

Abstract
Eukaryotic genomes often contain more potential replication origins than are actually used during S phase. The molecular mechanisms that prevent some origins from firing are unknown. Here we show that dormant replication origins on the left arm of budding yeast chromosome III become activated when both passive replication through them is prevented and the Mec1/Rad53 checkpoint that blocks late-origin firing is inactivated. Under these conditions, dormant origins fire very late relative to other active origins. These experiments show that some dormant replication origins are competent to fire during S phase and that passage of a replication fork through such origins can inactivate them.
Keywords: DNA replication, S. cerevisiae, ORC
DNA replication initiates from multiple origins in eukaryotic cells (Cairns 1966; Huberman and Riggs 1968). The usage of these replication origins changes dramatically during metazoan development. For example, in early Drosophila cleavage embryos, S phase lasts just a few minutes, whereas it takes 8 hr for the same process to occur in somatic cells (Dolfini et al. 1970; Blumenthal et al. 1973). This increase in the length of S phase is achieved both by inactivating a large number of origins, thus increasing the distance that each replication fork must travel, and by imposing asynchrony in the time of activation of the remaining active origins (Blumenthal et al. 1973). Similar observations have been made in amphibian development (Callan 1973). Recently, it has been shown that the inactivation of origins within the rDNA locus during Xenopus development is not a random process. Early in development, initiation occurs at many sites across the locus. However, as the embryo approaches the midblastula transition, initiation events become increasingly localized to the nontranscribed spacer region between each transcription unit (Hyrien et al. 1995). In Xenopus egg extracts it has been shown that origin inactivation is not due to limiting amounts of known initiator proteins such as the origin recognition complex (ORC), Cdc6, or minichromosome maintenance proteins (MCM) (Walter and Newport 1997). A similar restriction of potential origin use occurs during the G1 phase of the mammalian cell cycle. Prior to an origin decision point (ODP), mammalian G1 nuclei initiate replication randomly across the dihydrofolate reductase (DHFR) locus in Xenopus egg extracts; after this ODP, initiation occurs more specifically at the DHFR origin (Wu and Gilbert 1996).
Several dormant origins of replication have been documented in the budding yeast Saccharomyces cerevisiae (Dubey et al. 1991; Newlon et al. 1993). These sequences, when removed from their chromosomal context and placed in a plasmid, can initiate replication efficiently; however, they do not initiate in their normal chromosomal context. We have shown previously that the ORC efficiently binds one of these origins, ARS301, in vivo. Additionally, a Cdc6p-dependent prereplicative complex (pre-RC) is assembled at ARS301 during G1 and displaced some time after Start but before mitosis. Comparison of the genomic footprinting pattern of ARS301 in the active (plasmid) and inactive (chromosome) states indicated that there were no apparent differences in the architecture of either the pre- or post-RCs (Santocanale and Diffley 1996). In this paper we describe conditions that allow activation of these origins in their normal chromosomal location.
Results and Discussion
When replication forks from early-firing origins are stalled by dNTP depletion using hydroxyurea (HU), late-firing origins are actively prevented from firing by the Mec1/Rad53 checkpoint pathway (Santocanale and Diffley 1998). A similar phenomenon is seen when DNA is damaged (Shirahige et al. 1998; M. Weinberger, P.A. Trabold, M. Lu, K. Sharma, J.A. Huberman, and W.C. Burhans, in prep.). This ‘origin-firing checkpoint’ blocks the pre-RC-to-post-RC conversion at late origins, thus maintaining these origins in an initiation-competent state for a long period of time (Santocanale and Diffley 1998). Pre-RCs at ARS301 are distinguished by Cdc6p-dependent protection of ORC-induced DNase I-hypersensitive sites (Fig. 1, cf. lanes 4–6 and 7–9, asterisks). Figure 1 (lanes 10–12) shows that the chromosomal ARS301 remains prereplicative in HU in wild-type cells. In a rad53 mutant, however, the pre-RC at ARS301 is lost in HU as evidenced by reappearance of the hypersensitive sites (Fig. 1, cf. lanes 17 and 18 and 13–16).
Figure 1.
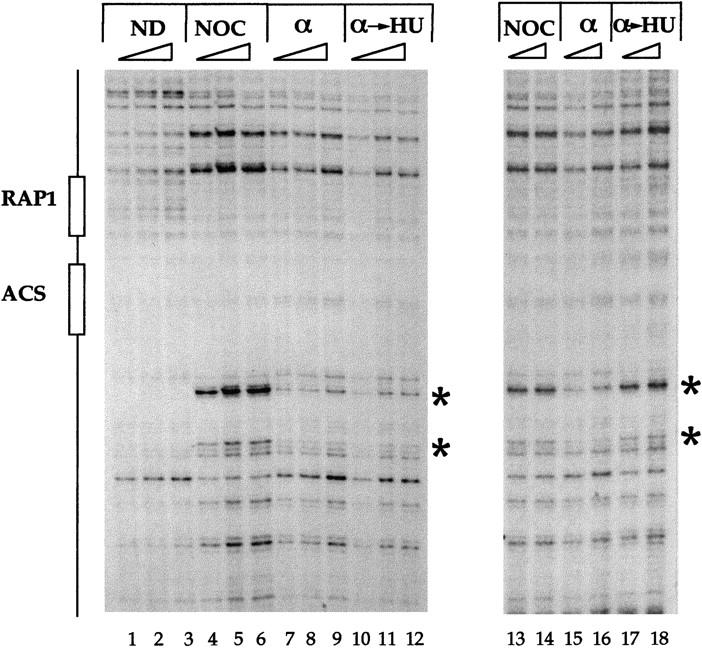
Chromosomal ARS301 conversion from pre- to postreplicative state in HU is blocked by the MEC1/RAD53 checkpoint. Genomic-footprinting analysis of the chromatin structure of ARS301. Wild-type (y300; left) or rad53 (y301; right (Allen et al. 1994) cells were arrested in G2 with nocodazole (NOC) or in G1 with α factor (α) or released from α-factor block into HU for 90 min (α → HU). Naked DNA (ND) was included as control. (*) ORC-induced hypersensitive sites. Cell cycle blocks and genomic footprinting protocols were as described previously (Santocanale and Diffley 1996, 1998). Samples were treated with increasing amounts of DNase I.
ARS301 is normally passively replicated by a fork originating from ARS305 (Dubey et al. 1991), located ∼30 kb away from ARS301. We have shown previously using both density substitution and alkaline gel electrophoresis that replication initiates efficiently at ARS305 but replication forks stall within the first ∼10 kb when G1-arrested cells are released into HU (Bousset and Diffley 1998; Santocanale and Diffley 1998). Abrogation of the Mec1/Rad53 checkpoint does not promote detectably more DNA synthesis in the presence of HU nor does it appear to change the position at which the replication fork from ARS305 stalls (Santocanale and Diffley 1998). Thus, the loss of pre-RCs at ARS301 (Fig. 1, lanes 17,18) was unlikely to be the result of passive replication from ARS305.
An alternative possibility is that ARS301 might be activated in HU in the rad53 mutant. We therefore looked for replication intermediates (RIs) originating from ARS301 itself. Wild-type and rad53 or mec1 mutant cells were released from a G1 block into HU-containing medium and small nascent RIs were examined by alkaline gel electrophoresis and DNA blot hybridization using a probe to ARS301 (Santocanale and Diffley 1998). Figure 2A shows that RIs are not detected in wild-type cells but accumulate in both checkpoint mutants with time. To further characterize this we examined RIs by neutral–neutral two-dimensional gel electrophoresis (Brewer and Fangman 1987). Figure 2B shows that RIs can be seen in both mutants but not wild-type cells after 90 min in HU, confirming the results obtained with the alkaline gels. In the two-dimensional gels both bubble arcs and Y arcs can be seen. Detection of a bubble arc in both the rad53 and mec1 mutants demonstrates that the dormant origin ARS301 becomes active in at least a fraction of these checkpoint-deficient cells.
Figure 2.

Activation of the dormant chromosomal ARS301 and ARS302/303. (A) Detection of RIs from ARS301. α factor-arrested cells were released from the G1 block into medium containing HU. At the indicated times replication intermediates that originated from ARS301 were studied using the alkaline gel electrophoresis method as described (Santocanale and Diffley 1998). The relevant genotype is indicated. A 207-bp HindIII–BamHI DNA fragment containing the ARS301 sequence prepared from plasmid pCS1 (Santocanale and Diffley 1996) was used as a probe. (B) Characterization of RIs from ARS301. DNAs prepared from α factor-arrested cells (α) or cells released into HU containing medium for 90 min (HU 90′) were analyzed as described in Materials and Methods. (C) Detection of replication intermediates from ARS302. Cells were grown and RIs analyzed as in A. A 1-kb DNA fragment of chromosome III (14,000–15,000 nucleotides) containing the ARS302 sequence was used as a probe for hybridization (lanes 1–6). In lanes 7–12 a 0.8-kb fragment of chromosome III (21,200–22,000 nucleotides) that does not contain origin sequences and lies between ARS303 and ARS304 was used as a probe.
The presence of Y arcs in these gels, indicative of passive replication in some cells, suggests that other dormant origins located nearby may also have been activated. ARS302 and ARS303 are very close together (within 500 bp) and are only ∼3.5 kb from ARS301. Figure 2C (lanes 1–6) shows that we were also able to detect small RIs that hybridize with a probe spanning ARS302/303 in HU in the rad53 mutant but not in wild-type cells. As an additional control, Figure 2C (lanes 7–12) shows that small RIs were not detected with a probe between ARS302/303 and ARS304 (≥ 6 kb from each ARS), consistent with the notion that these RIs are only found near replication origins. In conclusion, both ARS301 and ARS302/303 become active in HU in checkpoint-mutant cells.
The activation of ARS301 in the previous experiments occurred in checkpoint mutants but not in wild-type cells when replication was blocked with HU. Therefore, activation of ARS301 requires abrogation of the Mec1/Rad53 checkpoint pathway. Next, we asked whether inactivation of this pathway was sufficient to allow ARS301 activation or whether the replication fork block was also required. DNA prepared from logarithmically growing cells in the absence of HU was analyzed by neutral–neutral two-dimensional gels, and RIs from ARS301 were visualized by autoradiography. Unlike Figure 2B, Figure 3 shows that only Y arcs are seen in these experiments. Therefore, ARS301 remains inactive in normally cycling checkpoint-deficient cells. As a control for the integrity of RIs in these experiments the same filters were reprobed with an ARS305 probe, and a full transition from bubble to Y arc was observed (data not shown). Recently it has been shown that the firing of some origins is accelerated in mutants that are defective in transcriptional silencing. In addition, the telomeric X element ARS becomes activated in a sir3 mutant (Stevenson and Gottschling 1999). We tested whether inactivation of both the Mec1/Rad53 pathway and transcriptional silencing could activate ARS301 in the absence of the replication fork block. Figure 3 (bottom panels) show that even in a rad53, sir4 double mutant, ARS301 remains inactive in the absence of HU. The bottom right panel shows that ARS305 is efficiently activated in these cells.
Figure 3.
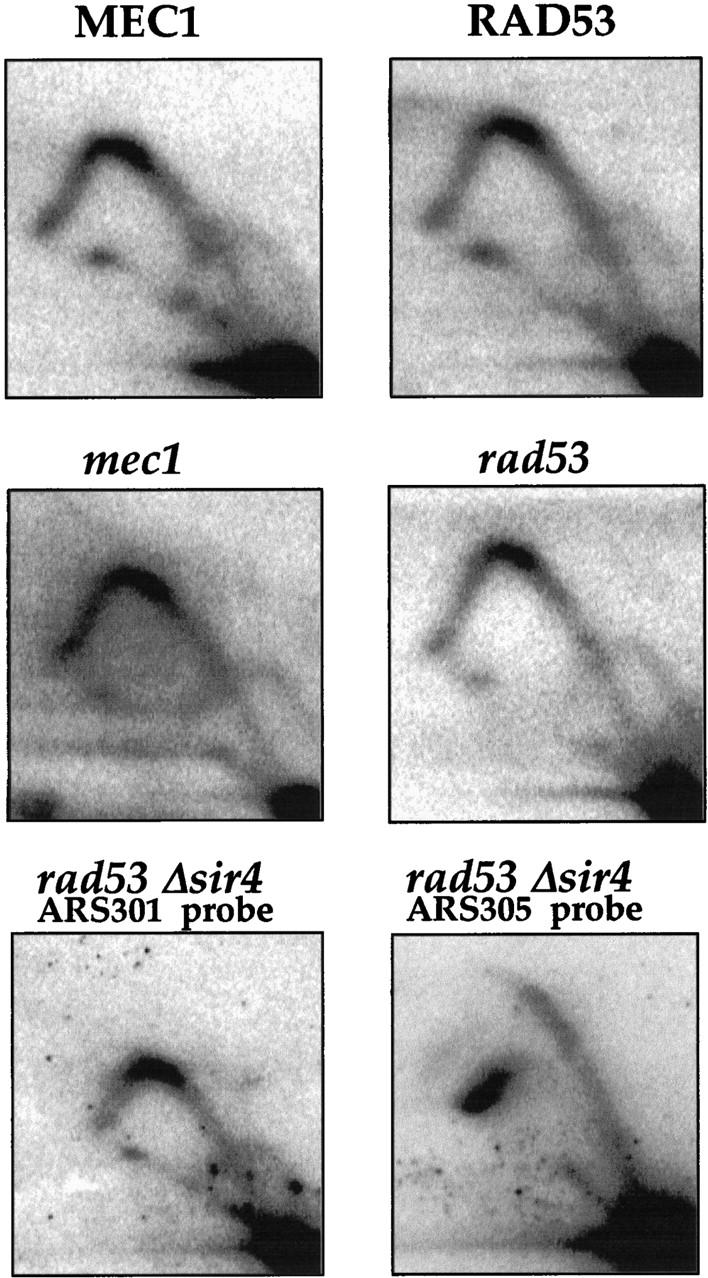
ARS301 is not activated in an unperturbed S phase in checkpoint mutants. Genomic DNA was prepared from logarithmically growing wild-type, rad53, mec1, or rad53/sir4 mutant cells. RIs around ARS301 (or, where indicated, ARS305) were studied as described in Materials and Methods. The relevant genotype of yeast strains Y10860, Y10848 (Paulovich et al. 1997), Y300, Y301 (Allen et al. 1994), and YCS38 (MATa, can1-100, ade2-1, his3-11,15, leu2-3,112, trp1-1, ura3-1, Δsir4::kanR ΔARS1::HIS3, sad1-1) is indicated.
We conclude that both checkpoint deficiency and replication fork arrest are required to activate ARS301 in its normal chromosomal location. HU might act by preventing the replication fork that normally originates from the early-firing ARS305 from passively replicating ARS301 before ARS301 can be activated. A prediction of this hypothesis is that the firing of ARS301 should be considerably delayed relative to that of ARS305 in HU. We have found previously that in the rad53 mutant treated with HU, the relative order and timing of origin firing appears to be maintained (Santocanale and Diffley 1998). We therefore compared the time of ARS301 firing to that of well-characterized early (ARS305)- and late (ARS501)-firing origins. rad53 cells were released from G1 arrest into S phase in the presence of HU and at different times the accumulation of RIs from the different origins was determined. Figure 4 shows that in this experiment, RIs from ARS305 first appear ∼30 min after G1 release and peak at 45 min. The accumulation of RIs from ARS501 is delayed by 15–30 min relative to ARS305. Strikingly, RIs from ARS301 are not detected until 75–90 min after release into HU, a full 45 min after ARS305. The relative times of ARS305 and ARS501 activation obtained in this experiment are similar to those determined for the same origins in wild-type cells without the use of HU (Ferguson et al. 1991; Bousset and Diffley 1998).
Figure 4.
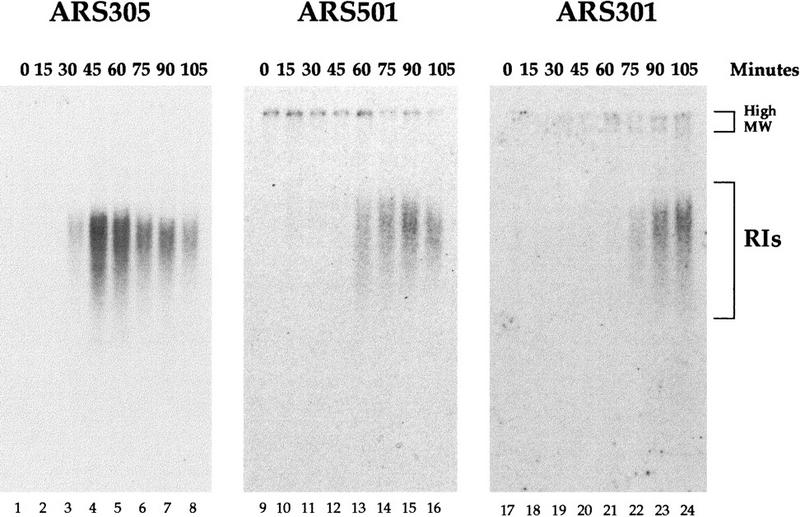
Activation timing of ARS301 in a rad53 mutant in HU. α factor-arrested rad53 cells were released from the G1 block into medium containing HU. At the indicated times aliquots of the culture were collected and genomic DNA prepared as in Fig. 1. Kinetics of RI accumulation from early-firing ARS305, late-firing ARS501, and chromosomal ARS301 were compared using the alkaline gel electrophoresis method.
Accurate measurement of the replication timing of chromosome III has shown that a replication fork originating from early-firing ARS305 takes ∼15 min to passively replicate the ARS301 sequence (Reynolds et al. 1989). Therefore, ARS301 would always be passively replicated before it could be activated. The firing of ARS301 is so late that it would probably be passively replicated by even more distant origins in the absence of ARS305 firing. Previous experiments studying replication fork direction suggested that ARS301 does not become activated when other active origins on chromosome III are deleted (Newlon et al. 1993). More recently, it has been shown directly that ARS301 is weakly activated when both ARS305 and ARS306 are deleted (Vujcic et al. 1999).
Why ARS301 should be such a late-firing replication origin in its chromosomal context is uncertain. We note that a plasmid containing a 207-bp ARS301 fragment replicates after chromosomal ARS305 (Bousset and Diffley 1998), suggesting that some determinants of late ARS301 firing reside in or near the origin. However, on a plasmid, ARS301 fires at or before the time that an active, late-firing origin (ARS501) fires (Bousset and Diffley 1998), suggesting that the time of ARS301 activation is advanced on a plasmid. This is similar to ARS501, which replicates earlier on a plasmid than it does in its normal chromosomal location (Ferguson and Fangman 1992).
One prediction of the licensing factor model is that passage of a replication fork through a potentially active origin should cause its inactivation (Harland and Laskey 1980; Harland 1981; Blow and Laskey 1988). The experiments described in this paper and in our previous work (Santocanale and Diffley 1996) strongly support this view; S-phase progression promotes the conversion of the inactive ARS301 from pre-RC to post-RC. In addition, activation of ARS301 (in the checkpoint mutants) only occurs if replication fork progression is blocked. Together, these results suggest that origin inactivation by passive replication may be a highly conserved mechanism involved in ensuring once-per-cell cycle replication.
Experiments in both budding and fission yeast have shown that when two origins are located close together, it is rare that both origins are active in the same cell during S phase (Brewer and Fangman 1993, 1994; Dubey et al. 1994; Marahrens and Stillman 1994). We have shown previously that this ‘origin interference’ or ‘replicator dominance’ is unlikely to be related to ORC or pre-RC assembly, as these factors bind efficiently to all origins in a very closely spaced tandem array of origins (Santocanale and Diffley 1996). An alternative explanation for this phenomenon is that these closely spaced origins might not fire at the same time. Forks from the first origin to fire might then pass through the second origin, rendering it inactive. This possibility is clearly speculative and warrants further examination.
Origin inactivation during development occurs at the same time that embryonic cells acquire the capacity to inhibit mitosis in response to drugs such as HU in both Xenopus and Drosophila (Kimelman et al. 1987; Raff and Glover 1988; Newport and Dasso 1989). For example, at the mid-blastula transition in Drosophila interphase increases in length mainly because S-phase progression is much slower as a result of both origin inactivation and asynchronous origin firing (Blumenthal et al. 1973; McKnight and Miller 1977). At this point a functional S-phase checkpoint becomes essential (Fogarty et al. 1997; Sibon et al. 1997). It is tempting to speculate that in these organisms, the inactivation of at least some replication origins may be a consequence of the acquisition of a temporal program of origin firing.
Materials and methods
Genomic footprinting, alkaline gel electrophoresis, and detection of replication intermediates were performed as described (Santocanale and Diffley 1998). Two-dimensional electrophoresis was performed as described (Brewer and Fangman 1987); before separation the DNA was digested with EcoRV and HindIII endonucleases. For the experiments in Figure 2, 100-ml log-phase cultures were treated with 6 μm α factor for 3.5 hr at 25°C to arrest in G1. Cells were washed once with YPD medium and released into fresh YPD containing 0.2 m HU. Cells were collected at either 0 or 90 min after G1 release. Genomic DNA was prepared and run directly on the first dimension after digestion, without benzoylated napthoylated DEAE (BND)–cellulose fractionation. For the experiments in Figure 3, genomic DNA was prepared from 2 liters of log-phase cultures. After digestion with restriction enzymes, RIs were enriched using BND–cellulose fractionation as described (Huberman et al. 1987).
All of the two-dimensional gels center ARS301 in a 4.6-kb EcoRV–HindIII fragment, probed with a 1.2-kb XbaI fragment covering ARS301. The gels that show only Y arcs were stripped and reprobed for ARS305 (using a 2-kb BamHI–PstI probe covering ARS305) as a positive control for intact bubble-containing RIs on the same membrane.
The DNA probe for ARS302 was amplified from genomic DNA using
oligonucleotides 5′-CAATCAAAGAAATCTTTCACTGCTC-3′ and 5′-GAAGACCCCAGTATTTTAGC-3′. The probe for the interorigin sequence in chromosome III was amplified using oligonucleotides 5′- CCCGAATTTTTAATGTGTGTGG-3′ and 5′-GCTTCAATAGCATTTCAAACAC-3′.
Acknowledgments
We thank Joel A. Huberman and Alain Verreault for critically reading this manuscript and William Burhans and David Kowalski for communicating results prior to publication. We also thank J.A.H. for advice and encouragement during this project. This work was supported by the Imperial Cancer Research Fund and National Institutes of Health grant GM49294 to J.A.H.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL J.Diffley@icrf.icnet.uk; FAX 44-171-269-3801.
References
- Allen JB, Zhou Z, Siede W, Friedberg EC, Elledge SJ. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes & Dev. 1994;8:2401–2415. doi: 10.1101/gad.8.20.2401. [DOI] [PubMed] [Google Scholar]
- Blow JJ, Laskey RA. A role for the nuclear envelope in controlling DNA replication within the cell cycle. Nature. 1988;332:546–548. doi: 10.1038/332546a0. [DOI] [PubMed] [Google Scholar]
- Blumenthal AB, Kriegstein HJ, Hogness DS. The units of DNA replication in Drosophila melanogaster chromosomes. Cold Spring Harb Symp Quant Biol. 1973;38:205–223. doi: 10.1101/sqb.1974.038.01.024. [DOI] [PubMed] [Google Scholar]
- Bousset K, Diffley JFX. The Cdc7 protein kinase is required for origin firing during S phase. Genes & Dev. 1998;12:480–490. doi: 10.1101/gad.12.4.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer BJ, Fangman WL. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell. 1987;51:463–471. doi: 10.1016/0092-8674(87)90642-8. [DOI] [PubMed] [Google Scholar]
- ————— Initiation at closely spaced replication origins in a yeast chromosome. Science. 1993;262:1728–1731. doi: 10.1126/science.8259517. [DOI] [PubMed] [Google Scholar]
- ————— Initiation preference at a yeast origin of replication. Proc Natl Acad Sci. 1994;91:3418–3422. doi: 10.1073/pnas.91.8.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J. Autoradiography of HeLa cell DNA. J Mol Biol. 1966;15:372–373. doi: 10.1016/s0022-2836(66)80233-4. [DOI] [PubMed] [Google Scholar]
- Callan HG. DNA replication in the chromosomes of eukaryotes. Cold Spring Harb Symp Quant Biol. 1973;38:195–203. doi: 10.1101/sqb.1974.038.01.023. [DOI] [PubMed] [Google Scholar]
- Dolfini S, Courgeon AM, L T. The cell cycle of an established line of Drosophila melanogaster cells in vitro. Experientia. 1970;26:1020–1021. doi: 10.1007/BF02114171. [DOI] [PubMed] [Google Scholar]
- Dubey DD, Davis LR, Greenfeder SA, Ong LY, Zhu JG, Broach JR, Newlon CS, Huberman JA. Evidence suggesting that the ARS elements associated with silencers of the yeast mating-type locus HML do not function as chromosomal DNA replication origins. Mol Cell Biol. 1991;11:5346–5355. doi: 10.1128/mcb.11.10.5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey DD, Zhu J, Carlson DL, Sharma K, Huberman JA. Three ARS elements contribute to the ura4 replication origin region in the fission yeast, Schizosaccharomyces pombe. EMBO J. 1994;13:3638–3647. doi: 10.1002/j.1460-2075.1994.tb06671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson BM, Fangman WL. A position effect on the time of replication origin activation in yeast. Cell. 1992;68:333–339. doi: 10.1016/0092-8674(92)90474-q. [DOI] [PubMed] [Google Scholar]
- Ferguson BM, Brewer BJ, Reynolds AE, Fangman WL. A yeast origin of replication is activated late in S phase. Cell. 1991;65:507–515. doi: 10.1016/0092-8674(91)90468-e. [DOI] [PubMed] [Google Scholar]
- Fogarty P, Campbell SD, Abu-Shumays R, Phalle BS, Yu KR, Uy GL, Goldberg ML, Sullivan W. The Drosophila grapes gene is related to checkpoint gene chk1/rad27 and is required for late syncytial division fidelity. Curr Biol. 1997;7:418–426. doi: 10.1016/s0960-9822(06)00189-8. [DOI] [PubMed] [Google Scholar]
- Harland R. Initiation of DNA replication in eukaryotic chromosomes. Trends Biochem Sci. 1981;6:71–74. [Google Scholar]
- Harland RM, Laskey RA. Regulated replication of DNA micro-injected into eggs of Xenopus laevis. Cell. 1980;21:761–771. doi: 10.1016/0092-8674(80)90439-0. [DOI] [PubMed] [Google Scholar]
- Huberman JA, Riggs AD. On the mechanism of DNA replication in mammalian chromosomes. J Mol Biol. 1968;32:327–341. doi: 10.1016/0022-2836(68)90013-2. [DOI] [PubMed] [Google Scholar]
- Huberman JA, Spotila LD, Nawotka KA, El Assouli SM, Davis LR. The in vivo replication origin of the yeast 2 micron plasmid. Cell. 1987;51:473–481. doi: 10.1016/0092-8674(87)90643-x. [DOI] [PubMed] [Google Scholar]
- Hyrien O, Maric C, Mechali M. Transition in specification of embryonic metozoan DNA replication origins. Science. 1995;270:994–997. doi: 10.1126/science.270.5238.994. [DOI] [PubMed] [Google Scholar]
- Kimelman D, Kirschner M, Scherson T. The events of the midblastula transition in Xenopus are regulated by changes in the cell cycle. Cell. 1987;48:399–407. doi: 10.1016/0092-8674(87)90191-7. [DOI] [PubMed] [Google Scholar]
- Marahrens Y, Stillman B. Replicator dominance in a eukaryotic chromosome. EMBO J. 1994;13:3395–3400. doi: 10.1002/j.1460-2075.1994.tb06642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight SL, Miller OL. Electron microscopic analysis of chromatin replication in the cellular blastoderm Drosophila melanogaster embryo. Cell. 1977;12:795–804. doi: 10.1016/0092-8674(77)90278-1. [DOI] [PubMed] [Google Scholar]
- Newlon CS, Collins I, Dershowitz A, Deshpande AM, Greenfeder SA, Ong LY, Theis JF. Analysis of replication origin function on chromosome III of Saccharomyces cerevisiae. Cold Spring Harb Symp Quant Biol. 1993;58:415–423. doi: 10.1101/sqb.1993.058.01.048. [DOI] [PubMed] [Google Scholar]
- Newport J, Dasso M. On the coupling between DNA replication and mitosis. J Cell Sci Suppl. 1989;12:149–160. doi: 10.1242/jcs.1989.supplement_12.13. [DOI] [PubMed] [Google Scholar]
- Paulovich AG, Margulies RU, Garvik BM, Hartwell LH. RAD9, RAD17, and RAD24 are required for S phase regulation in Saccharomyces cerevisiae in response to DNA damage. Genetics. 1997;145:45–62. doi: 10.1093/genetics/145.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff JW, Glover DM. Nuclear and cytoplasmic mitotic cycles continue in Drosophila embryos in which DNA synthesis is inhibited with aphidicolin. J Cell Biol. 1988;107:2009–2019. doi: 10.1083/jcb.107.6.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, McCarroll RM, Newlon CS, Fangman WL. Time of replication of ARS elements along yeast chromosome III. Mol Cell Biol. 1989;9:4488–4494. doi: 10.1128/mcb.9.10.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santocanale C, Diffley JFX. ORC- and Cdc6-dependent complexes at active and inactive chromosomal replication origins in Saccharomyces cerevisiae. EMBO J. 1996;15:6671–6679. [PMC free article] [PubMed] [Google Scholar]
- ————— A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. 1998;395:615–618. doi: 10.1038/27001. [DOI] [PubMed] [Google Scholar]
- Shirahige K, Hori Y, Shiraishi K, Yamashita M, Takahashi K, Obuse C, Tsurimoto T, Yoshikawa H. Regulation of DNA-replication origins during cell-cycle progression. Nature. 1998;395:618–621. doi: 10.1038/27007. [DOI] [PubMed] [Google Scholar]
- Sibon OCM, Stevenson VA, Theurkauf WE. DNA-replication checkpoint control at the midblastula transition. Nature. 1997;388:93–97. doi: 10.1038/40439. [DOI] [PubMed] [Google Scholar]
- Stevenson JB, Gottschling DE. Telomeric chromatin modulates replication timing near chromosome ends. Genes & Dev. 1999;13:146–151. doi: 10.1101/gad.13.2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujcic, M., C.A. Miller, and D. Kowalski. 1999. Activation of silent replication origins at ARS elements near the HML locus in budding yeast. Mol. Cell. Biol. (in press). [DOI] [PMC free article] [PubMed]
- Walter J, Newport JW. Regulation of replicon size in Xenopus egg extract. Science. 1997;275:993–995. doi: 10.1126/science.275.5302.993. [DOI] [PubMed] [Google Scholar]
- Wu JR, Gilbert DM. A distinct G1 step required to specify the Chinese hamster DHFR replication origin. Science. 1996;271:1270–1272. doi: 10.1126/science.271.5253.1270. [DOI] [PubMed] [Google Scholar]