

Abstract
Cyclin E is an unstable protein that is degraded in a ubiquitin- and proteasome- dependent pathway. Two factors stimulate cyclin E ubiquitination in vivo: when it is free of its CDK partner, and when it is phosphorylated on threonine 380. We pursued the first of these pathways by using a two-hybrid screen to identify proteins that could bind only to free cyclin E. This resulted in the isolation of human Cul-3, a member of the cullin family of E3 ubiquitin–protein ligases. We found that Cul-3 was bound to cyclin E but not to cyclin E-Cdk2 complexes in mammalian cells, and that overexpression of Cul-3 increased ubiquitination of cyclin E but not other cyclins. Conversely, deletion of the Cul-3 gene in mice caused increased accumulation of cyclin E protein, and had cell-type-specific effects on S-phase regulation. In the extraembryonic ectoderm, in which cells undergo a standard mitotic cycle, there was a greatly increased number of cells in S phase. In the trophectoderm, in which cells go through endocycles, there was a block to entry into S phase. The SCF pathway, which targets cyclins for ubiquitination on the basis of their phosphorylation state, and the Cul-3 pathway, which selects cyclin E for ubiquitination on the basis of its assembly into CDK complexes, may be complementary ways to control cyclin abundance.
Keywords: Cullins, ubiquitin degradation, cyclins, S phase
Cyclin E is an evolutionarily conserved protein whose essential function is to promote the cell cycle transition from G1 to S phase (Knoblich et al. 1994; Ohtsubo et al. 1995). Cyclin E binds to and activates the cyclin-dependent kinase Cdk2, and it is the catalytic activity of this holoenzyme that mediates the effects of cyclin E on cell cycle (Fang and Newport 1991; Koff et al. 1991, 1992, 1993; Dulic et al. 1992; Jackson et al. 1995). Hence, mutants of cyclin E that cannot bind to CDK2 are biologically inert (Kelly et al. 1998), and ectopic overexpression of a catalytically inactive mutant of CDK2 prevents a cell from entering S phase (Heuvel and Harlow 1993). Conversely, elevated amounts of active cyclin E–CDK2 accelerate entry into S phase (Ohtsubo and Roberts 1993; Resnitzky and Reed 1995). The substrates of cyclin E–Cdk2 are not fully defined, but are thought to include inhibitors of S phase such as pRb (Hinds et al. 1992; Connell-Crowley et al. 1997; Zarkowska and Mittnacht 1997; Kelly et al. 1998) and p27Kip1 (Sheaff et al. 1997), which are inactivated by CDK2-directed phosphorylation. Additionally, proteins that stimulate DNA synthesis might be phosphorylated and thereby activated by CDK2 (Blow and Nurse 1990; D’Urso et al. 1990; Zhao et al. 1998).
The abundance of the cyclin E protein and cyclin E–Cdk2 catalytic activity oscillate in parallel during the cell cycle, reaching a peak as cells begin chromosome replication and a trough at G2/M (Koff et al. 1991; Dulic et al. 1992; Ohtsubo et al. 1995). This is achieved, in part, by cell cycle-dependent gene transcription (Ohtani et al. 1995; Geng et al. 1996). The cyclin E promoter contains E2F-binding sites, and activation of the E2F transcriptional program during G1 increases cyclin E gene expression. Phosphorylation of the Retinoblastoma (Rb) protein by the cyclin D-associated kinases is initially responsible for the release of E2F and increased expression of E2F-responsive genes (Weinberg 1995). Hence, the D-type cyclins function upstream of cyclin E in a pathway that leads to cyclin E gene expression (Lukas et al. 1995; Geng et al. 1999; Leng et al. 1997). Expression and activation of cyclin D–CDK complexes is positively controlled by mitogens, which indirectly establishes the dependency of cyclin E accumulation on mitogenic signaling (Sherr 1995). Therefore, increased expression of cyclin E in pre-S-phase cells reflects the fact that the mitogen requirement for cell proliferation has been satisfied and also represents a necessary step for beginning DNA synthesis.
A second essential component of cyclin E periodicity is post-transcriptional regulation by ubiquitin-dependent proteolysis (Clurman et al. 1996; Won and Reed 1996). Cyclin E has a short half life of less than 30 min, which can be increased to greater than 2 hr by pharmacologic inhibition of the proteasome (Clurman et al. 1996; Won and Reed 1996). Rapid turnover of cyclin E protein ensures that its levels closely parallel the changing abundance of its mRNA, and therefore underlies the strict dependence of cyclin E protein expression on the cyclin D/Rb/E2F pathway. Turnover of cyclin E by the ubiquitin–proteasome pathway is regulated both by the binding of cyclin E to CDK2, and by cyclin E phosphorylation (Clurman et al. 1996; Won and Reed 1996). Thus, unbound cyclin E is readily ubiquitinated and degraded by the proteasome, whereas cyclin E within cyclin E–CDK2 complexes is protected from ubiquitination. However, the protection afforded by CDK2 is reversed in a process that involves phosphorylation of cyclin E on threonine 380, which triggers ubiquitination of cyclin E and degradation of cyclin E in the proteasome.
The regular rise and fall of cyclin E protein levels is an essential feature of normal cell cycle regulation. Firstly, the upswing in cyclin E expression is one means by which exit from G1 is coupled to the receipt of extracellular mitogenic cues or other proliferative signals. Secondly, the timing of S phase is determined by the abundance of the cyclin E protein. Thirdly, the decline in cyclin E abundance later in S and G2 resets the cell cycle program to its initial state, and thereby reestablishes the dependency of G1 progression on mitogens in the ensuing cell cycle. Finally, cyclin E oscillation appears to be essential for endocycles, cell cycles in which sequential S phases occur without intervening mitoses. It is thought that each cycle of chromosome replication must be accompanied by a decrease and then a rise in cyclin E activity (Folette et al. 1998; Weiss et al. 1998).
The proteins that select cyclin E for ubiquitination are not known. Identifying these proteins will be essential for understanding how cyclin E is recognized by the ubiquitin–proteasome pathway, for determining whether the activities of the relevant ubiquitinating enzymes are regulated during the cell cycle or modulated by extracellular signals, and for addressing whether the pathways responsible for cyclin E turnover are altered in tumorigenic cells that display deregulated cyclin E expression. Using a combination of molecular and genetic approaches, we have identified a member of the cullin family, Cul-3, as being one component of a pathway that controls cyclin E ubiquitination. Homozygous deletion of the Cul-3 gene is shown to cause overexpression of the cyclin E protein and to disrupt normal cell cycle regulation in vivo.
Results
Cloning and characterization of human Cullin-3
Our previous work indicated that one pathway for ubiquitination of cyclin E was critically affected by the binding of cyclin E to a CDK (Clurman et al. 1996). Thus, free (unbound) cyclin E was readily ubiquitinated, whereas cyclin E bound to a CDK was protected from ubiquitination. To identify molecules that might be involved in targeting free cyclin E for ubiquitination, we performed a two-hybrid screen in which a mutant version of cyclin E (cyclin E R130A) was used as bait. In both mammalian cells and yeast wild type, cyclin E binds to and activates CDKs, whereas cyclin E(R130A) cannot. Clones that scored positively for an interaction with cyclin E(R130A) were rescreened against wild-type cyclin E. From 1.5 × 106 transformants we identified a single protein that was able to bind to cyclin E R130A but could not bind to wild-type cyclin E (Fig. 1A), properties that were consistent with it having a role in targeting cyclin E for ubiquitination. The DNA sequence of this interactor revealed that it was a portion of the protein Cullin-3 (Cul-3) (amino acids 395–768). These binding properties were not an artifact of using a truncated Cul-3 protein, because reconstruction experiments demonstrated that full-length Cul-3 also bound to cyclin E R130A, and not to wild-type cyclin E in this assay (not shown). Cul-3 is a member of the cullin family of genes, defined as homologs of the Cul-1 gene from nematodes (Kipreos et al. 1996; Du et al. 1998). This family includes the Cdc53 protein in budding yeast, which has been shown to be part of an E3 ubiquitin–protein ligase (Patton et al. 1998).
Figure 1.

Cloning of human Cullin 3. (A) Two-hybrid interaction between cyclin E and Cul-3. (Left) Growth of Saccharomyces cerevisae when selected for the presence of the cyclin (either wild-type cyclin E or a CDK nonbinding mutant) and cullin plasmids; (right) growth of same strains when selection is applied for a two-hyrid interaction between the cyclin and the cullin. (B) Graphical representation of the two Cul-3 cDNAs identified. Cul-3 long contains 768 amino acids and Cul-3 short is missing one exon of 66 amino acids and is therefore only 702 amino acids long.
The partial Cul-3 clone obtained in the two-hybrid screen was used as a probe to isolate cDNAs from a human B-cell library containing the complete Cul-3 ORF. The complete Cul-3 ORF is 2307 bp and is predicted to encode a protein of 768 amino acids. The amino acid sequence shows homology to all the cullins in the databases with the greatest similarity in the cullin domain, which includes amino acids 741–768 in the Cul-3 sequence. Cul-3, like other cullins, lacks a HECT domain, a sequence found in a subset of E3 enzymes.
A second cDNA also apparently containing a complete Cul-3 ORF was isolated in this same screen. However, this clone encoded a version of Cul-3 with an in-frame deletion of amino acid 23 through amino acid 88 (Fig. 1B). Subsequent sequencing of the Cul-3 gene (not shown) revealed that the deleted region precisely corresponded to an exon, and that the two cDNAs (hereafter referred to as Cul-3 long and Cul-3 short) therefore represented alternatively spliced forms of Cul-3 mRNA.
Pattern of Cul-3 protein expression
Portions of Cul-3 corresponding to the amino, middle, and carboxy parts of the protein were individually expressed as recombinant proteins in Escherichia coli and used separately to immunize rabbits, thereby generating three distinct antisera that recognize different regions of the Cul-3 protein (see Materials and Methods). All three antisera were affinity purified against the immunizing antigen and when used for immunoblotting of whole cell extracts they were found to recognize a single protein with the predicted molecular size of Cul-3 (Fig. 2; data not shown). Each of the antibodies detected increased expression of full-length Cul-3 protein in whole cell extracts from mammalian cells that had been transfected with a CMV promoter-driven mammalian expression vector containing the Cul-3 cDNA, and none of the antibodies recognized Cul-1. Both the transfected and endogenous protein run as a doublet in all cells examined (see below). Finally, no immunostaining was detected in Cul-3−/− mice (see Fig. 5, below), confirming the specificity of the antibodies for Cul-3.
Figure 2.
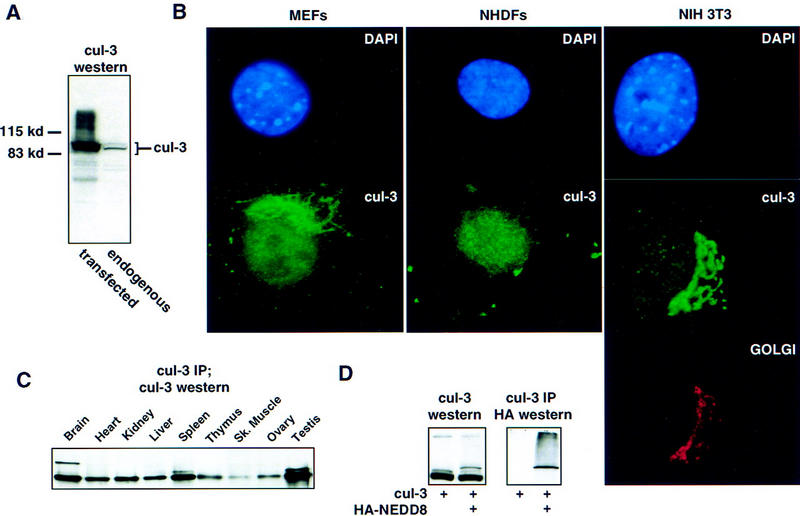
Characterization of human Cul-3. (A) Immunoblot of Cul-3 in whole cell extracts from h293 cells. (Left) Extract from cells transfected with full-length Cul-3 cDNA; (right) untransfected cell extract. (B) Immunolocalization of Cul-3. The indicated cells were stained with DAPI and Cul-3 antibodies. In addition, the NIH-3T3 cells were stained with rhodamine-conjugated wheat germ agglutinin to identify the Golgi apparatus. (C) Cul-3 expression in mouse tissues. Cul-3 was immunoprecipitated from tissue extracts with antibodies against the carboxy-terminal portion of Cul-3,and then analyzed by Western blots with antibodies against the amino-terminal portion of Cul-3. (D) NEDD8 modification of Cul-3 protein. (Left) Cul-3 was transfected into h293 cells with or without HA-NEDD8. Extracts were blotted with anti-Cul-3 antibodies. Inclusion of epitope-tagged NEDD8 shifted the mobility of higher molecular weight form of Cul-3. (Right) Cul-3 protein was immunoprecipitated from the same extracts and the material was analyzed on a Western blot with anti-HA antibody to detect NEDD8. The higher molecular weight form of Cul-3 was immunoreactive with the anti-HA antibody.
Figure 5.
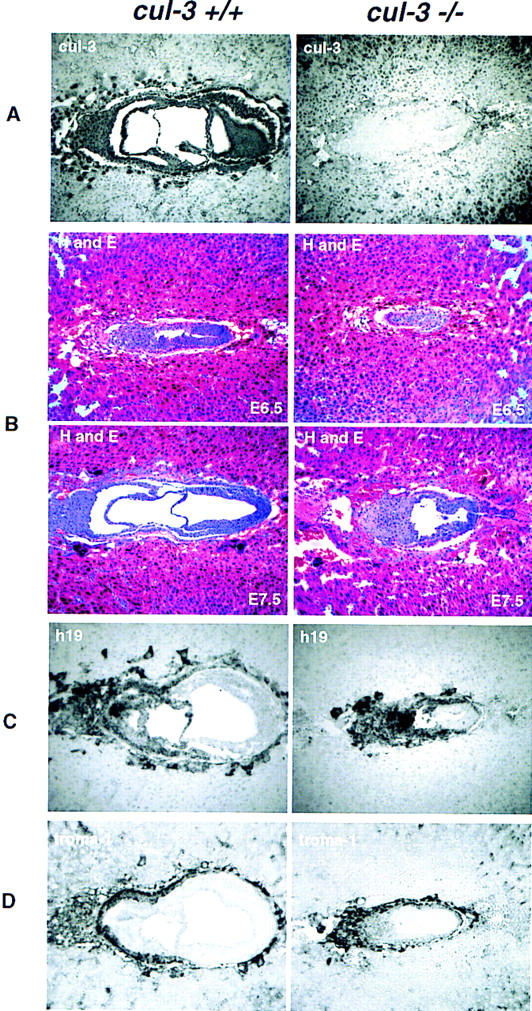
Embryonic lethal phenotype of Cul-3−/− mice. (A) Affinity-purified anti-Cul-3 antibodies were used to stain sections from E7.5 embryos. (Left) Cul-3 staining in a wild-type embryo. Note that the most intense nuclear staining is present in the extraembryonic ectoderm and the trophoblast giant cells. (Right) The Cul-3−/− embryo does not stain with Cul-3 antibodies. (B) H and E staining of embryos at either E6.5 (top) or E7.5 (bottom). (C) RNA in situ hybridization to detect expression of the H19 gene. H19-positive cells represent the extraembryonic ectoderm and trophectoderm lineages. (D) Detection of the trophectoderm lineage with Troma-1 antibodies.
Various cell types, both mortal and immortal, from mice and humans, were stained with each of the Cul-3 antibodies to determine the subcellular location of the Cul-3 protein (see Materials and Methods for complete listing of all cell types examined). All three antibodies, and all of the cell types we examined, demonstrated the same pattern of Cul-3 localization to both the nucleus and the Golgi (Fig. 2B). Golgi staining was confirmed by use of rhodamine-tagged wheat germ agglutinin to visualize the Golgi, and by specific dissolution of the Golgi with Brefelden A (not shown). The Golgi localization of Cul-3 was more dramatic in murine compared with human cells, but this appeared to represent a difference in the elaboration of the Golgi in mouse versus human cells rather than a difference in Cul-3 itself.
In asynchronously proliferating cells there was no obvious cell to cell heterogeneity in the Cul-3 staining pattern that would suggest its distribution changed during the cell cycle; the one exception being the pancellular distribution seen in mitotic cells (not shown). Also, MANCA cells were separated according to position in the cell cycle by centrifugal elutriation and whole cell extracts immunoblotted for Cul-3 protein. This revealed no cell cycle-dependent changes in Cul-3 protein expression (not shown).
These same antibodies were used to determine the tissue distribution of the Cul-3 protein in adult mice. As shown in Figure 2C, Cul-3 protein is expressed in all tissues examined with the greatest amount of Cul-3 expressed in brain, spleen, and testis. Cul-3 expression was also detected in all cell lines examined, and its abundance did not differ substantially between primary and immortalized cell lines (data not shown).
Cul-3 is modified by NEDD8
Mammalian cullins (Osaka et al. 1998; Wada et al. 1999) and the yeast homolog Cdc53 (Lammer et al. 1998; Liakopoulos et al. 1998) have been shown to be modified by NEDD8, a ubiquitin homolog (Kamitani et al. 1997; Gong and Yeh 1999). When the Cul-3 cDNA was cotransfected into h293 cells with HA-tagged NEDD8, the more slowly migrating of the two Cul-3 isoforms showed a decrease in its electrophoretic mobility that was consistent with its modification by the epitope-tagged NEDD8 as opposed to the endogenous (untagged) NEDD8. Cul-3 was then immunoprecipitated with anti-Cul-3 antibodies, and immunoblotted with anti-HA antibodies. We found that the more slowly migrating form of Cul-3 was specifically recognized by the anti-HA antibodies, and is therefore directly conjugated to NEDD8 (Fig. 2D).
Cul-3 binds to cyclin E in human cells
The interaction between Cul-3 and cyclin E was examined in mammalian cells. Expression vectors encoding full-length Cul-3 and myc-epitope-tagged cyclins E, D1, A, or B were cotransfected into h293 cells (Fig. 3A). Cul-3 could be coimmunoprecipitated with cyclins D1 and E, but not cyclins A and B. The binding of Cul-3 to cyclin E was confirmed with multiple different antisera that recognized different parts of the cullin or cyclin, in reciprocal immuneprecipitations, and in experiments with epitope-tagged or untagged proteins (not shown). In these experiments, the cyclins were expressed in excess over their endogenous CDK partners, so the binding interactions we detected were between Cul-3 and free (unbound) cyclins. This was confirmed in parallel transfection experiments that showed that Cul-3 also bound to cyclin E R130A (not shown). In accord with the results of our two-hybrid screen, overexpression of Cdk2 together with wild-type cyclin E prevented the binding of Cul-3 to cyclin E (Fig. 3B). However, overexpression of Cdk2 did not decrease binding of cyclin E R130A to Cul-3 (not shown). Thus, assembly of cyclin E into complexes with Cdk2 inhibits its interaction with Cul-3.
Figure 3.

Cul-3 binds to cyclin E in mammalian cells. (A) Myc-epitope-tagged cyclins A, B, D, and E were cotransfected into cells with a Cul-3 cDNA. (Left) Western blot with the anti-myc tag antibody shows that the four cyclins were expressed at equivalent levels; (right) immuneprecipitates of Cul-3 contained cyclins E and D1, and not A or B. (B) Cotransfection of cells with cyclin E and Cdk2 prevented the binding of Cul-3 to cyclin E. (C) Cyclin E cDNA was cotransfected into h293 cells with either Cul-3 long cDNA or Cul-3 short cDNA. Only Cul-3 long binds to cyclin E. (D) A complex between Cul-3 and cyclin E can be detected in extracts from untransfected h293 cells. Binding of Cul-1 to cyclin E was not seen. (E) Epitope-tagged Cul-1 and Cul-3 were expressed in h293 cells together with cyclin E. Western blotting with an anti-epitope tag antibody showed that equivalent amounts of the two cullins were expressed (not shown). Immuneprecipitations showed that only Cul-3 bound to cyclin E. (HC) Position of immunoglobulin heavy chain.
The abilities of the long and short alternatively spliced forms of Cul-3 to bind to cyclin E were tested by cotransfecting into h293 cells expression vectors encoding either form of Cul-3 together with myc-epitope-tagged cyclin E (Fig. 3C). Using anti-myc tag antibodies to specifically immunoprecipitate cyclin E, we found that the long form but not the short form of Cul-3 bound to cyclin E. This identified a region of Cul-3 in the amino terminus of the protein as being important for its interaction with cyclin E in mammalian cells. This region did not appear to be important for the interaction in yeast, because it was absent from our initial isolate of Cul-3 obtained in the two-hybrid screen. It is important to point out that in all tissues and cell lines that we have examined, the endogenous Cul-3 protein had an apparent molecular size consistent with that of the long form of Cul-3. Thus far we have not detected expression of the short form of Cul-3 protein. RT–PCR analyses confirmed that Cul-3 long was the major form of Cul-3 expressed in cells and tissues (not shown).
For technical reasons, it is often advantageous to study interactions between transfected, overexpressed proteins. Nevertheless, it is always important to confirm the relevance of those interactions by examining the state of the corresponding endogenous cellular proteins. To this end, extracts from untransfected h293 cells were prepared and endogenous cyclin E was immunoprecipitated. Immunoblotting the bound material with affinity-purified Cul-3 antibodies, demonstrated the presence of cyclin E-associated Cul-3 protein. (Fig. 3D). We do not yet know if this interaction is direct, or mediated by other proteins as has been seen in other cullin complexes (Feldman et al. 1997; Lyapina et al. 1989; Yu et al. 1998).
We also examined the ability of cyclin E to associate with Cul-1. This was of particular interest because Cul-1 is the closest homolog among the mammalian cullins to the yeast Cdc53 protein. Cdc53 has been shown to be involved in the ubiquitination of the yeast Cln G1 cyclins, and therefore mammalian Cul-1 was considered to be a candidate for being involved in cyclin E ubiquitination in mammalian cells (Koepp et al. 1999). However, no binding of Cul-1 to cyclin E could be detected (Fig. 3D). The relative abilities of Cul-1 and Cul-3 to bind to cyclin E were also tested in a transfection assay. Expression vectors encoding Cul-3 and Cul-1 were cotransfected with myc-epitope-tagged cyclin E into h293 cells, and cyclin E immunoprecipitates tested for the presence of associated cullins (Fig. 3E). Just as we had seen with the endogenous proteins, transfected Cul-3 bound to cyclin E and Cul-1 did not. Therefore, Cul-1 does not seem to be involved in the pathway that recognizes free cyclin E.
Cul-3 stimulates ubiquitination of cyclin E
To study the effects of Cul-3 on the ubiquitination of cyclin E, expression vectors encoding Cul-3 and cyclin E were cotransfected into h293 cells. The presence of Cul-3 stimulated the accumulation of higher molecular weight forms of cyclin E, which for the following reasons are likely to be cyclin E-ubiquitin conjugates. First, these high molecular forms of cyclin E were similar to those that accumulated when the turnover of ubiquitin-conjugated proteins was prevented by pharmacological inhibition of the proteasome with MG-132 (Fig. 4A, left). Second, cyclin E and Cul-3 were cotransfected into h293 cells together with a plasmid expressing an HA-epitope-tagged form of ubiquitin. Cyclin E was immunoprecipitated with anti-cyclin E antibodies, and the recovered protein was immunoblotted with antibodies that recognize the HA epitope tag that was present on the cotransfected ubiquitin (Fig. 4A, right). This approach directly demonstrated that Cul-3 stimulated the accumulation of ubiquitin-conjugated cyclin E. When the same experiment was performed with cyclin A, Cul-3 had no effect on the accumulation of cyclin A-ubiquitin conjugates, although proteosomal inhibition with MG-132 readily caused accumulation of cyclin A-ubiquitin conjugates (Fig. 4B). Therefore, the effects of Cul-3 were specific for cyclin E, and overexpression of Cul-3 did not result in nonspecific inhibition of the proteasome.
Figure 4.

Cul-3 stimulates ubiquitination of cyclin E. (A) Cyclin E and Cul-3 cDNAs were cotransfected into h293 cells. (Left) Higher molecular weight forms of cyclin E are observed either in the presence of the proteasome inhibitor MG-132, or in the presence of Cul-3; (right) the same experiment was performed in the presence of HA-tagged ubiquitin. Both Cul-3 and MG-132 stimulate the conjugation of ubiquitin to cyclin E (B) Same as part A except that cyclin A was used instead of cyclin E. Cul-3 had no effect on the ubiquitination of cyclin A.
Mutation of the CDK phosphorylation sites in cyclin E, including the critical threonine 380 residue, had no effect on either binding of Cul-3 to cyclin E (not shown) or on the ability of Cul-3 to stimulate cyclin E ubiquitination (Fig. 4A, middle). Therefore, the Cul-3 pathway for ubiquitination of free cyclin E is independent of cyclin E phosphorylation.
Construction of a Cul-3 knockout mouse
The Cul-3 gene was disrupted by electroporation of murine embryonic stem cells (ES cells) with a targeting vector, pJS1052, in which amino acids 127–293 of the Cul-3 gene were deleted and replaced with the neomycin gene under the control of the PGK promoter (see Materials and Methods for details). The targeting construct contained 6.8 kb of Cul-3 genomic DNA as the upstream arm, and 1.4 kb of Cul-3 genomic DNA as the downstream arm flanking the neomycin gene. Upstream of the long arm was the PGK promoter driving the thymidine kinase gene, which was used for counterselection to increase recovery of homologous integration events. Among the first 20 neomycin resistant ES cell colonies, 5 were shown by polymerase chain reaction to have undergone homologous recombination between pJS1052 and the chromosomal Cul-3 gene. This was confirmed by Southern blot hybridization.
These five ES cell clones were microinjected into C57/BL blastocysts. Chimeric males were backcrossed to wild-type C57/BL females and two independent ES cell clones that successfully contributed to the germ line were used for subsequent experiments. The effects of the internal Cul-3 deletion on Cul-3 gene expression were assessed in embryonic fibroblasts prepared from E16 heterozygous mice. We used antibodies that had been specifically raised against amino acids 1–286 (which includes the part remaining in the Cul-3 knockout), and antibodies raised against the carboxy-terminal end of Cul-3 (see Materials and Methods) to assay for expression of Cul-3 protein by immunoblotting of whole cell extracts. Both antibodies detected full-length Cul-3 protein in the heterozygous MEFs, but neither detected any expression of a truncated form of Cul-3 protein that might have arisen from the mutated allele (not shown). We concluded that the deletion of amino acids 127–296 results in a null allele.
F1 heterozygous mice containing one intact allele of Cul-3 were intercrossed to obtain F2 generation mice lacking Cul-3 protein. Among the first 100 progeny, no viable Cul-3−/− mice were obtained, whereas Cul-3+/+ and Cul-3+/− animals were obtained at the expected frequencies. We concluded that homozygous deletion of the Cul-3 gene caused an embryonic lethal phenotype.
Characterization of Cul-3−/− embryos
To determine the effects of the Cul-3 deletion on development, we analyzed embryos obtained from pregnant females at various days of gestation following mating of F1 heterozygous animals. Homozygous Cul-3−/− embryos were identified either by PCR of DNA prepared from yolk sacs or, for embryos at E7.5 and younger, by immunostaining with anti-Cul-3 antibodies (Fig. 5A). Note that we were unable to distinguish between Cul-3+/+ and Cul-3+/− embryos at stage E7.5 or younger, because Cul-3 immunostaining does not differentiate between these two genotypes. However, no systematic abnormalities were observed among the embryos that stained positively for Cul-3 protein and Cul-3 heterozygous mice were represented in the expected proportion among the viable progeny of Cul-3+/− intercrosses, suggesting that Cul-3+/− embryos developed normally.
Cul-3−/− embryos were found at the expected Mendelian ratio until E7.5, after which Cul-3−/− yolk sacs contained partially degenerating or fully resorbed embryos. To further characterize the phenotype of Cul-3 mutant mice, we performed histological analyses of serial sections prepared from E6.5 and E7.5 embryos. E6.5 Cul-3−/− embryos were substantially smaller than wild-type embryos, and displayed markedly abnormal patterning both in the embryonic and extraembryonic tissues (Fig. 5B). E7.5 embryos were characterized in greater detail. Cul-3−/− embryos showed complete disorganization of the extraembryonic tissues, which were identified by RNA in situ hybridization with a probe for the H19 gene (Poirier et al. 1991; Fig. 5C), including the absence of an amnion and absence of the extraembryonic cavities. The trophectoderm, identified by staining with Troma-1 antibodies (Brulet et al. 1980), was present, but abnormally developed and trophoblast giant cells were more sparsely represented than in wild-type embryos (Fig. 5D). Gastrulation was also abnormal without clear evidence for the presence of embryonic mesoderm and endoderm.
Increased abundance of cyclin E protein in Cul-3−/− embryos
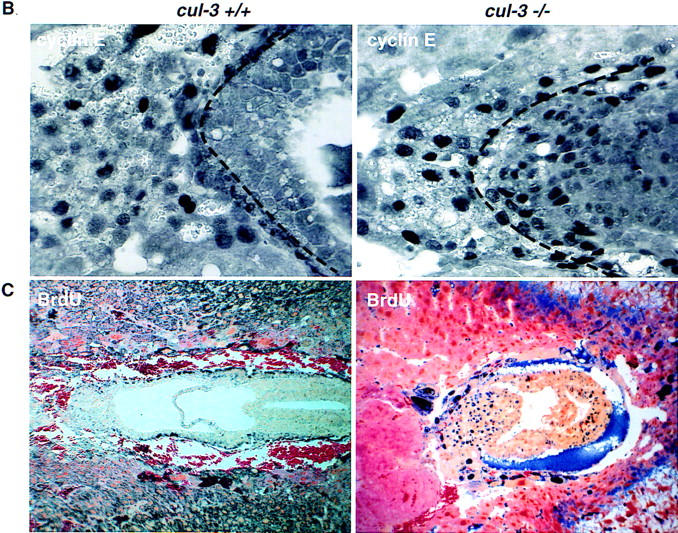
Serial sections of E7.5 embryos were stained with affinity-purified antibodies against cyclin E (Fig. 6—note that these images are not the same scale). In embryos expressing wild-type Cul-3 protein, cyclin E was most abundantly expressed in the trophoblast giant cells, and occasional, more weakly staining cells were detected scattered throughout the embryonic and extraembryonic tissues. Cul-3−/− embryos also expressed abundant cyclin E protein in the trophoblast cells, but equally high amounts were detected in the majority of cells in the ectoplacental cone and the extraembryonic ectoderm. In situ hybridization with cyclin E antisense RNA as probe did not reveal increased expression of cyclin E mRNA in Cul-3−/− embryos (not shown). Therefore, post-transcriptional events were the cause of elevated cyclin E protein expression in the Cul-3−/− embryos. Immunostaining did not detect increased expression of either cyclin A or cyclin D1 protein in the Cul-3 mutant embryos (not shown).
Figure 6.
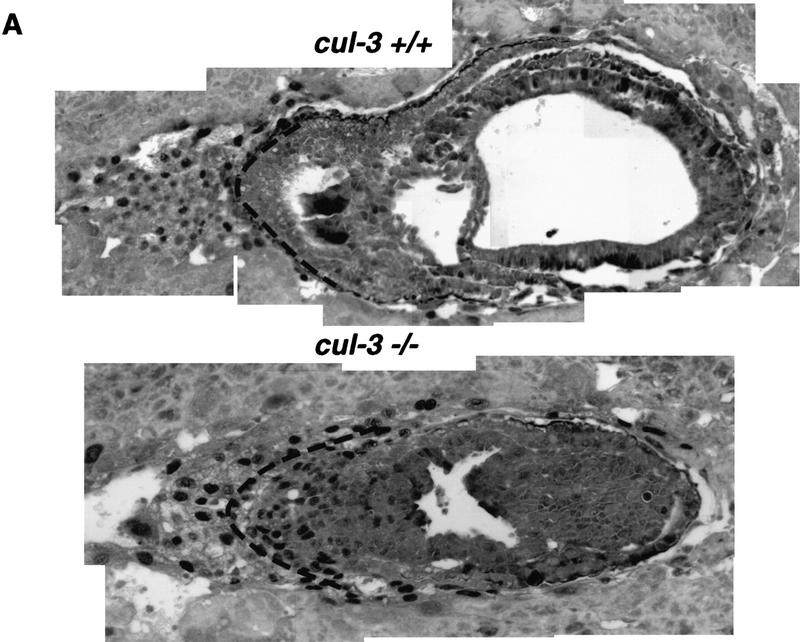
Expression of cyclin E protein in wild-type and Cul-3−/− embryos. (A) Montage images of either wild-type (top) or Cul-3−/− embryos (bottom) stained with anti-cyclin E antibodies. The images are not to scale. A broken line has been placed on both photos to delineate the border between the extraembryonic tissue and the trophectoderm as determined by staining with Troma-1 antibodies. Increased cyclin E protein is detected specifically in the extraembryonic ectoderm. (B) A high-magnification picture of the same embryos, to scale, to highlight the extraembryonic region in which cyclin E expression is increased in the Cul-3−/− embryo. (C) Wild-type (left) and mutant embryos (right) isolated from BrdUinjected animals were stained for BrdU incorporation. The Cul-3−/− embryo showed many more BrdU-positive cells, with the greatest number being in the same region shown to overexpress cyclin E.
Abnormal regulation of S phase in Cul-3−/− embryos
We studied the effects of the Cul-3 mutation on patterns of DNA replication in E7.5 embryos. Pregnant mice were injected intraperitoneally with BrdU. Four hours later embryos were removed, and sections stained with anti-BrdU antibodies to identify S-phase cells. A dramatic increase in the number of cells synthesizing DNA was detected in the extraembryonic ectoderm and ectoplacental cone of the Cul-3−/− embryos, the same cell types that expressed increased amounts of cyclin E protein (Fig. 6C).
Trophoblastic cells also expressed high amounts of cyclin E. Unlike the cells in the extraembryonic ectoderm, these cells undergo endoreduplicative cell cycles, increasing their DNA content many fold (MacAuley et al. 1998). DNA synthesis in these cells was studied in vitro, in cultured living blastocysts. Blastocysts were isolated 3.5 d.p.c., cultured on cover slips for 4 days, then fixed and immunostained for the expression of various proteins (Fig. 7). Also, 2 hr prior to fixing and staining, BrdU was added to the culture medium to label S-phase nuclei. Wild-type blastocysts developed both an inner cell mass, and migratory trophoblast giant cells (Fig. 7A); the latter were readily identifiable by their large nuclei, which result from genome endoreduplication, and also by their positive cytoplasmic immunostaining with Troma-1 antibodies (Fig. 7B). The trophoblast giant cells stained heterogeneously for cyclin E protein, consistent with the fact that cyclin E is known to oscillate in abundance during cycles of endoreduplication. BrdU staining revealed a good correspondence between the cyclin E positive cells and the cells that were in S phase (Fig. 7B).
Figure 7.
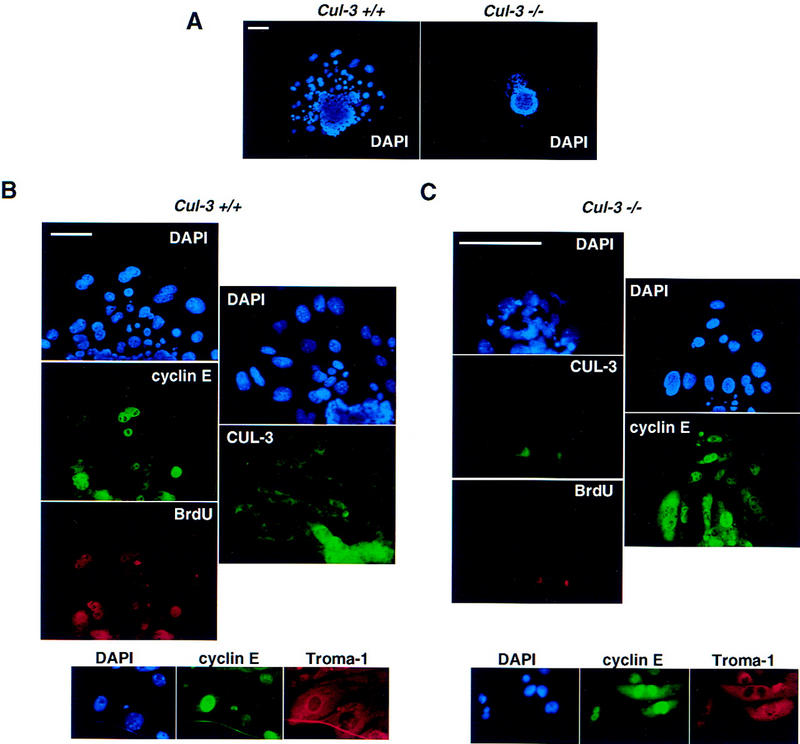
Analysis of wild-type and Cul-3−/− blastocysts. Bar, 100 μm. (A) DAPI staining of wild-type (left) and Cul-3−/− blastocysts. Images are to scale. Note the much smaller nuclei of the migratory Cul-3−/− trophoblasts. (B) Wild-type trophoblast cells are stained with DAPI, anti-cyclin E, and anti-BrdU antibodies (left). (Right) Blastocysts stained with DAPI and Cul-3 antibody to show that the giant cells normally express Cul-3. (Bottom) The giant cells in wild-type blastocysts also express Troma-1. (C) Same as B, except that Cul-3−/− blastocysts are shown. The mutant trophoblasts do not stain for Cul-3 or BrdU.
Cul-3−/− blastocysts were identified either by PCR or by immunostaining for expression of the Cul-3 protein. These mutant blastocysts also developed an inner cell mass and migratory trophoblast cells. But, unlike the trophoblasts that expressed Cul-3 protein, the Cul-3−/− trophoblasts had much smaller nuclei, often the size of a normal diploid cell (Fig. 7A). Troma-1 immunostaining confirmed that these cells with small nuclei were trophoblasts (Fig. 7C). Every one of these small, Cul-3−/− trophoblast cells exhibited strong nuclear cyclin E immunostaining, suggesting that the normal cell cycle-dependent change in cyclin E levels was attenuated in these cells. Often the cyclin E staining was cytoplasmic as well as nuclear, and was more intense than in wild-type cells. Remarkably, none of the Cul-3−/− trophoblasts were positive for BrdU despite the high levels of cyclin E protein. Thus, their small nuclear size correlated with an absence of ongoing genome duplication. The disparate effects of cyclin E overexpression on S phase in mitotic versus endoreduplicative cell cycles is consistent with what has been observed previously in other model systems (Ohtsubo and Roberts 1993; Follette et al. 1998; Weiss et al. 1998).
Discussion
Ubiquitination of cyclin E by Cul-3
Both molecular and genetic approaches show that Cul-3 is important for the ubiquitination and degradation of mammalian cyclin E. Knockout of the Cul-3 gene in mice caused an early embryonic lethal phenotype that was associated with increased amounts of cyclin E protein in the extraembryonic ectoderm and in the trophectoderm. The effect of the Cul-3 deletion was specific to cyclin E, as the expression of neither cyclin A nor cyclin D1 were increased.
The increased expression of cyclin E in cells lacking Cul-3 protein probably reflected a direct effect of Cul-3 on cyclin E turnover. We found that Cul-3 was tightly bound to cyclin E in vivo, and that increased expression of Cul-3 increased cyclin E ubiquitination. In contrast, Cul-3 neither bound to cyclin A nor had any effect on its ubiquitination. These observations support the idea that Cul-3 is part of an E3 protein–ubiquitin ligase that selects certain proteins, including cyclin E, for ubiquitination.
We do not mean to imply that cyclin E is the only target of Cul-3, nor that cyclin E is selected for ubiquitination only by Cul-3. First, localization of Cul-3 to the Golgi apparatus suggests that Cul-3 may have additional substrates. These might include misfolded or improperly assembled proteins that arise during intracellular trafficking, proteins that are processed in preparation for export, or proteins that are modified by ubiquitin for retrograde transport (Hicke 1999). Second, overexpression of cyclin E is restricted to certain cell types in Cul-3−/− embryos. Other turnover pathways (perhaps involving other cullins) may be operative in those cells in which cyclin E levels are unaffected by the absence of Cul-3. This possibility is discussed further below.
More than one pathway for cyclin E ubiquitination?
Ubiquitination of cyclin E depends on two parameters; its binding to a CDK, and its state of phosphorylation on threonine 380 (Clurman et al. 1996; Won and Reed 1996). These two pathways may be governed by distinct ubiquitinating enzymes, one which recognizes a feature unique to unbound cyclin E, and the other which directly recognizes phosphorylated T380. We have shown previously that phosphorylation of T380 is required for ubiquitination of cyclin E bound to Cdk2, but not for ubiquitination of unbound cyclin E. Ubiquitination of a budding yeast G1 cyclin, Cln2, occurs by a phosphorylation-triggered pathway in which an E3 protein–ubiquitin ligase, the SCF complex, binds directly to the phosphorylated cyclin (Deshaies et al. 1995; Willems et al. 1996; Skowyra et al. 1997, 1999). The fact that phosphorylation of T380 in cyclin E promotes the ubiquitination of cyclin E is consistent with the SCF paradigm, but direct evidence for the involvement of this pathway in the turnover of cyclin E is, thus far, lacking.
A second pathway, one which involves ubiquitination and degradation of proteins that are separated from their normal binding partners, may also be critically important. This pathway was initially recognized as being crucial for the rapid turnover of proteins within the endoplasmic reticulum that are either misfolded or incorrectly assembled into multiprotein complexes (Hurtley and Helenius 1989; Bonifacino and Weissman 1998). One example is the rapid destruction of T-cell receptor α chains that fail to assemble into complexes with other receptor subunits (Bonifacino et al. 1989, 1990). This general idea was then extended to include nuclear proteins including cyclin E, which is protected from ubiquitination when assembled with Cdk2 (Clurman et al. 1996), and E2F-1, which is protected from ubiquitination when bound to Rb (Hofmann et al. 1996). The α2 transcription factor in budding yeast is another example of a protein whose ubiquitination is controlled in this way (Johnson et al. 1998; Laney and Hochstrasser 1999). Within the α2 protein is the Deg1 sequence, which is a recognition motif for an E3 ubiquitin–protein ligase. When the α2 protein is monomeric, the Deg1 sequence is exposed, and α2 is rapidly ubiquitinated and turned over by the proteasome. However, when α2 binds to its partner, the a1 transcription factor, the Deg1 motif is buried in the heterodimer interface, and the protein is protected from ubiquitination. In the case of cyclin E, Cul-3 recognizes and stimulates the ubiquitination of unbound cyclin E, not cyclin E within cyclin E–Cdk2 complexes. Cyclin E, like α2, might contain an instability determinant that is masked in the cyclin E–Cdk2 complex. Alternatively, it is possible that other features of the cyclin E–Cdk2 complex, such as its kinase activity, might downregulate the interaction between cyclin E and Cul-3. It is important to emphasize that this pathway for cyclin E ubiquitination is unlikely to be limited to the destruction of unfolded or otherwise nonfunctional protein. Cells lacking Cul-3 appear to accumulate excess, biologically active cyclin E as evidenced by the misregulation of S phase in Cul-3−/− embryos. It therefore seems that the unbound cyclin E is at least potentially active, and it is crucial for the cell to limit the size of this pool.
Other features of Cul-3 also suggest that there may be significant differences between its mechanism of action and the phosphorylation-dependent pathway controlled by the SCF. Among the mammalian cullins, Cul-1, not Cul-3, is most closely related to Cdc53 (the cullin component of the budding yeast SCF). Cul-1, like Cdc53, binds to Skp1 and to the E2 ubiquitin-conjugating enzyme Cdc34 (Lisztwan et al. 1998; Michel and Xiong 1998; Yu et al. 1998). Moreover, human Cul-1 can complement mutations in budding yeast Cdc53 and Cul-1 is involved in the phosphorylation-triggered ubiquitination of proteins in mammalian cells, including E2F-1, B-catenin, and IkB (Hatakeyama et al. 1999; Kroll et al. 1999; Latres et al. 1999; Suzuki et al. 1999; Winston et al. 1999). Cul-3, on the other hand, does not bind to Skp-1 (Michel and Xiong 1998) or Cdc34 (our unpublished observations), does not complement mutations in Cdc53 (J. Singer et al., unpubl.), and does not require substrate phosphorylation for binding and ubiquitination. It therefore seems possible that these two pathways for ubiquitinating protein substrates will be governed by very distinct E3 enzymes. In some embryonic tissues, like the embryonic ectoderm, loss of Cul-3 had no apparent effect on cyclin E abundance, suggesting that the relative importance of different pathways for controlling cyclin E abundance may vary among different cell types. Phosphorylation-triggered ubiquitination of cyclin E, perhaps mediated by Cul-1, may be a counterpart to the Cul-3 pathway that specifically recognizes unbound cyclin E. The above discussion is not intended to exclude the possibility that Cul-3 is also involved in the ubiquitination of phosphorylated cyclin E. We have suggested that phosphorylation may trigger the separation of cyclin E from its CDK partner, in which case the two ubiquitination pathways would converge.
S-phase regulation by Cul-3
Regulation of S phase is abnormal in Cul-3−/− embryos. In extraembryonic ectodermal cells, which undergo a standard mitotic cycle, the loss of Cul-3 results in a greatly elevated frequency of cells in S phase. In the trophoblast giant cells, which endoreduplicate their genomes, the loss of Cul-3 has the opposite effect of imposing a block to S-phase entry. These paradoxical results can both be explained by elevated expression of cyclin E.
Most current models of cell cycle regulation incorporate the idea that each round of DNA replication (i.e., each S phase) is regulated by two CDK-dependent steps. The first step requires that CDK activity be low or absent, creating an environment permissive for the assembly of initiation complexes at replication origins. The second step requires active CDKs to initiate DNA synthesis from those primed origins and to simultaneously establish an environment refractory to assembly of new initiation complexes. Rounds of genome duplication, as occur either during endocycles or in consecutive mitotic cycles, would then each require that CDK activity oscillate between a state permissive for assembly of initiation complexes and a state permissive for starting DNA synthesis.
This model predicts that constitutively high CDK activity would interrupt the replication cycle by not allowing assembly of new initiation complexes at replication origins. This was tested in the salivary gland cells of developing Drosophila embryos (Follette et al. 1989; Weiss et al. 1998). These cells endoreduplicate their genomes, each round of S phase being preceded by a fall in cyclin-E activity and then initiated in concert with a rise in cyclin E activity. Overexpression of cyclin E prevented this oscillation and the endoreduplication cycles were blocked. We suggest that S-phase entry in Cul-3−/− trophoblasts is similarly prevented by the constitutively elevated amounts of cyclin E in these cells.
In contrast to what happens in cells undergoing endoreduplication, increased expression of cyclin E in cells undergoing a typical mitotic cycle decreases the duration of G1 and increases the percentage of cells in S phase (Ohtsubo and Roberts 1993; Resnitzky and Reed 1995). This result is consistent with what we observed in the Cul-3−/− cells of the extraembryonic ectoderm, which have increased cyclin E and extra S-phase cells. There are various ways to reconcile the seemingly contradictory effects of elevated cyclin E in mitotic cycles and endocycles. Most explanations focus on a key difference between these two types of cell cycles—the presence or absence of an M phase—and postulate that cyclin E somehow becomes functionally inactivated during mitosis, permitting the replication cycle to continue despite the constitutive presence of the cyclin. One interesting idea is that during mitosis, nuclear cyclin-CDK enzymes are dispersed into the cytoplasm by nuclear envelope breakdown, resulting in a de facto oscillation in CDK activity (Hua et al. 1997).
Materials and methods
Two-hybrid screen
The R130A cyclin E cDNA was cloned in frame to the LexA gene in the vector BTM116. Yeast cells containing LexA-binding sites in the HIS3 promoter and carrying a plasmid containing a LexA-dependent promoter driving lacZ expression were transformed with the cyclin E bait as well as a mouse embryonic cDNA library. Transformed cells were grown under selection for the plasmids overnight and plated for the two-hybrid interaction the next day. Potential candidates were screened for β-galactosidase activity and positive clones were rechecked with a LexA lamin bait for specificity.
Antibodies
hCul-3 was subcloned as three parts in frame with a polyhistidine tag in the vector pET16; the amino-terminal portion contains amino acids 1–286, the middle portion amino acids 287–553, and the carboxy-terminal portion amino acids 554–768. All three peptides were expressed in the bacterial strain BL21(DE3) and cell lysates were either passed over nickel columns, after which Cul-3 was eluted with imidazole, or the lysates were mixed with SDS sample buffer and separated from other cellular proteins by electrophoresis followed by electroelution of the Cul-3 protein. Purified protein was then used for inoculation into rabbits. To affinity purify antibodies, a strip of membrane containing the Cul-3 peptide was incubated with serum and the bound antibodies eluted with low pH glycine. Cells studied included human diploid fibroblasts, human diploid microvascular endothelial cells, human diploid umbilical vein endothelial cells, HeLa cells, h293 cells, U2-OS cells, NIH-3T3 cells, and primary mouse embryo fibroblasts (MEFs).
The following antibodies were used in these experiments: monoclonal anti-myc tag (9E10) and rabbit polyclonal anti-cyclin E (Clurman et al. 1996); monoclonal anti-cyclin E (M20) (Santa Cruz Biotechnology); monoclonal anti-Troma-1 (Developmental Studies Hybridoma Bank, University of Iowa); rabbit polyclonal anti-HA tag (HA.11) (Berkeley Antibody Company); rabbit polyclonal anti Cul-1 (J. Michel and Y. Xiong, University of North Carolina, Chapel Hill).
Transient transfections, cell lysis, Western blots, and immunoprecipitations were performed as described previously (Clurman et al. 1996). Immunoprecipitations were routinely checked for the presence of the immuneprecipitated protein, and all interactions between two transfected proteins were shown to be dependent on, or stimulated by, transfection of both proteins.
Immunofluorescence
Immunofluorescence was performed on cells adhering to coverslips by fixing in 4% paraformaldehyde in PBS for 10 min followed by treatment with 0.2% Triton X-100 for an additional 10 min. Cells were then blocked with 1% BSA and 20% goat serum for 30 min. Coverslips were inverted onto 20 μl of primary antibody and incubated for 1 hr. The coverslips were washed and placed on 20 μl of secondary antibody for an additional hour. They were then incubated with DAPI (4′,6-diamidino-2-phenylindole) for 5 min, dehydrated in 100% MeOH, followed by mounting in 6 μl of Vectashield and sealed with nail polish. Blastocysts were fixed in 3.7% formaldehyde for 15 min, permeabilized in 0.3% Triton X-100, and stained as above. Images were obtained on a Nikon E800 fluorescent microscope with a digital camera (SenSys) and Metamorph software.
Immunohistochemistry
Embryos (7.5 day) were prepared by timed matings with Cul-3 heterozygous animals. The pregnant uterus was surgically removed and the individual decidua were separated and fixed in 4% paraformaldehyde overnight at 4°C. Embryos were then embedded in paraffin blocks and cut into 4 μm sections. The sections were placed on slides to be used for either antibody staining or RNA in situ hybridization.
For antibody staining, the sections were deparaffinized and placed into a 3% solution of hydrogen peroxide in methanol for 10 min. Slides were immersed in 10 mm citrate buffer and boiled for 10 min in a microwave oven. The slides were allowed to cool and were then treated with 5% serum followed by an overnight incubation at 4°C with primary antibody. Sections were then incubated with biotinylated secondary for 1 hr and avidin–HRP complex for an additional 30 min. The slides where then immersed in DAB (3,3′-diaminobenzidine)/NiCl2 solution for 3 min, rinsed, dehydrated, and mounted.
In situ hybridizations were performed by deparaffinizing the sections and treating them with proteinase K for 5 min. The sections were then fixed in 4% paraformaldehyde for 15 min. The slides were then placed in prehybridization solution for 2 hr at 65°C in a sealed humidified container. The sections were then hybridized overnight with digoxigenin-labeled riboprobe. The slides were washed, blocked, and incubated overnight with alkaline phosphatase conjugated anti-digoxigenin antibody. The RNA was visualized by staining with a NBT/BCIP solution for 8 hr followed by dehydration and mounting.
For the H19 in situs a 2-kb EcoRI fragment from the carboxyl terminus of the cDNA was used for making the riboprobe. The cyclin E riboprobe was made from the last 600 bp of the mouse cyclin E cDNA. The antibodies used for cyclin A (c19), cyclin D1 (72–13G), and cyclin E (M20) immunohistochemistry were from Santa Cruz Biotechnology, or cyclin E antibody described previously was used (Clurman et al. 1996).
Targeted mouse gene disruption
A 22-kb NotI fragment containing a portion of the Cul-3 gene was obtained from a mouse 129/Sv λ genomic library with portions of the Cul-3 cDNA as a probe. Sequencing of the genomic insert was done partially by shotgun cloning HaeIII–AluI partial digest fragments of the 22-kb NotI fragment into the EcoRV site of pBSII (Stratagene) and sequencing 100 individual clones with both the T7 and T3 sequencing primers. The sequencing data was then assembled into multiple contigs with Sequencher software. Alignments with the Cul-3 cDNA sequence and sequence analysis was performed with DNA Strider. pJS1052 was constructed by cloning a 6.8-kb EcoRI fragment as the upstream arm that ended at amino acid number 126 of the Cul-3 coding region and a downstream 1.4-kb XbaI–EcoRV Cul-3 genomic fragment that contained coding regions starting with amino acid number 294 of the Cul-3 protein into the targeting vector pPNT. The vector was linearized with NotI and transfected into XY AK7 ES cells with electroporation. The ES cells were then selected in 400 μg/ml G418 and 0.2 μm FIAU. ES cell colonies with homologous recombination were identified by PCR amplification of a 2-kb fragment with a primer from the neomycin gene (pgk2, CCCTTCCCAGCCTCTGAG) and a primer from Cul-3 genomic DNA (cul3PCR2, CAACTCATACATTCACACATGG). PCR reactions were performed for 40 cycles (93°C for 30 sec; 57°C for 30 sec; 65°C for 2 min). ES cells were introduced into 5 d.p.c. C57/B6J mouse embryos. Germ-line transmission, as determined by PCR, was identified in chimeric males obtained from two independent clones that were used for subsequent experiments.
Acknowledgments
We thank members of the Roberts laboratory, especially Holly Sundberg, Robert Sheaff, and Nisar Malek for advice and discussion. We also thank Ray Deshaies for Cul-1 materials, Phil Soriano for advice, and the 129 genomic library, Keesook Lee for her help with the mouse knockout, and Mike Tyers and Randy Johnson for communicating results prior to their publication. This work was supported in part by a grant from Mitotix Incorporated, and by grants from the National Institutes of Health to J.M.R. J.D.S. is supported by a fellowship from the Department of Defense, grant no. DADM17-98-1-8085. J.M.R. is an investigator of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jroberts@fred.fhcrc.org; FAX (206) 667-6877.
References
- Blow JJ, Nurse P. A cdc2-like protein is involved in the initiation of DNA replication in Xenopus egg extracts. Cell. 1990;62:855–862. doi: 10.1016/0092-8674(90)90261-c. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Weissman AM. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol. 1998;14:19–57. doi: 10.1146/annurev.cellbio.14.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Suzuki CK, Lippincott-Schwartz J, Weissman AM, Klausner RD. Pre-Golgi degradation of newly synthesized T-cell antigen receptor chains: Intrinsic sensitivity and the role of subunit assembly. J Cell Biol. 1989;109:73–83. doi: 10.1083/jcb.109.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Cosson P, Klausner RD. Colocalized transmembrane determinants for ER degradation and subunit assembly explain the intracellular fate of TCR chains. Cell. 1990;63:503–513. doi: 10.1016/0092-8674(90)90447-m. [DOI] [PubMed] [Google Scholar]
- Brulet P, Babinet C, Kemler R, Jacob F. Monoclonal antibodies against trophectoderm-specific markers during mouse blastocyst formation. Proc Natl Acad Sci. 1980;77:4113–4117. doi: 10.1073/pnas.77.7.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes & Dev. 1996;10:1979–1990. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- Connell-Crowley L, Harper JW, Goodrich DW. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol Biol Cell. 1997;8:287–301. doi: 10.1091/mbc.8.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Chau V, Kirschner MW. Ubiquitination of the G1 cyclin Cln2 by a Cdc34p dependent pathway. EMBO J. 1995;14:303–312. doi: 10.1002/j.1460-2075.1995.tb07004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Sansores-Garcia L, Zu Z, Wu KK. Cloning and expression analysis of a novel salicylate suppressible gene, Hs-CUL-3, a member of cullin/Cdc53 family. J Biol Chem. 1998;273:24289–24292. doi: 10.1074/jbc.273.38.24289. [DOI] [PubMed] [Google Scholar]
- Dulic V, Lees E, Reed SI. Association of human cyclin E with a periodic G1-S phase protein kinase. Science. 1992;257:1958–1961. doi: 10.1126/science.1329201. [DOI] [PubMed] [Google Scholar]
- D’Urso G, Marraccino RL, Marshak DR, Roberts JM. Cell cycle control of DNA replication by a homologue from human cells of the p34cdc2 protein kinase. Science. 1990;250:786–791. doi: 10.1126/science.2173140. [DOI] [PubMed] [Google Scholar]
- Fang F, Newport JW. Evidence that the G1-S and G2-M transitions are controlled by different cdc2 proteins in higher eukaryotes. Cell. 1991;66:731–742. doi: 10.1016/0092-8674(91)90117-h. [DOI] [PubMed] [Google Scholar]
- Feldman RM, Correll CC, Kaplan KB, Deshaies RJ. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- Follette PJ, Duronio RJ, O’Farrell PH. Fluctuations in cyclin E levels are required for multiple rounds of endocycle S phase in Drosophila. Curr Biol. 1998;8:235–238. doi: 10.1016/s0960-9822(98)70089-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Eaton EN, Picon M, Roberts JM, Lundberg AS, Gifford A, Sardet C, Weinberg RA. Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene. 1996;12:1173–1180. [PubMed] [Google Scholar]
- Geng Y, Whoriskey W, Park MY, Bronson RT, Medema RH, Li T, Weinberg RA, Sicinski P. Rescue of cyclin D1 deficiency by knockin cyclin E. Cell. 1999;97:767–777. doi: 10.1016/s0092-8674(00)80788-6. [DOI] [PubMed] [Google Scholar]
- Gong L, Yeh ET. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J Biol Chem. 1999;274:12036–12042. doi: 10.1074/jbc.274.17.12036. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Kitagawa M, Nakayama K, Shirane M, Matsumoto M, Hattori K, Higashi H, Nakano H, Okumura K, Ono K, et al. Ubiquitin-dependent degradation of IkappaBalpha is mediated by a ubiquitin ligase Skp1/Cul 1/F-box protein FWD1. Proc Natl Acad Sci. 1999;96:3859–3863. doi: 10.1073/pnas.96.7.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuvel Svd, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- Hicke L. Gettin’ down with ubiquitin: Turning off cell-surface receptors, transporters and channels. Trends Cell Biol. 1999;9:107–112. doi: 10.1016/s0962-8924(98)01491-3. [DOI] [PubMed] [Google Scholar]
- Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Martelli F, Livingston DM, Wang Z. The retinoblastoma gene product protects E2F-1 from degradation by the ubiquitin-proteasome pathway. Genes & Dev. 1996;10:2949–2959. doi: 10.1101/gad.10.23.2949. [DOI] [PubMed] [Google Scholar]
- Hua XH, Yan H, Newport J. A role for Cdk2 kinase in negatively regulating DNA replication during S phase of the cell cycle. J Cell Biol. 1997;137:183–192. doi: 10.1083/jcb.137.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtley SM, Helenius A. Protein oligomerization in the endoplasmic reticulum. Annu Rev Cell Biol. 1989;5:277–307. doi: 10.1146/annurev.cb.05.110189.001425. [DOI] [PubMed] [Google Scholar]
- Jackson PK, Chevalier S, Philippe M, Kirschner MK. Early events in DNA replication require cyclin E and are blocked by p21CIP1. J Cell Biol. 1995;130:755–769. doi: 10.1083/jcb.130.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PR, Swanson R, Rakhilina L, Hochstrasser M. Degradation signal masking by heterodimerization of MATalpha2 and MATa1 blocks their mutual destruction by the ubiquitin-proteasome pathway. Cell. 1998;94:217–227. doi: 10.1016/s0092-8674(00)81421-x. [DOI] [PubMed] [Google Scholar]
- Kamitani T, Kito K, Nguyen HP, Yeh ET. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem. 1997;272:28557–28562. doi: 10.1074/jbc.272.45.28557. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Wolfe KG, Roberts JM. Identification of a substrate-targeting domain in cyclin E necessary for phosphorylation of the retinoblastoma protein. Proc Natl Acad Sci. 1998;95:2535–2540. doi: 10.1073/pnas.95.5.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM. cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell. 1996;85:829–839. doi: 10.1016/s0092-8674(00)81267-2. [DOI] [PubMed] [Google Scholar]
- Knoblich JA, Sauer K, Jones L, Richardson H, Saint R, Lehner CF. Cyclin E controls S phase progression and its down-regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell. 1994;77:107–120. doi: 10.1016/0092-8674(94)90239-9. [DOI] [PubMed] [Google Scholar]
- Koepp DM, Harper JW, Elledge SJ. How the cyclin became a cyclin: Regulated proteolysis in the cell cycle. Cell. 1999;97:431–434. doi: 10.1016/s0092-8674(00)80753-9. [DOI] [PubMed] [Google Scholar]
- Koff A, Cross F, Schumacher J, Leguellec K, Philippe M, Roberts JM. Human Cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell. 1991;66:1217–1228. doi: 10.1016/0092-8674(91)90044-y. [DOI] [PubMed] [Google Scholar]
- Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, Nishimoto T, Morgan DO, Franza BR, Roberts JM. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science. 1992;257:1689–1694. doi: 10.1126/science.1388288. [DOI] [PubMed] [Google Scholar]
- Koff A, Ohtsuki M, Polyak K, Roberts JM, Massague J. Negative regulation of G1 in mammalian cells: Inhibition of cyclin E-dependent kinase by TGF-beta. Science. 1993;260:536–539. doi: 10.1126/science.8475385. [DOI] [PubMed] [Google Scholar]
- Kroll M, Margottin F, Kohl A, Renard P, Durand H, Concordet JP, Bachelerie F, Arenzana-Seisdedos F, Benarous R. Inducible degradation of IkappaBalpha by the proteasome requires interaction with the F-box protein h-betaTrCP. J Biol Chem. 1999;274:7941–7945. doi: 10.1074/jbc.274.12.7941. [DOI] [PubMed] [Google Scholar]
- Lammer D, Mathias N, Laplaza JM, Jiang W, Liu Y, Callis J, Goebl M, Estelle M. Modification of yeast Cdc53p by the ubiquitin-related protein rub1p affects function of the SCFCdc4 complex. Genes & Dev. 1998;12:914–926. doi: 10.1101/gad.12.7.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laney JD, Hochstrasser M. Substrate targeting in the ubiquitin system. Cell. 1999;97:427–430. doi: 10.1016/s0092-8674(00)80752-7. [DOI] [PubMed] [Google Scholar]
- Latres E, Chiaur DS, Pagano M. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene. 1999;18:849–854. doi: 10.1038/sj.onc.1202653. [DOI] [PubMed] [Google Scholar]
- Leng X, Connell-Crowley L, Goodrich D, Harper JW. S-Phase entry upon ectopic expression of G1 cyclin-dependent kinases in the absence of retinoblastoma protein phosphorylation. Curr Biol. 1997;7:709–712. doi: 10.1016/s0960-9822(06)00301-0. [DOI] [PubMed] [Google Scholar]
- Liakopoulos D, Doenges G, Matuschewski K, Jentsch S. A novel protein modification pathway related to the ubiquitin system. EMBO J. 1998;17:12208–2214. doi: 10.1093/emboj/17.8.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisztwan J, Marti A, Sutterluty H, Gstaiger M, Wirbelauer C, Krek W. Association of human CUL-1 and ubiquitin-conjugating enzyme CDC34 with the F-box protein p45(SKP2): Evidence for evolutionary conservation in the subunit composition of the CDC34-SCF pathway. EMBO J. 1998;17:368–383. doi: 10.1093/emboj/17.2.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J, Bartkova J, Rohde M, Strauss M, Bartek J. Cyclin D1 is dispensable for G1 control in retinoblastoma gene-deficient cells independently of cdk4 activity. Mol Cell Biol. 1995;15:2600–2611. doi: 10.1128/mcb.15.5.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyapina SA, Correll CC, Kipreos ET, Deshaies RJ. Human CUL1 forms an evolutionarily conserved ubiquitin ligase complex (SCF) with SKP1 and an F-box protein. Proc Natl Acad Sci. 1998;95:7451–7456. doi: 10.1073/pnas.95.13.7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAuley A, Cross JC, Werb Z. Reprogramming the cell cycle for endoreduplication in rodent trophoblast cells. Mol Biol Cell. 1998;9:795–807. doi: 10.1091/mbc.9.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel JJ, Xiong Y. Human CUL-1, but not other cullin family members, selectively interacts with SKP1 to form a complex with SKP2 and cyclin A. Cell Growth Differ. 1998;9:435–449. [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo M, Roberts JM. Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science. 1993;259:1908–1912. doi: 10.1126/science.8384376. [DOI] [PubMed] [Google Scholar]
- Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol. 1995;15:2612–2624. doi: 10.1128/mcb.15.5.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaka F, Kawasaki H, Aida N, Saeki M, Chiba T, Kawashima S, Tanaka K, Kato S. A new NEDD8-ligating system for cullin-4A. Genes & Dev. 1998;12:2263–2268. doi: 10.1101/gad.12.15.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton EE, Willems AR, Sa D, Kuras L, Thomas D, Craig KL, Tyers M. Cdc53 is a scaffold protein for multiple Cdc34/Skp1/F-box protein complexes that regulate cell division and methionine biosynthesis in yeast. (Published erratum appears in 12: 3144.) Genes & Dev. 1998;12:692–705. doi: 10.1101/gad.12.5.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier F, Chan C-TJ, Timmons PM, Robertson EJ, Evans MJ, Rigby PWJ. The mirine H19 gene is activated during embryonic stem cell differentiation in vitro and at the time of implantation in the developing embryo. Development. 1991;113:1105–1114. doi: 10.1242/dev.113.4.1105. [DOI] [PubMed] [Google Scholar]
- Resnitzky D, Reed SI. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol Cell Biol. 1995;15:3463–3469. doi: 10.1128/mcb.15.7.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E–CDK2 is a regulator of p27Kip1. Genes & Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. D-type cyclins. Trends Biochem Sci. 1995;20:187–190. doi: 10.1016/s0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Skowyra D, Koepp DM, Kamura T, Conrad MN, Conaway RC, Conaway JW, Elledge SJ, Harper JW. Reconstitution of G1 cyclin ubiquitination with complexes containing SCFGrr1 and Rbx1. Science. 1999;284:662–665. doi: 10.1126/science.284.5414.662. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Chiba T, Kobayashi M, Takeuchi M, Suzuki T, Ichiyama A, Ikenoue T, Omata M, Furuichi K, Tanaka K. IkappaBalpha ubiquitination is catalyzed by an SCF-like complex containing Skp1, cullin-1, and two F-box/WD40-repeat proteins, betaTrCP1 and betaTrCP2. Biochem Biophys Res Commun. 1999;256:127–132. doi: 10.1006/bbrc.1999.0289. [DOI] [PubMed] [Google Scholar]
- Wada H, Yeh ET, Kamitani T. Identification of NEDD8-conjugation site in human cullin-2. Biochem Biophys Res Comm. 1999;257:100–105. doi: 10.1006/bbrc.1999.0339. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Weiss A, Herzig A, Jacobs H, Lehner CF. Continuous Cyclin E expression inhibits progression through endoreduplication cycles in Drosophila. Curr Biol. 1998;8:239–242. doi: 10.1016/s0960-9822(98)70090-9. [DOI] [PubMed] [Google Scholar]
- Willems AR, Lanker S, Patton EE, Craig KL, Nason TF, Mathias N, Kobayashi R, Wittenberg C, Tyers M. Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway. Cell. 1996;86:453–463. doi: 10.1016/s0092-8674(00)80118-x. [DOI] [PubMed] [Google Scholar]
- Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW. The SCFβ-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBβ in vitro. Genes & Dev. 1999;13:270–283. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won KA, Reed SI. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J. 1996;15:4182–4193. [PMC free article] [PubMed] [Google Scholar]
- Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci. 1998;95:11324–11329. doi: 10.1073/pnas.95.19.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem. 1997;272:12738–12746. doi: 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
- Zhao J, Dynlacht B, Imai T, Hori T, Harlow E. Expression of NPAT, a novel substrate of cyclin E–CDK2, promotes S-phase entry. Genes & Dev. 1998;12:456–461. doi: 10.1101/gad.12.4.456. [DOI] [PMC free article] [PubMed] [Google Scholar]