporter protein green fluorescent protein. The 293 cells were transfected with these plasmids, trypsinized 48 hr post transfection, and sorted into nontransfected, low-GFP expressors, and high-GFP expressors with a FACS Vantage sorter (Becton Dickinson). Each group of cells was pelleted, resuspended in lysis buffer, and extracted as described (Harrington et al. 1997b). Cell extracts were then assayed for protein expression by Western analysis with a anti-hTERT peptide antibody (Harrington et al. 1997b) and for telomerase activity.
For stably expressing wild type and mutant in an inducible way, these plasmids were subcloned into pIND ecdysone-inducible expression vector by the provided protocol (Invitrogen). A431 and 293 cells stably expressing ecdysone receptor (EcR) were first isolated with Zeocin (Invitrogen) as selective marker. The EcR cells were then used to generate clones that incorporated ecdysone response elements followed by wild-type and mutant TERTs with G418 (GIBCO-BRL) as selective marker. Each clone was screened for expression of TERT by inducing with muristerone for 24 hr followed by Western analysis. The resulting clones were designated as A clones for TERT 3-1 mutants, B clones for TERT 5-1 mutants, C clones for TERT 5-1,2 mutants, D clones for wild-type TERT transfectants, and E clones for TERT 5-2 mutants.
Cell growth and apoptosis assays
A431 and 293 cells were maintained in DMEM supplemented with 10% fetal calf serum, 100 U/ml of penicillin, 100 μg/ml of streptomycin, 400 μg/ml of Zeocin, and 700 μg/ml of G418. For induction of wild-type and mutant TERTs, cells were seeded at a density of 1.25 × 104 cells/cm2, allowed to attach and grow overnight, and changed to the medium containing muristerone (1 μm for A431 cells, and 2 μm for 293 cells). The cells were then allowed to grow in this medium for a length of time, depending on each individual experiment.
For long-term growth of 293 stable clones, each cell line was plated at 106 cells per 100-mm dish either in the presence of absence of muristerone. Cells were trypsinized every 3 days and counted with a hemocytometer. Each time 106 cells were replated onto a new 100-mm dish. The rest of the cells in each passage were pelleted and used to prepare cell extract, genomic DNA, and total RNA by standard protocol.
On induction, apoptotic cells were detected by the TUNEL method with an In Situ Cell Death Detection Kit (Boehringer Mannheim). For quantitative analysis of apoptosis, living A431 cells were trypsinized, washed once in ice-cold PBS, and incubated with annexin-V–fluoroescein/PI (Boehringer Mannheim) in calcium-containing HEPES buffer; the cells were then immediately analyzed with a FACScan machine (Becton Dickinson).
For cell cycle analysis, cells were fixed and stained by propidium iodide. The DNA content of each cell population was then analyzed by flow cytometry. DNA synthesis was measured by BrdU incorporation. Briefly, cells were pulse labeled in medium containing BrdU (Becton Dickinson) for 2 hr, then fixed in 70% ethanol, followed by staining with fluoroescein-conjugated anti-BrdU antibody (Becton Dickinson) and subsequent microscopic and flow cytometry analysis.
Colony formation of 293 cells was performed by plating 2 × 103 cells into six 60-mm dishes in the presence or absence of muristerone. The cells were allowed to grow for 2 weeks with medium changed at day 7, then stained with Giemsa to visualize colonies. The number of colonies was calculated and the representative plates were photographed.
Telomerase assay and measurement of TRF
Cell lysates were prepared as described previously (Harrington et al. 1997). Protein concentration of the lysates was measured by Bradford assay (Bio-Rad). Telomerase activity was assayed by telomere repeat amplification protocol (TRAP) using the TRAPENE telomerase detection kit (Oncor, Gaithersburg, MD) following provided procedures. To measure TRF, genomic DNA was digested with restriction enzymes HinfI and RsaI (New England BioLabs), fractionized on 0.6%–0.7% agarose gel and transferred onto a nylon membrane. The membrane was then hybridized with an oligonucleotide (TTAGGGTTAGGGTTAGGG) end labeled with [γ-32P]ATP (Amersham, Arlington Heights, IL) by standard protocol and exposed to a PhosphorImager screen (Molecular Dynamcs, Sunnyvale, CA) for 4–12 hr. The screen was scanned with a Strom820 phosphor screen scanner (Molecular Dynamics).
Acknowledgments
We thank T. Beattie for experimental assistance, H. Yamane and D. Yeung for antibody preparation, L. Chiu and G. Elliot for assistance with FACS analysis, and S. Coats for helpful advice.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mrobinso@amgen.com; FAX (805) 447-1982.
References
- Beattie TL, Zhou W, Robinson MO, Harrington L. Reconstitution of human telomerase activity in vitro. Curr Biol. 1998;8:177–180. doi: 10.1016/s0960-9822(98)70067-3. [DOI] [PubMed] [Google Scholar]
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Bryan TM, Reddel RR. Telomere dynamics and telomerase activity in in vitro immortalised human cells. Eur J Cancer. 1997;33:767–773. doi: 10.1016/S0959-8049(97)00065-8. [DOI] [PubMed] [Google Scholar]
- Bryan TM, Sperger JM, Chapman KB, Cech TR. Telomerase reverse transcriptase genes identified in Tetrahymena thermophila and Oxytricha trifallax. Proc Natl Acad Sci. 1998;95:8479–8484. doi: 10.1073/pnas.95.15.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Artand SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomerase dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. doi: 10.1016/s0092-8674(00)80762-x. [DOI] [PubMed] [Google Scholar]
- Collins K, Gandhi L. The reverse transcriptase component of the Tetrahymena telomerase ribonucleoprotein complex. Proc Natl Acad Sci. 1998;95:8485–8490. doi: 10.1073/pnas.95.15.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins K, Kobayashi R, Greider CW. Purification of Tetrahymena telomerase and cloning of genes encoding the two protein components of the enzyme. Cell. 1995;81:677–686. doi: 10.1016/0092-8674(95)90529-4. [DOI] [PubMed] [Google Scholar]
- Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counter CM, Meyerson M, Eaton EN, Weinberg RA. The catalytic subunit of yeast telomerase. Proc Natl Acad Sci. 1997;94:9202–9207. doi: 10.1073/pnas.94.17.9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- Greenberg RA, Allsopp RC, Chin L, Morin GB, DePinho RA. Expression of mouse telomerase reverse transcriptase during development, differentiation and proliferation. Oncogene. 1998;16:1723–1730. doi: 10.1038/sj.onc.1201933. [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, DePinho RA. Short dysfunctional telomeres impair tumorigenesis in the INK4a2/3 cancer-prone mouse. Cell. 1999;97:515–525. doi: 10.1016/s0092-8674(00)80761-8. [DOI] [PubMed] [Google Scholar]
- Greider CW. Telomere length regulation. Annu Rev Biochem. 1996;65:337–365. doi: 10.1146/annurev.bi.65.070196.002005. [DOI] [PubMed] [Google Scholar]
- ————— Telomeres and senescence: The history, the experiment, the future. Curr Biol. 1998;8:178–181. doi: 10.1016/s0960-9822(98)70105-8. [DOI] [PubMed] [Google Scholar]
- Harley CB. Telomere loss: Mitotic clock or genetic time bomb? Mutat Res. 1991;256:271–282. doi: 10.1016/0921-8734(91)90018-7. [DOI] [PubMed] [Google Scholar]
- Harley CB, Sherwood SW. Telomerase, checkpoints and cancer. Cancer Surv. 1997;29:263–284. [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Harrington L, McPhail T, Mar V, Zhou W, Oulton R, Bass MB, Arruda I, Robinson MO. A mammalian telomerase-associated protein. Science. 1997a;275:973–977. doi: 10.1126/science.275.5302.973. [DOI] [PubMed] [Google Scholar]
- Harrington L, Zhou W, McPhail T, Oulton R, Yeung DS, Mar V, Bass MB, Robinson MO. Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes & Dev. 1997b;11:3109–3115. doi: 10.1101/gad.11.23.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L. The limited in vitro lifetime of human diploid strains. Exp Cell Res. 1961;25:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- Karlseder J, Broccoli D, Dai Y, Hardy S, DeLange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- Kilian A, Bowtell DD, Abud HE, Hime GR, Venter DJ, Keese PK, Duncan EL, Reddel RR, Jefferson RA. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet. 1997;6:2011–2019. doi: 10.1093/hmg/6.12.2011. [DOI] [PubMed] [Google Scholar]
- Kondo S, Tanaka Y, Kondo Y, Hitomi M, Barnett GH, Ishizaka Y, Liu J, Haqqi T, Nishiyama A, Villeponteau B, et al. Antisense telomerase treatment: Induction of two distinct pathways, apoptosis and differentiation. FASEB J. 1998a;12:801–811. doi: 10.1096/fasebj.12.10.801. [DOI] [PubMed] [Google Scholar]
- Kondo S, Kondo Y, Li GY, Silverman RH, Cowell JK. Targeted therapy of human malignant glioma in a mouse model by 2-5A antisense directed against telomerase RNA. Oncogene. 1998b;16:3323–3330. doi: 10.1038/sj.onc.1201885. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Kondo S, Tanaka Y, Haqqi T, Barna BP, Cowell JK. Inhibition of telomerase increases the susceptibility of human malignant glioblastoma cells to cisplatin-induced apoptosis. Oncogene. 1998;16:2243–2248. doi: 10.1038/sj.onc.1201754. [DOI] [PubMed] [Google Scholar]
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- Lendvay TS, Morris DK, Sah J, Balasubramanian B, Lundblad V. Senescence mutants of Saccharomyces cerevisiae with a defect in telomere replication identify three additional EST genes. Genetics. 1996;144:1399–1412. doi: 10.1093/genetics/144.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276:561–567. doi: 10.1126/science.276.5312.561. [DOI] [PubMed] [Google Scholar]
- Lundblad V. The end replication problem: More than one solution. Nat Med. 1997;3:1198–1199. doi: 10.1038/nm1197-1198. [DOI] [PubMed] [Google Scholar]
- Lundblad V, Szostak JW. A mutant with a defect in telomere elongation leads to senescence in yeast. Cell. 1989;57:633–643. doi: 10.1016/0092-8674(89)90132-3. [DOI] [PubMed] [Google Scholar]
- Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Saito M, Nakamura H, Matsuura A, Ishikawa F. TLP1: A gene encoding a protein component of mammalian telomerase is a novel member of WD repeats family. Cell. 1997;88:875–884. doi: 10.1016/s0092-8674(00)81933-9. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Tahara H, Tahara E, Saito M, Ito K, Nakamura H, Nakanishi T, Tahara E, Ide T, Ishikawa F. Telomerase activation by hTRT in human normal fibroblasts and hepatocellular carcinomas. Nat Genet. 1998;18:65–68. doi: 10.1038/ng0198-65. [DOI] [PubMed] [Google Scholar]
- Niida H, Matsumoto T, Satoh H, Shiwa M, Tokutake Y, Furuichi Y, Shinkai Y. Severe growth defect in mouse cells lacking the telomerase RNA component. Nat Genet. 1998;19:203–206. doi: 10.1038/580. [DOI] [PubMed] [Google Scholar]
- Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Benchimol S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol. 1998;8:279–282. doi: 10.1016/s0960-9822(98)70109-5. [DOI] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Meth. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Weinrich SL, Pruzan R, Ma L, Ouellette M, Tesmer VM, Holt SE, Bodnar AG, Lichtsteiner S, Kim NW, Trager JB, et al. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat Genet. 1997;17:498–502. doi: 10.1038/ng1297-498. [DOI] [PubMed] [Google Scholar]