

Abstract
The TOR protein kinases (TOR1 and TOR2 in yeast; mTOR/FRAP/RAFT1 in mammals) promote cellular proliferation in response to nutrients and growth factors, but their role in development is poorly understood. Here, we show that the Drosophila TOR homolog dTOR is required cell autonomously for normal growth and proliferation during larval development, and for increases in cellular growth caused by activation of the phosphoinositide 3-kinase (PI3K) signaling pathway. As in mammalian cells, the kinase activity of dTOR is required for growth factor-dependent phosphorylation of p70 S6 kinase (p70S6K) in vitro, and we demonstrate that overexpression of p70S6K in vivo can rescue dTOR mutant animals to viability. Loss of dTOR also results in cellular phenotypes characteristic of amino acid deprivation, including reduced nucleolar size, lipid vesicle aggregation in the larval fat body, and a cell type-specific pattern of cell cycle arrest that can be bypassed by overexpression of the S-phase regulator cyclin E. Our results suggest that dTOR regulates growth during animal development by coupling growth factor signaling to nutrient availability.
Keywords: Rapamycin, TOR/FRAP, cell growth, cell cycle, Drosophila
During metazoan evolution, control of cell growth has evolved from a simple cell autonomous response to nutrient levels to a complex network of intercellular growth factor-mediated signals. Despite these additional layers of control, individual cells of higher eukaryotes have retained the ability to sense and respond directly to levels of nutrients such as amino acids (for review, see Kimball and Jefferson 2000). This more primal mode of regulation may serve as a checkpoint to gauge the appropriateness of intercellular growth signals, and coordination of these two types of regulatory inputs is likely to be essential for normal cell growth, proliferation, and survival.
The mammalian target of rapamycin (mTOR; also known as FRAP or RAFT1) has been implicated in such coordination. mTOR is a large protein kinase of the phosphoinositide kinase (PIK)-related family, whose other members include the DNA damage checkpoint proteins ATM, ATR, and DNA–PK (for review, see Keith and Schreiber 1995). Inactivation of mTOR with the macrolide rapamycin results in G1 arrest and an attendant dephosphorylation of two of mTOR's targets, p70S6K and 4E–BP1 (for review, see Brown and Schreiber 1996; Dennis et al. 1999). These proteins regulate ribosome biogenesis and cap-dependent translation, respectively, and their phosphorylation is required to achieve the high levels of protein synthesis necessary for cell cycle entry.
Binding of growth factors such as insulin to their receptors in mammalian cultured cells causes rapid, PI3K-dependent phosphorylation of p70S6K and 4E–BP1 through a series of serine/threonine kinases including PDK1 and Akt/PKB (for a recent review, see Toker 2000). Although mTOR is required for PI3K-dependent p70S6K and 4E–BP1 phosphorylation, how it interacts with this signaling pathway is unclear. Akt can stimulate the kinase activity of mTOR immunoprecipitates and can phosphorylate mTOR in vitro (Scott et al. 1998; Navé et al. 1999; Sekulic et al. 2000). However, recent experiments have revealed that mutation of the Akt phosphorylation sites on mTOR has no effect on its kinase activity (Sekulic et al. 2000). Furthermore, addition of growth factors to serum-starved cells causes a robust increase in p70S6K and 4E–BP1 phosphorylation, but only a marginal increase in mTOR kinase activity (Burnett et al. 1998; Scott et al. 1998). In addition, specific truncation mutants of p70S6K have been identified whose PI3K-mediated phosphorylation is mTOR independent (Cheatham et al. 1995; Weng et al. 1995). These results suggest that signals from PI3K to p70S6K and 4E–BP1 do not pass through mTOR, and hence that mTOR and PI3K may converge on the same substrates through distinct pathways.
Loss of TOR activity in the yeast Saccharomyces causes a group of phenotypes nearly identical to those of cells starved for nutrients, including severely reduced protein synthesis, accumulation of glycogen, enlargement of the vacuole, induction of autophagy, and a specific pattern of gene expression (Kunz et al. 1993; Barbet et al. 1996; Noda and Ohsumi 1998; Cardenas et al. 1999; Hardwick et al. 1999). This has led to the idea that the yeast TOR proteins act as part of a nutrient-sensing mechanism. Remarkably, this function of TOR may also be conserved in higher eukaryotes. Withdrawal of amino acids in vitro or starvation in vivo results in rapid, reversible dephosphorylation of p70S6K and 4E–BP1, and rephosphorylation of these proteins after readdition of nutrients is blocked by rapamycin (Svanberg et al. 1997; Fox et al. 1998; Hara et al. 1998; Wang et al. 1998; Xu et al. 1998; Iiboshi et al. 1999). In contrast, activity of the PI3K/Akt or MAPK pathways is unaffected by amino acid levels in cell culture (Hara et al. 1998; Wang et al. 1998; Iiboshi et al. 1999). Furthermore, rapamycin-resistant mutants of p70S6K are also resistant to dephosphorylation by amino acid withdrawal (Hara et al. 1998). These data support a model in which full activation of p70S6K and eIF4E requires two distinct signals, one in response to growth factors and another from an amino acid sensing pathway, and thus provide a mechanism whereby individual cells can coordinate their responses to growth factors with nutrient availability. However, changes in TOR kinase activity in response to amino acids or other nutrients have not been observed (Kleijn and Proud 2000), and thus the mechanisms by which TOR is regulated remain unclear.
To date, studies of mammalian TOR have relied on inactivation by rapamycin, as TOR mutations have not yet been reported in multicellular organisms. Although rapamycin mimics many of the effects of TOR mutational inactivation in yeast, some essential functions of TOR2, such as polarization of the actin cytoskeleton, are not inhibited by rapamycin (Zheng et al. 1995; Schmidt et al. 1996). This may be the case in higher eukaryotes as well, as it has been shown that rapamycin does not fully inhibit the kinase activity of mTOR (Burnett et al. 1998; Peterson et al. 2000). Furthermore, the kinase domain of mTOR constitutes only ∼10% of the total protein, and the in vivo function of the remainder is essentially unknown.
To initiate a genetic analysis of TOR in a multicellular organism, we have generated mutations in the Drosophila TOR homolog, dTOR, and have used these mutants to study the role of dTOR during development. The PI3K/Akt/p70S6K signaling module is conserved in Drosophila, where it acts to regulate cell, organ, and organismal growth (for review, see Coelho and Leevers 2000). Mutational inactivation of this pathway reduces cell size, hinders proliferation, and delays or arrests development, and its activation leads to autonomous increases in cell and organ size. We find that dTOR mutant phenotypes recapitulate aspects of both PI3K-dependent signaling and nutritional sensing, consistent with dTOR acting at the junction of these pathways.
Results
Identification of mutations in dTOR
The Drosophila genome encodes four members of the PIK-related family: mei41, which encodes an ATR/ATM homolog required for cell cycle arrest in response to DNA damage (Hari et al. 1995); CG6535, a second ATM-related gene of unknown function (Adams et al. 2000); CG4549, whose closest relative encodes the nonsense-mediated mRNA decay protein SMG-1 in C. elegans (O'Connor and Anderson 1999; Adams et al. 2000); and the dTOR gene described here. A fifth member of this family, the DNA-dependent protein kinase, is not found in Drosophila or C. elegans.
Using a combination of cDNA library screening and RACE (5′ rapid amplification of cDNA ends; see Materials and Methods), we isolated overlapping cDNAs that together contain a large ORF of 2471 amino acids with strong similarity to mammalian mTOR and to TOR1 and TOR2 from budding yeast. Subsequently, the identical ORF was identified by computational analysis of the annotated Drosophila genome (CG5092, GenBank accession no. AE003638; Adams et al. 2000). Sequence comparisons revealed that the predicted protein, which we have named dTOR, is 56% and 38% identical to human mTOR and yeast TOR2, respectively, with the highest levels of identity in the carboxy-terminal region containing the putative kinase and rapamycin/FKBP12-binding domains (73% identity with mTOR over the carboxy-terminal 675 amino acids; Fig. 1). Additional structural motifs were also found to be well-conserved, including a series of HEAT repeats in the amino-terminal half of the protein, a domain shown to bind the peripheral membrane protein gephyrin (Sabatini et al. 1999), and a short sequence at the extreme carboxyl terminus of essential but unknown function that is highly conserved amongst PIK-related family members (Keith and Schreiber 1995; Peterson et al. 2000). Interestingly, previously described sites of autophosphorylation (Peterson et al. 2000) and phosphorylation by Akt/PKB (Scott et al. 1998; Navé et al. 1999; Sekulic et al. 2000) are not conserved in dTOR (Fig. 1). Genomic DNA blot analysis and in situ hybridization to larval polytene chromosomes revealed that dTOR is a single copy gene (data not shown), as was confirmed by Drosophila genomic sequencing (Adams et al. 2000).
Figure 1.

Comparison of Drosophila and human TOR proteins. The ClustalW 1.8 algorithm was used to align the amino acid sequences of dTOR and human mTOR/FRAP (GenBank accession no. 744518). Dark boxes indicate identical amino acids; gray boxes indicate similarity. The conserved FKBP12–rapamycin-binding domain (amino acids 2025–2114 in mTOR; Vilella-Bach et al. 1999) is underlined, and the essential serine in this domain is indicated by an asterisk. Sites of mTOR autophosphorylation (Ser-2481) and phosphorylation by Akt/PKB (Ser-2448) are marked with carats.
To begin a mutational analysis of dTOR, we searched the Berkeley Drosophila Genome Project P-element database and identified two independent homozygous lethal lines bearing P insertions in the dTOR gene (designated here as dTORP1 and dTORP2). Mobilization of these elements restored viability to each chromosome, indicating that the insertions are responsible for the associated lethality. Comparison of the insertion sites of dTORP1 and dTORP2 with the dTOR transcription unit revealed that the P-elements are inserted at 24 and 74 bases downstream of the dTOR transcription start site, respectively (Fig. 2A), and would likely interfere with normal dTOR expression.
Figure 2.

Mutations in dTOR inhibit larval growth. (A) The structure of the 8.0-kb dTOR transcript is shown. Coding sequence is indicated by filled boxes; 5′ and 3′ untranslated regions are represented by open boxes. Breaks in the boxes indicate introns. Sites of the two P-element insertions in dTOR are indicated, and the range of the dTORΔP deletion is shown. The stippled bar at the bottom of the figure indicates the DNA fragment used for genomic rescue. Vha68-1 encodes a mitochondrial ATPase transcribed in the opposite direction as dTOR. (B) Size comparison of wild-type and dTOR homozygous mutant larvae at 126 h of development. Wild-type larvae pupate within 12 h of this time point, whereas dTOR mutants remain arrested in the larval stage with little or no further growth. (C) Heterozygosity for dTOR sensitizes larvae to rapamycin. On normal food, wild-type (black circles) and dTOR/+ larvae (black diamonds) grow at indistinguishable rates. Addition of 1 μM rapamycin delays development by ∼3 d in wild-type (open circles) and ∼6 d in dTOR/+ larvae (open diamonds).
To generate additional dTOR alleles, a series of deletions spanning the dTOR gene was generated by imprecise mobilization of the P-elements. One such mutant, designated dTORΔP, was selected for further analysis. Sequence analysis revealed that the dTORΔP deletion originates at the dTORP2 insertion site and extends 3514 bp downstream, removing the dTOR translation start site and amino-terminal 902 codons, and thus likely represents a null allele of dTOR (Fig. 2A). This was confirmed by the absence of detectable dTOR protein in immunoblots of dTORΔP larval extracts (data not shown). A 9.4-kb genomic rescue construct encompassing the dTOR gene and no other predicted transcription units (Fig. 2A) restored full viability and fertility to dTORΔP homozygotes. Therefore, the phenotypes described below are due to loss of dTOR function.
dTOR is required for normal growth
dTORΔP homozygotes were found to hatch at normal rates, but grew more slowly than normal and eventually arrested during larval development, reaching only 24% the mass of wild-type controls (Fig. 2B). Larvae homozygous for dTORP1 or dTORP2 alleles displayed a less severe phenotype, eventually growing to approximately 40% and 79% the mass of wild type, respectively, indicating that these alleles likely retain partial dTOR function. In each case, the mutants remained viable and active during an extended larval period of ∼30 d, and eventually died without pupating. Larvae heterozygous for dTOR grew at a rate indistinguishable from wild-type controls under normal culture conditions, but were hypersensitive to low concentrations of rapamycin (Fig. 2C). Thus, dTOR encodes a rapamycin-sensitive protein required for normal growth during larval development.
Cells lacking dTOR are reduced in size and proliferative capacity
Overall growth of an organism is generally accompanied by increases in cell number (proliferation), cell size (hypertrophy), or both (for review, see Conlon and Raff 1999). To determine how mutations in dTOR inhibit growth, we examined these parameters in a number of tissues.
The effect of dTOR on cell size was analyzed in marked clones of dTORΔP homozygous cells, which were generated by FLP/FRT-mediated mitotic recombination in dTORΔP heterozygous animals. Examination of adult cuticular structures revealed that dTOR homozygous mutant cells were markedly reduced in size. For example, bristles of the wing margin that lack dTOR were both thinner and shorter than adjacent wild-type cells (Fig. 3, cf. A and B). Area measurements of mutant clones in the wing epithelium (Fig. 3C) showed that dTORΔP mutant cells were approximately half (56%) the size of controls (n = 498 cells). Similar effects were observed in the eye, abdomen, and notum (data not shown).
Figure 3.

dTOR clonal phenotypes. (A–C) Loss of dTOR reduces the size of cells in the adult wing. Clones of y−-marked wild-type (A) or dTORΔP (B,C) cells were induced during the first larval instar by FLP/FRT-mediated mitotic recombination. Representative clones including bristles of the wing margin (A,B) and epithelial cells of the wing blade (C) are shown. The red trace in C outlines a dTORΔP mutant clone. Each hairlike structure is a trichome emanating from a single epithelial cell. Note the reduced size and increased density of dTOR cells. Genotype of A–C: y,w, HS-FLP122/+; dTORΔP FRT40A/Py+ FRT40A. (D–F) Phenotypes of dTOR mutant cells during proliferation. (D) Confocal image of wing imaginal disc containing dTORΔP clones marked with ubiquitin–GFP. By 72 h after induction, dTORΔP mutant clones (indicated by arrows; cells lack marker) contain fewer cells than their wild-type twinspot clones (indicated by arrowheads; cells carry two copies of GFP marker). (E,F) FACS histograms of dissociated cells from wing discs containing dTORΔP clones, as in D. dTORΔP cells and wild-type control cells are indicated by heavy and light traces, respectively. Cells lacking dTOR have reduced forward light scatter (FSC) values, indicating a smaller cell size (E). Populations of dTOR mutant cells also have a lower fraction of cells in S and G2 phases of the cell cycle (F). Genotype of D–F: y, w, HS-FLP122/+; dTORΔP FRT40A/Ubi-GFP FRT40A.
To determine whether loss of dTOR affected the size of actively proliferating cells, we examined dTOR mutant clones in the developing imaginal discs, epithelial primordia that proliferate mitotically to give rise to adult structures. Imaginal wing discs containing GFP-marked clones of dTORΔP homozygous cells (Fig. 3D) were dissociated into single cells, which were then analyzed by flow cytometry. The mean forward light scatter value (a measure of cell size) of dTOR mutant cells was decreased by 30% compared to wild-type control cells from the same discs (Fig. 3E). This decrease in cell size was observed in all phases of the cell cycle (data not shown). Thus, loss of dTOR causes a cell autonomous reduction in the size of both proliferating and postmitotic cells.
Fluorescence-activated cell sorter (FACS) analysis also revealed that the cell cycle phasing of dTOR cells differed significantly from that of controls (Fig. 3F), with relatively more cells in G1, and fewer in S and G2 phases. This is consistent with the ability of rapamycin to induce G1 arrest in yeast and in mammalian cell culture (Heitman et al. 1991; Chung et al. 1992). To measure proliferation rates of dTOR mutant cells, we compared the number of cells in dTORΔP clones with that of their wild-type sister clones (twin spots). Clones of dTORΔP mutant cells were similar in size to their twin spots at 48 h after induction (data not shown), but by 72–96 h they contained significantly fewer cells (Fig. 3D), indicating that loss of dTOR leads to a reduced rate of cell proliferation. In addition, lone twin spots lacking a corresponding mutant sister clone were occasionally observed at 96 h after induction, indicating that at some frequency dTORΔP homozygous cells were eliminated from the disc epithelium. Because dTORΔP cells remain viable for weeks in the context of a homozygous mutant animal (see above and Fig. 2B), the loss of dTORΔP mutant clones is likely the result of cell competition with adjacent wild-type cells, as has been described previously for cells with a growth disadvantage (Morata and Ripoll 1975).
Growth properties of cells in the salivary glands of homozygous dTORΔP larvae were also examined. The salivary gland is comprised of two cell types: polytene gland cells that undergo multiple rounds of endoreduplication to generate giant nuclei with a ploidy of up to 2048 C, and imaginal ring cells that remain diploid and cycle mitotically. Loss of dTOR affected both cell types. The endoreplicative cells in dTORΔP salivary glands underwent only four to five rounds of replication before entering quiescence (see below), reaching a ploidy of 16–32C and a size ∼10% that of wild type (Fig. 4A,B). The imaginal rings in dTORΔP larvae contained approximately fivefold fewer cells than wild type (Fig. 4C,D). Together, our results indicate that dTOR is required to promote cell cycle progression in both mitotic and endoreplicative cells, and acts primarily at the G1/S transition.
Figure 4.
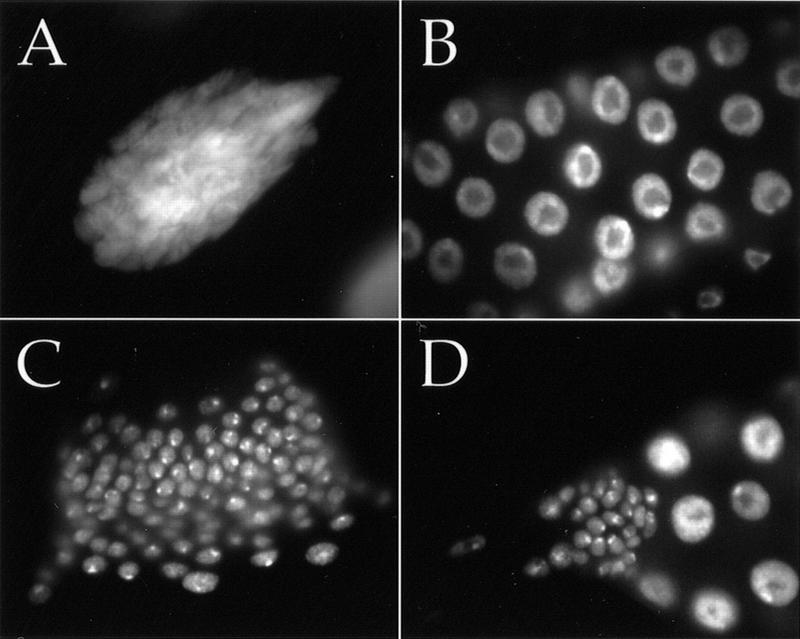
Both mitotic and endoreplicative cell cycles require dTOR. Salivary gland nuclei stained with Hoechst 33258 are shown for wild type (A,C) and dTORΔP (B,D) at 5–6 d of development. (A–D) Same magnification. (A) A single endoreplicative nucleus of ∼1024 C from a wild-type salivary gland. Endoreplicative salivary gland nuclei from dTORΔP larvae reach a ploidy of only 16–32 C (B). Proliferative diploid cells of the imaginal ring reach ∼fivefold greater numbers in wild-type (C) than dTORΔP (D) salivary glands.
Growth stimulation by the PI3K pathway requires dTOR
The cell autonomous reduction in the size of dTOR mutant cells is reminiscent of mutations in components of the PI3K/S6K signaling pathway (Coelho and Leevers 2000). Mutations in dPTEN, the fly homolog of the PTEN tumor suppressor, cause activation of this pathway, leading to increased cell growth. To determine whether dTOR is required for PI3K-dependent signaling, we examined the growth properties of cells lacking both dPTEN and dTOR. As we have shown previously (Gao et al. 2000), clonal loss of dPTEN caused enlargement of imaginal and adult cells, and increased the percentage of cells in the S and G2 phases of the cell cycle (Fig. 5A–C). In contrast, cells carrying null alleles of both dPTEN and dTOR were indistinguishable from cells lacking dTOR alone, with a similar reduction in cell size and accumulation in G1 (Fig. 5G–I). Loss of dTOR also prevented the increased proliferation caused by mutations in dPTEN (data not shown). Cells mutant for weaker alleles of dPTEN and dTOR (MGH1 and P2, respectively) were intermediate in size (data not shown), indicating that dTORP2 cells retain partial signaling function. We conclude that dTOR is epistatic to dPTEN, and therefore, that dTOR functions at a step downstream of or in parallel to PI3K signaling.
Figure 5.
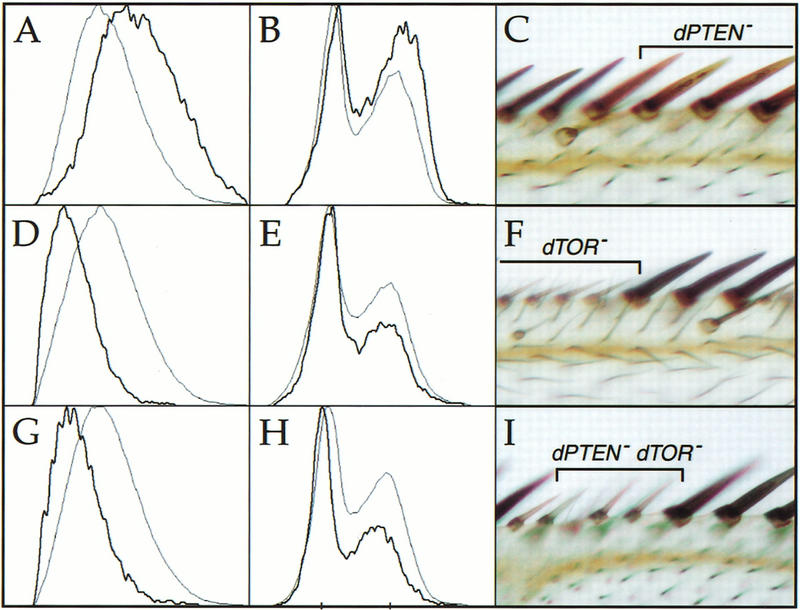
dTOR acts downstream of dPTEN. Clones of cells mutant for null alleles of dPTEN (A–C), dTOR (D–F), or dPTEN and dTOR (G–I) were induced by FLP/FRT-mediated mitotic recombination. Mutant cells were marked by the absence of GFP or y+ as in Fig. 3. The leftmost columns depict histograms of wing imaginal disc cells analyzed by flow cytometry; dark traces indicate mutant cells, and light traces represent wild-type control cells from the same discs. The right column shows photomicrographs of mutant clones in the anterior margin of the adult wing. Loss of dPTEN causes cell enlargement (A,C), and increases the proportion of cells in the S and G2 phases of the cell cycle (B). Loss of dTOR decreases cell size (D,E) and reduces the S and G2 populations (F). Cells lacking dTOR and dPTEN are indistinguishable from cells lacking dTOR alone (G–I). Genotypes: (A,B) y, w, HS-FLP122/+; dPTENDJ189FRT40A/Ubi-GFP FRT40A; (C) y, w, HS-FLP122/+; dPTENDJ189FRT40A/Py+ FRT40A. (D,E) y, w, HS-FLP122/+; dTORΔP FRT40A/Ubi-GFP FRT40A; (F) y, w, HS-FLP122/+; dTORΔP FRT40A/Py+ FRT40A. (G,H) y, w, HS-FLP122/ +; dTORΔP dPTENDJ189 FRT40A/Ubi-GFP FRT40A; (I) y, w, HS-FLP122/+; dTORΔP dPTENDJ189 FRT40A/Py+ FRT40A.
p70S6K is a critical effector of dTOR in vivo
In further tests of dTOR's role in PI3K/S6K signaling, we found that rapamycin inhibited the serum-dependent phosphorylation of Drosophila p70S6K (dS6K) expressed in S2 cells (Fig. 6A, lane 2), as reported previously (Stewart et al. 1996). Dephosphorylation of dS6K by rapamycin was prevented by cotransfection of a dTOR point mutant containing a Ser1956 to Thr substitution (dTORRR; Fig. 6A, lane 6), which confers rapamycin resistance to mammalian and yeast TOR proteins. Expression of dTORRR carrying an additional point mutation in a residue crucial for kinase activity (dTORRRKD; Fig. 6A, lane 5) failed to protect dS6K from rapamycin-induced dephosphorylation, indicating that the kinase function of dTOR is required to maintain dS6K phosphorylation.
Figure 6.

dTOR interacts with dS6K. (A) Immunoblot of extracts from Drosophila S2 cells transfected with HA-tagged dS6K, visualized with anti-HA antibodies. dS6K migrates as a doublet, and the slower migrating band (top), which represents phosphorylated dS6K, is abolished by treatment with rapamycin (lane 2; Stewart et al. 1996). Phosphorylation of dS6K is maintained in the presence of rapamycin when dTORS1956T (dTORRR, lane 6) but not kinase-inactive dTORS1956T (dTORRRKD, lane 5) is cotransfected with dS6K. (B) Constitutive expression of activated human p70S6K1 rescues dTOR flies to viability. Genotypes: (Left) dTORP2 UAS-p70S6K1-D4/dTORP1; Act5c-Gal4/+; (right) dTORP2 UAS-p70S6K1-D4/CyO; Act5c-Gal4/+. Note that the rescued dTOR fly (left) is smaller than the control. (C) Constitutive expression of dS6K provides rapamycin resistance. UAS-dS6K/+; Act5c-Gal4/+ flies cultured with 1 μM rapamycin eclose ∼3 d earlier than wild-type controls.
To determine whether these biochemical interactions between dTOR and dS6K were relevant to their functions in vivo, we tested for genetic interactions between them. Remarkably, constitutive overexpression of Drosophila dS6K or human p70S6K1 was able to rescue dTORP2/P2 and dTORP1/P2 flies to viability (Fig. 6B). The greatest degree of rescue was provided by a mutant version of p70S6K1, in which four mitogen-induced phosphorylation sites are mutated to aspartate and glutamate residues (mutant D4; Cheatham et al. 1995). Expression of this construct allowed 74% of expected dTORP1/P2 progeny to survive to adulthood, whereas no dTORP1/P2 animals survived in the absence of S6K overexpression. dTOR flies rescued by S6K overexpression were slightly smaller than wild-type controls (Fig. 6B), but were fertile and developed at a similar rate as wild type. Although overexpression of S6K did not rescue dTORΔP null mutants to adulthood, it did enable them to progress to the pupal stage (data not shown). Overexpression of S6K in wild-type larvae also conferred significant resistance to rapamycin (Fig. 6C). Again, constitutively active p70S6K1 provided the greatest degree of rapamycin resistance. Together, these results indicate that a major function of dTOR is to maintain levels of active S6K sufficient for normal growth.
Amino acid withdrawal phenocopies of dTOR mutant phenotypes
Like dTOR mutants, wild-type larvae deprived of amino acids enter an extended larval period with little or no growth (Britton and Edgar 1998). Amino acid deprivation also causes a series of distinctive cellular phenotypes including a reduction in nucleolar area, changes in morphology of the larval fat body, and a cell type-specific cell cycle arrest (Butterworth et al. 1965; Nanya and Bicudo 1995; Britton and Edgar 1998; Zinke et al. 1999). As TOR proteins have been proposed to be regulated in response to amino acid levels, we wished to examine whether loss of dTOR mimics these cellular effects.
The nucleolus is the major cellular site of ribosomal assembly, and its size has been shown to correlate with protein synthetic capacity and proliferation rate (Derenzini et al. 1998). To measure nucleolar size in wild-type and dTOR mutant cells, wing imaginal discs containing clones of dTORΔP homozygous cells were labeled with an antibody against the nucleolar protein fibrillarin (Aris and Blobel 1988), and examined by confocal sectioning (Fig. 7A–C). We found that the nucleolar area in clones of dTOR mutant cells in the wing imaginal disc was approximately half that of surrounding wild-type cells (dTOR nucleoli, 27.9 ± 5.5 pixels2, n = 95; wild-type nucleoli, 52.3 ± 11.1 pixels2, n = 100), consistent with a role for dTOR in ribosome biogenesis.
Figure 7.

Loss of dTOR mimics starvation. (A–C) Wing imaginal disc containing a clone of dTORΔP cells marked by the absence of GFP (A) and stained with antifibrillarin to highlight nucleoli (B). The extent of the dTOR mutant clone is outlined in yellow. (C) An overlay of GFP (green) and antifibrillarin (red). Loss of dTOR causes a reduction in nucleolar area. (D–F) Differential interference contrast images of salivary glands (s.g.) and associated fat body (f.b.) from normally fed larvae (D), larvae that were deprived of amino acids for 4 d (E), and normally fed dTORΔP larvae (F). Starvation and loss of dTOR each cause aggregation of lipid vesicles in the fat body, as well as accumulation of proteinaceous granules in salivary gland cells.
During metamorphosis or starvation, stores of protein, lipid, and glycogen are mobilized from adipose cells of the larval fat body and are used by other tissues as an energy source in place of dietary nutrients. These metabolic effects are visible as changes in appearance of fat body cells (Butterworth et al. 1965; Britton and Edgar 1998; Zinke et al. 1999). The major visible change in fat body cells in larvae deprived of amino acids is an aggregation of lipid vesicles (Fig. 7E), and this effect was indistinguishable from that caused by loss of dTOR (Fig. 7F).
Within 48–72 h of amino acid withdrawal, endoreplicative cells become quiescent, whereas mitotic neuroblasts of the central nervous system continue to cycle for at least 8 d in the absence of amino acids (Britton and Edgar 1998). This pattern of cell cycle responses is distinct from that caused by complete inhibition of protein synthesis, which causes all larval cells to arrest DNA synthesis (Britton and Edgar 1998). To determine whether loss of dTOR causes a cell cycle response similar to that elicited by starvation, we examined the cell cycle behavior of these cell types in dTORΔP larvae at multiple stages of development. At 3–4 d after egg deposition (AED), both endoreplicative and mitotic tissues were found to cycle normally in dTORΔP homozygotes, as measured by incorporation of the nucleotide analog BrdU (Fig. 8A–D). In contrast, by 5–6 d AED all endoreplicative tissues including the gut (data not shown), fat body, and salivary glands failed to incorporate BrdU, whereas neuroblasts continued to cycle (Fig. 8E,F). A similar pattern was observed at 10 d AED (data not shown). Presumably the appearance of this cell cycle arrest at 4–5 d AED results from the perdurance of maternal stores of dTOR mRNA or protein until this time. We found that the cell cycle arrest of dTOR endoreplicative cells could be bypassed by overexpression of the G1/S regulators cyclin E (Fig. 8H) or dE2F/dDP (data not shown), as was demonstrated previously for endoreplicative cells arrested by amino acid withdrawal (Britton and Edgar 1998). Thus, amino acid insufficiency and loss of dTOR each cause similar growth arrests, changes in cell morphology, and cell type-specific patterns of G1 arrest.
Figure 8.
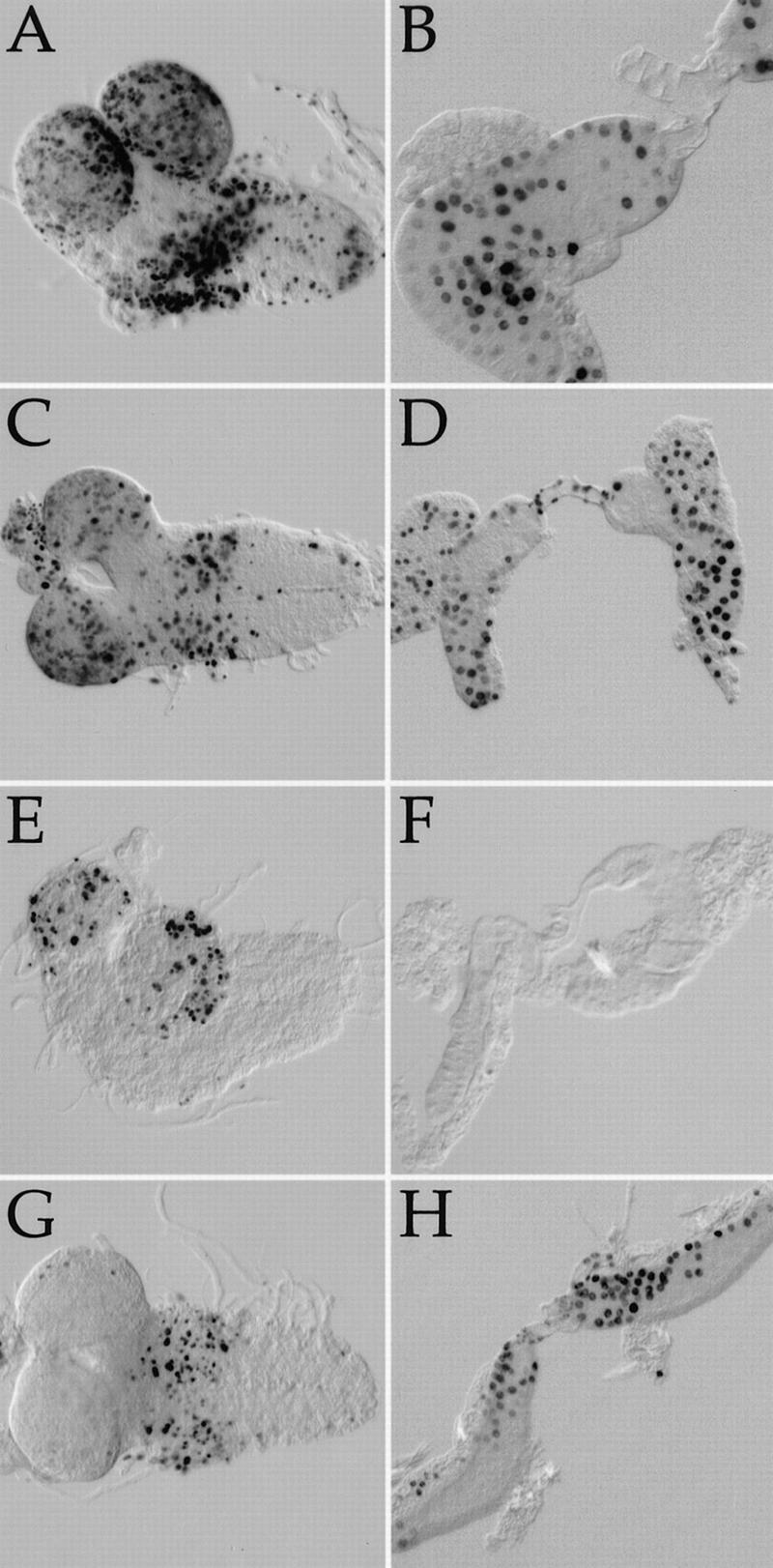
Cell cycle behavior of dTOR cells. BrdU incorporation patterns in larval brains (A,C,E,G) and salivary glands (B,D,F,H). Darkly stained nuclei indicate cells that have incorporated BrdU and thus were in S-phase during the experiment. (A,B) Wild type at 3–4 d AED. (C,D) dTORΔP at 3–4 d AED. (E,F) dTORΔP at 5–6 d AED. Note that salivary gland cells no longer incorporate BrdU, whereas CNS neuroblasts continue to cycle. (G,H) dTORΔP carrying a single copy of a HS–cyclin E transgene at 6–7 d AED, after a 2-h heat shock. Ectopic expression of cyclin E drives quiescent dTORΔP salivary gland cells into S-phase.
dTOR regulates G1/S progression through cyclin E
In budding yeast, TOR proteins govern S-phase entry in response to nutrient levels by regulating translation of the G1/S regulator Cln3 (Barbet et al. 1996; Polymenis and Schmidt 1997). Drosophila cyclin E has been proposed to play a role analogous to Cln3, and its abundance increases in response to growth stimuli such as overexpression of activated Ras (Johnston et al. 1999; Prober and Edgar 2000). Because cyclin E overexpression was able to bypass the cell cycle arrest in dTOR mutants (Fig. 8H), we examined whether loss of dTOR affected cyclin E expression. Immunoblot analysis of whole larval extracts revealed that the level of cyclin E protein was reduced ∼30-fold in dTORΔP mutants compared to wild-type larvae of similar stage (Fig. 9A).
Figure 9.

Cyclin E expression is reduced in dTOR mutants. (A) Immunoblot of total larval extracts from dTORΔP and wild-type second instar larvae, probed with anti-cyclin E. The blot was also probed with anti-β-tubulin as a loading control. (B–D) Shown is a region of a wing imaginal disc containing dTORΔP clones 72 h after induction, marked by the absence of GFP (B) and stained with anti-cyclin E antibody (C). dTOR mutant clones are outlined in yellow. These images are superimposed in D, with GFP shown in green and cyclin E in red. The arrow indicates a single dTOR mutant cell expressing cyclin E; the arrowhead indicates a dTOR clone lacking cyclin E expression.
We have shown previously that clonal induction of cells lacking cyclin E in the wing imaginal disc results in a G1 arrest within one to two cell divisions (Neufeld et al. 1998). In contrast, cells mutant for dTOR continue to cycle slowly for several days, and give rise to clones containing multiple cells (see Fig. 3D), indicating that dTOR mutant cells retain at least partial cyclin E activity. Accordingly, we found that cyclin E protein was reduced but not eliminated in dTOR mutant clones in the wing disc. Although 72-h dTORΔP clones containing little or no detectable cyclin E immunoreactivity were often observed (Fig. 9B–D, arrowhead; data not shown), we also found many dTOR clones containing cells with apparently normal cyclin E levels (Fig. 9B–D, arrow). We conclude that the observed reduction in cyclin E protein levels in dTORΔP larval extracts is due largely to reduced cyclin E levels in endoreplicating cells, which comprise the majority of larval mass, resulting in a G1 arrest in this cell type. In contrast, dTOR may be required in imaginal cells to maintain normal rates of cyclin E accumulation, rather than absolute levels, consistent with the reduced rate of cell division and extended G1 phase observed in dTOR mutant clones.
Discussion
Studies in Drosophila have revealed that a critical role of the PI3K/Akt/p70S6K signaling pathway is to regulate size at the cellular, organ, and organismal levels (Coelho and Leevers 2000). A role for this pathway in size control is conserved in mammals, as it was recently shown that PI3K regulates cell and organ size in mice (Shioi et al. 2000). The results presented here indicate that the relationship of TOR to this pathway is also conserved between flies and mammals. dTOR is required for phosphorylation of dS6K in cell culture, and mutant animals with reduced dTOR activity can be rescued to viability by overexpression of dS6K. Moreover, dTOR is required for the changes in cell size and cell cycle phasing caused by activation of PI3K signaling in vivo. Like PI3K pathway mutants, loss of dTOR function reduces cell size and proliferation, with little apparent disruption of normal patterning.
Regulation of dTOR activity
Although these phenotypic similarities underscore the role of dTOR in PI3K signaling, dTOR mutants differ from the known PI3K pathway mutants in several important respects. First, the growth defects caused by loss of dTOR are more severe than those arising from mutations in components of the PI3K signaling pathway. Null mutations in the PI3K subunits Dp110 or p60 allow growth to the third instar larval stage, and chico (IRS-1) and dS6K mutants survive to adulthood (Bohni et al. 1999; Montagne et al. 1999; Weinkove et al. 1999). Although BrdU incorporation was not analyzed in these studies, the results imply that at least in the case of chico and dS6K, many cells are able to cycle normally throughout development. In contrast, animals lacking dTOR reach only the size of second instar larvae, at which point they undergo cell cycle arrest. Moreover, whereas overexpression of Dp110, Akt, or dS6K leads to increased growth rate and cell size (Montagne et al. 1999; Verdu et al. 1999; Weinkove et al. 1999), we find that dTOR overexpression inhibits growth and reduces cell size (T.P. Neufeld, unpubl.). Although variations in genetic background may partially account for some of these phenotypic differences, these results are inconsistent with dTOR acting as an integral component of a linear PI3K/Akt/S6K pathway, and instead argue that dTOR may converge upon this pathway in response to a distinct set of cues.
In yeast, TOR1 and TOR2 regulate cell growth directly in response to levels of nutrients such as amino acids, rather than in response to intercellular signals (Barbet et al. 1996). Similarly, it has recently been shown that human mTOR may also be regulated by nutrient levels (Fox et al. 1998; Hara et al. 1998; Wang et al. 1998; Xu et al. 1998; Iiboshi et al. 1999). These considerations prompted us to compare the phenotypes of dTOR mutant animals with the physiological changes caused by nutrient deprivation. By the three criteria we examined—nucleolar size, fat body vesicle formation, and endoreplicative cell cycle arrest—loss of dTOR precisely mirrored the effects of starvation. An efficient explanation of these results is that dTOR is required for normal responses to changes in nutrient levels. This would be consistent with a model in which full activation of growth targets such as p70S6K requires two distinct inputs: growth factor-mediated intercellular signals through PI3K, and nutrient-sensing signals through TOR. In this view, TOR proteins may act as part of a checkpoint to attenuate growth factor signaling when local conditions are unfavorable for cell growth.
How TOR proteins might be regulated by nutrient levels is unclear. In the case of amino acids, recent evidence suggests that the primary signal may be uncharged tRNA, which increases in abundance when amino acid levels are low (Iiboshi et al. 1999). Interestingly, other members of the PIK-related kinase family such as ATM and DNA–PK also act as checkpoint proteins that are regulated by specific nucleic acid structures. In addition, a recently described member of this family, SMG-1, is involved in the degradation of aberrant mRNAs containing inappropriate nonsense codons (O'Connor and Anderson 1999). Thus, regulation by nucleic acid may be a common feature of this kinase family.
Effectors of dTOR function
Activation of p70S6K is a common response to virtually all mitogenic stimuli (Chou and Blenis 1995). Phosphorylation of the target of p70S6K, ribosomal protein S6, leads to the selective increase in translation of a subset of transcripts that contain an oligopyrimidine tract at their 5′ termini (5′ TOP). This class of messages encodes ribosomal proteins and translation elongation factors, and thus p70S6K activation leads to increased ribosomal biogenesis (for review, see Brown and Schreiber 1996). Our demonstration here that overexpression of S6K can rescue dTOR mutants to viability indicates that dS6K is a critical effector of dTOR, and that one essential function of dTOR is to regulate the activity of dS6K.
Despite the central role of dS6K in mediating the functions of dTOR, several lines of evidence indicate that dTOR has additional dS6K-independent roles. First, dTOR mutant phenotypes are more severe than those of dS6K; lack of dTOR results in growth arrest at the second instar larval stage, whereas dS6K mutant animals survive to adulthood, albeit with a delayed development and decreased body size (Montagne et al. 1999). Second, although S6K overexpression suppressed the lethality of dTOR mutants, this was only the case for hypomorphic dTOR allelic combinations; dTOR null animals expressing S6K advanced only to the pupal stage. Third, dTOR flies rescued by S6K did not grow to the full size of wild-type controls (Fig. 6B). Thus, some dTOR functions are not fully rescued by S6K. As noted above, in mammalian cells mTOR also stimulates translation through phosphorylation and inactivation of 4E–BP1, an inhibitory binding factor of the translation initiation factor eIF4E. Drosophila eIF4E mutants display a severe growth arrest phenotype (Zhang et al., unpubl.), and thus aspects of dTOR function that are not rescued by dS6K may reflect diminished eIF4E activity. However, neither overexpression of eIF4E nor mutations in 4E–BP1 detectably alleviated dTOR mutant phenotypes (data not shown).
In addition to effects on translation, studies in budding yeast have found that inactivation of TOR modulates the level of a number of growth-related or nutrient-regulated transcripts, including those encoding ribosomal proteins (Barbet et al. 1996; Zaragoza et al. 1998; Beck and Hall 1999; Cardenas et al. 1999; Hardwick et al. 1999; Powers and Walter 1999). A transcriptional role for TOR in higher eukaryotes has been reported as well (Mahajan 1994). Moreover, recent studies have found that mTOR can interact with a number of additional signaling factors including STAT3, protein kinase C, c-Abl, and 14-3-3 (Parekh et al. 1999; Kumar et al. 2000a,b; Mori et al. 2000; Yokogami et al. 2000). Rapamycin has also recently been shown to disrupt microtubule assembly and function in yeast, independent of its effects on translation (Choi et al. 2000). Thus, the inability of S6K overexpression to fully supplant dTOR may be due to a requirement for dTOR in multiple cellular functions.
Coordination of cell division and growth
The reduced growth of dTOR mutants reflects reductions in both cell size and cell number. Because direct inhibition of proliferation increases rather than decreases cell size (Neufeld and Edgar 1998; Neufeld et al. 1998), we propose that the primary function of dTOR is to promote cell growth, and that the decreased proliferation of dTOR mutant cells is a secondary effect in response to their reduced rate of growth. The accumulation of dTOR mutant cells in the G1 phase of the cell cycle suggests that the G1/S transition is particularly sensitive to growth rate. This is consistent with previous observations that stimulation of cell growth by overexpression of dMyc, PI3K, or activated Ras promotes progression through the G1/S but not G2/M transition (Johnston et al. 1999; Weinkove et al. 1999; Prober and Edgar 2000).
A factor likely to be involved in coupling growth and division rates is the G1/S regulator cyclin E. Cyclin E is rate limiting for G1/S progression in imaginal discs (Neufeld et al. 1998), and its levels increase by a post-transcriptional mechanism in response to stimulation of cell growth (Prober and Edgar 2000). Our demonstration that dTOR is required for normal accumulation of cyclin E suggests that translational control is likely to play an important role in cyclin E regulation. In budding yeast, translation of the G1 cyclin Cln3 is regulated by a leaky scanning mechanism involving a small upstream ORF (Polymenis and Schmidt 1997). In this regard, it is interesting to note that the 5′-untranslated region of the Drosophila cyclin E message contains multiple upstream ORFs (Richardson et al. 1993). Thus, the mechanisms connecting G1/S progression to cell growth may be conserved between yeast and multicellular organisms.
Materials and methods
Fly strains
w; P[lacw]l(2)k17004/CyO (dTORP1) and w; EP(2)2353/CyO (dTORP2) are from the Bloomington Drosophila Stock Center and Berkeley Drosophila Genome Project, respectively. UbiGFP, FRT40A was a gift of Cristina Martin-Castellanos (Fred Hutchinson Cancer Research Center, Seattle, WA). UAS–dS6K and UAS–human p70S6K1 were gifts of Kelly Watson (Harvard University, Cambridge, MA). hs-cyclin E (Richardson et al. 1995), hs-dE2F hsdDP (Duronio et al. 1996), and dPTENDJ189 FRT40A (Gao et al. 2000) are as described. The Act5C–Gal4 line used for constitutive Gal4 expression was derived from Act5C>CD2>Gal4 (Pignoni and Zipursky 1997) by FLP/FRT-mediated germ-line excision of the CD2 insert.
dTOR deletion alleles were generated by imprecise mobilization of the dTORP1 and dTORP2 inserts. For each line, 100 potential excisions identified by loss of the w+ marker present on the P-element were tested for complementation with the starting P lines. Genomic DNA from lines that remained mutant for dTOR was used in PCR reactions (Expand Long Template PCR System, Roche Molecular Biochemicals) with primers flanking the P-insertion sites at −2970, −350, +2550, and +8470 relative to the dTORP1 insertion site. Ten lines bearing deletions in the dTOR gene were identified by increased electrophoretic mobility of PCR products. Sequence analysis revealed that these deletions each originate at the P-insertion site and extend downstream into the dTOR ORF, ranging in size from a 472-bp deletion that removes only the first exon, to an 8.0-kb deletion that removes all but the last 358 nucleotides of coding sequence.
Molecular biology
A small segment of dTOR was amplified by PCR using degenerate primers designed against conserved regions of mTOR and yeast TORs. This PCR fragment was then used to screen the LD Drosophila cDNA library (a gift of Ling Hong, Berkeley Drosophila Genome Project). Several overlapping cDNA clones were isolated, but did not represent the complete dTOR transcription unit. The remaining 5′ end of the dTOR transcript was amplified by RACE, using primers designed from the 5′ end of the assembled cDNA. Two independent RACE reactions gave the same size product. The assembled dTOR cDNA includes 284 bp of 5′ leader sequence, which contains an in-frame stop codon upstream of the dTOR ORF.
The dTOR genomic rescue construct was made as follows. The dTOR genomic region was subcloned as an 11.8-kb NsiI fragment from BAC48D10 (Berkeley Drosophila Genome Project) into the PstI site of pBluescript, to give pBS–dTOR-11.8. A 1.1-kb sequence upstream of the dTOR coding region was amplified by PCR and ligated into pET28a as an XbaI/SalI fragment to give pET–dTOR-1.1. An 8.3-kb SalI fragment from pBS–dTOR-11.8. was ligated into the SalI site of pET–dTOR-1.1., to give pET–dTOR-9.4. The 9.4-kb insert was liberated in an XbaI/XhoI partial digest, and transferred to pCasper4 to give pCasper–dTOR-9.4. This construct was injected into dTORΔP/CyO embryos by standard techniques.
Larval culture
To measure the effect of rapamycin on developmental timing, pools of 100 embryos from 4-h collections were placed in vials containing standard Drosophila medium with or without 1 μM rapamycin (Drug synthesis and chemistry branch, Developmental Therapeutics Program, Division of Cancer Treatment, National Cancer Institute), and cultured at 25°C until eclosion. Each experiment was conducted in triplicate, and the results averaged. For starvation experiments, larvae were cultured in 20% sucrose as described (Britton and Edgar 1998).
Histology
Clones of cells mutant for dTOR were generated by FLP/FRT-mediated mitotic recombination (Xu and Rubin 1993), and identified using a y+ transgene for adult bristles, and a ubiquitin–GFP marker in imaginal discs. Hair cell clones in the adult wing were identified on the basis of their small cell phenotype, and clonal areas were measured as described (Neufeld et al. 1998). For FACS analysis, mutant clones were induced at 48 h AED with a 2-h heat shock at 37°C. Wing imaginal disc cells were dissociated 72 h later and analyzed by flow cytometry as described (Neufeld et al. 1998). Cyclin E was detected on immunoblots with anti-Drosophila cyclin E monoclonal antibody (a gift of Helena Richardson, Adelaide University, South Australia), and in wing imaginal disc whole mounts with a guinea pig anti-Drosophila cyclin E polyclonal serum (a gift of Terry Orr-Weaver, MIT, Cambridge, MA).
For BrdU labeling, staged larvae were dissected in PBS and incubated for 9 h in M3 insect media (Sigma) containing 100 μg/mL BrdU (Sigma), then fixed on ice in 8% paraformaldehyde in PBS. For ectopic expression experiments, Drosophila cyclin E or dE2F/dDP were induced from heat shock-responsive transgenes with a 2-h heat shock at 37°C followed by 2 h of recovery at room temperature before dissection. BrdU incorporation was detected with mouse anti-BrdU primary (Pharmingen) and Texas Red-conjugated goat anti-mouse secondary antibodies (Jackson Immunolabs). Tissues were mounted in Fluorogard (Bio-Rad) and photographed under DIC optics on a Zeiss Axioscope 2 microscope.
To estimate ploidy of salivary gland polytene nuclei, tissues were fixed in 4% paraformaldehyde in PBS and stained with Hoechst 33258. Images were captured with a Zeiss Axioplan 2 microscope equipped with a CARV spinning disc optical module. Nuclear DNA content was quantitated by measuring fluorescent intensity in Adobe Photoshop.
dS6K phosphorylation
Drosophila S2 cells were cultured in Schneider's medium supplemented with 10% heat-inactivated FBS and antibiotics. S2 cells were seeded in a six-well plate at 75% confluency for 3 h (2 mL of medium in each well). Cells were rinsed twice with Schneider's medium without serum before transfection. DNA (10 mg) was incubated at room temperature for 20–30 min with 50 mg of lipofectin (GIBCO BRL) in 1 mL of Schneider's medium, then added to cells for 6–8 h. After recovering for 1 d in complete medium, cells were starved in serum-free medium for 4 h, then pretreated with 50 nM rapamycin for 20 min. Medium containing 20% serum was then added for 30 min, after which cells were lysed for band-shifting assays. For cotransfection experiments, 9 mg of dTOR variants and 1 mg of dS6K were used.
Acknowledgments
We are grateful to Kelly Watson, Cristina Martín-Castellanos, and Todd Laverty for fly stocks, to John Aris, Terry Orr-Weaver, and Helena Richardson for antibodies, to Jean-Karim Heriche for advice on rapamycin treatment, and to Mike O'Connor for support. We thank Doujia Pan, Bruce Edgar, David Prober, and Jessica Britton for comments on the manuscript. We also gratefully acknowledge the Drug Synthesis and Chemistry Branch of the National Cancer Institute (NCI) for rapamycin, and the University of Minnesota Cancer Center Flow Cytometry Core Facility for assistance with FACS analysis (NCI Grant P30 CA77598).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL neufeld@med.umn.edu; FAX (612) 626-7031.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.835000.
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Aris JP, Blobel G. Identification and characterization of a yeast nucleolar protein that is similar to a rat liver nucleolar protein. J Cell Biol. 1988;107:17–31. doi: 10.1083/jcb.107.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbet N-C, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN. TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell. 1996;7:25–42. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T, Hall MN. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999;402:689–692. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- Bohni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, Beckingham K, Hafen E. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1–4. Cell. 1999;97:865–875. doi: 10.1016/s0092-8674(00)80799-0. [DOI] [PubMed] [Google Scholar]
- Britton JS, Edgar BA. Environmental control of the cell cycle in Drosophila: Nutrition activates mitotic and endoreplicative cells by distinct mechanisms. Development. 1998;125:2149–2158. doi: 10.1242/dev.125.11.2149. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Schreiber SL. A signaling pathway to translational control. Cell. 1996;86:517–520. doi: 10.1016/s0092-8674(00)80125-7. [DOI] [PubMed] [Google Scholar]
- Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth FM, Bodenstein D, King RC. Adipose tissue of Drosophila melanogaster. I. An experimental study of larval fat body. J Exp Zool. 1965;158:141–154. doi: 10.1002/jez.1401580203. [DOI] [PubMed] [Google Scholar]
- Cardenas ME, Cutler NS, Lorenz MC, Di Como CJ, Heitman J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes & Dev. 1999;13:3271–3279. doi: 10.1101/gad.13.24.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatham L, Monfar M, Chou MM, Blenis J. Structural and functional analysis of pp70S6k. Proc Natl Acad Sci. 1995;92:11696–11700. doi: 10.1073/pnas.92.25.11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Adames NR, Chan TF, Zeng C, Cooper JA, Zheng XF. TOR signaling regulates microtubule structure and function. Curr Biol. 2000;10:861–864. doi: 10.1016/s0960-9822(00)00599-6. [DOI] [PubMed] [Google Scholar]
- Chou MM, Blenis J. The 70 kDa S6 kinase: Regulation of a kinase with multiple roles in mitogenic signalling. Curr Opin Cell Biol. 1995;7:806–814. doi: 10.1016/0955-0674(95)80064-6. [DOI] [PubMed] [Google Scholar]
- Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell. 1992;69:1227–12236. doi: 10.1016/0092-8674(92)90643-q. [DOI] [PubMed] [Google Scholar]
- Coelho CM, Leevers SJ. Do growth and cell division rates determine cell size in multicellular organisms? J Cell Sci. 2000;113:2927–2934. doi: 10.1242/jcs.113.17.2927. [DOI] [PubMed] [Google Scholar]
- Conlon I, Raff M. Size control in animal development. Cell. 1999;96:235–244. doi: 10.1016/s0092-8674(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Dennis PB, Fumagalli S, Thomas G. Target of rapamycin (TOR): Balancing the opposing forces of protein synthesis and degradation. Curr Opin Genet Dev. 1999;9:49–54. doi: 10.1016/s0959-437x(99)80007-0. [DOI] [PubMed] [Google Scholar]
- Derenzini M, Trere D, Pession A, Montanaro L, Sirri V, Ochs RL. Nucleolar function and size in cancer cells. Am J Pathol. 1998;152:1291–1297. [PMC free article] [PubMed] [Google Scholar]
- Duronio RJ, Brook A, Dyson N, O'Farrell PH. E2F-induced S phase requires cyclin E. Genes & Dev. 1996;10:2505–2513. doi: 10.1101/gad.10.19.2505. [DOI] [PubMed] [Google Scholar]
- Fox HL, Kimball SR, Jefferson LS, Lynch CJ. Amino acids stimulate phosphorylation of p70S6k and organization of rat adipocytes into multicellular clusters. Am J Physiol. 1998;274:C206–C213. doi: 10.1152/ajpcell.1998.274.1.C206. [DOI] [PubMed] [Google Scholar]
- Gao X, Neufeld TP, Pan D. Drosophila PTEN regulates cell growth and proliferation through PI3K-dependent and -independent pathways. Dev Biol. 2000;221:404–418. doi: 10.1006/dbio.2000.9680. [DOI] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- Hardwick JS, Kuruvilla FG, Tong JK, Shamji AF, Schreiber SL. Rapamycin-modulated transcription defines the subset of nutrient-sensitive signaling pathways directly controlled by the Tor proteins. Proc Natl Acad Sci. 1999;96:14866–14870. doi: 10.1073/pnas.96.26.14866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hari KL, Santerre A, Sekelsky JJ, McKim KS, Boyd JB, Hawley RS. The mei-41 gene of D. melanogaster is a structural and functional homolog of the human ataxia telangiectasia gene. Cell. 1995;82:815–821. doi: 10.1016/0092-8674(95)90478-6. [DOI] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- Iiboshi Y, Papst PJ, Kawasome H, Hosoi H, Abraham RT, Houghton PJ, Terada N. Amino acid-dependent control of p70(s6k). Involvement of tRNA aminoacylation in the regulation. J Biol Chem. 1999;274:1092–1099. doi: 10.1074/jbc.274.2.1092. [DOI] [PubMed] [Google Scholar]
- Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–790. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith CT, Schreiber SL. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science. 1995;270:50–51. doi: 10.1126/science.270.5233.50. [DOI] [PubMed] [Google Scholar]
- Kimball SR, Jefferson LS. Regulation of translation initiation in mammalian cells by amino acids. In: Sonenberg N, Hershey JWB, Mathews MB, editors. Translational control of gene expression. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2000. pp. 561–579. [Google Scholar]
- Kleijn M, Proud CG. Glucose and amino acids modulate translation factor activation by growth factors in PC12 cells. Biochem J. 2000;347:399–406. doi: 10.1042/0264-6021:3470399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Pandey P, Sabatini D, Kumar M, Majumder PK, Bharti A, Carmichael G, Kufe D, Kharbanda S. Functional interaction between RAFT1/FRAP/mTOR and protein kinase cdelta in the regulation of cap-dependent initiation of translation. EMBO J. 2000a;19:1087–1097. doi: 10.1093/emboj/19.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Sabatini D, Pandey P, Gingras AC, Majumder PK, Kumar M, Yuan ZM, Carmichael G, Weichselbaum R, Sonenberg N, et al. Regulation of the rapamycin and FKBP-target 1/mammalian target of rapamycin and cap-dependent initiation of translation by the c-Abl protein-tyrosine kinase. J Biol Chem. 2000b;275:10779–10787. doi: 10.1074/jbc.275.15.10779. [DOI] [PubMed] [Google Scholar]
- Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73:585–596. doi: 10.1016/0092-8674(93)90144-f. [DOI] [PubMed] [Google Scholar]
- Mahajan PB. Modulation of transcription of rRNA genes by rapamycin. Int J Immunopharmacol. 1994;16:711–721. doi: 10.1016/0192-0561(94)90091-4. [DOI] [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: A regulator of cell size. Science. 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- Morata G, Ripoll P. Minutes: Mutants of Drosophila autonomously affecting cell division rate. Dev Biol. 1975;427:211–221. doi: 10.1016/0012-1606(75)90330-9. [DOI] [PubMed] [Google Scholar]
- Mori H, Inoue M, Yano M, Wakabayashi H, Kido H. 14–3–3tau associates with a translational control factor FKBP12–rapamycin-associated protein in T-cells after stimulation by pervanadate. FEBS Lett. 2000;467:61–64. doi: 10.1016/s0014-5793(00)01126-1. [DOI] [PubMed] [Google Scholar]
- Nanya S, Bicudo HE. Variation of nucleolar area in fat tissue of Drosophila mulleri during development. Cytobios. 1995;81:73–86. [PubMed] [Google Scholar]
- Navé BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: Identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344:427–431. [PMC free article] [PubMed] [Google Scholar]
- Neufeld TP, Edgar BA. Connections between growth and the cell cycle. Curr Opin Cell Biol. 1998;10:784–790. doi: 10.1016/s0955-0674(98)80122-1. [DOI] [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93:1183–1193. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- O'Connor, S.L. and Anderson, R.P. 1999. smg-1, a PI-3-related kinase involved in nonsense-meditated mRNA decay in C. elegans. GenBank direct submission.
- Parekh D, Ziegler W, Yonezawa K, Hara K, Parker PJ. Mammalian TOR controls one of two kinase pathways acting upon nPKCdelta and nPKCepsilon. J Biol Chem. 1999;274:34758–3464. doi: 10.1074/jbc.274.49.34758. [DOI] [PubMed] [Google Scholar]
- Peterson RT, Beal PA, Comb MJ, Schreiber SL. FKBP12–rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J Biol Chem. 2000;275:7416–7423. doi: 10.1074/jbc.275.10.7416. [DOI] [PubMed] [Google Scholar]
- Pignoni F, Zipursky SL. Induction of Drosophila eye development by decapentaplegic. Development. 1997;124:271–278. doi: 10.1242/dev.124.2.271. [DOI] [PubMed] [Google Scholar]
- Polymenis M, Schmidt EV. Coupling of cell division to cell growth by translational control of the G1 cyclin CLN3 in yeast. Genes & Dev. 1997;11:2522–2531. doi: 10.1101/gad.11.19.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers T, Walter P. Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signaling pathway in Saccharomyces cerevisiae. Mol Biol Cell. 1999;10:987–1000. doi: 10.1091/mbc.10.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prober DA, Edgar BA. Ras promotes cellular growth in the Drosophila wing. Cell. 2000;100:435–436. doi: 10.1016/s0092-8674(00)80679-0. [DOI] [PubMed] [Google Scholar]
- Richardson HE, O'Keefe LV, Reed SI, Saint R. A Drosophila G1-specific cyclin E homolog exhibits different modes of expression during embryogenesis. Development. 1993;119:673–690. doi: 10.1242/dev.119.3.673. [DOI] [PubMed] [Google Scholar]
- Richardson H, O'Keefe LV, Marty T, Saint R. Ectopic cyclin E expression induces premature entry into S phase and disrupts pattern formation in the Drosophila eye imaginal disc. Development. 1995;121:3371–3379. doi: 10.1242/dev.121.10.3371. [DOI] [PubMed] [Google Scholar]
- Sabatini DM, Barrow RK, Blackshaw S, Burnett PE, Lai MM, Field ME, Bahr BA, Kirsch J, Betz H, Snyder SH. Interaction of RAFT1 with gephyrin required for rapamycin-sensitive signaling. Science. 1999;284:1161–1164. doi: 10.1126/science.284.5417.1161. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Kunz J, Hall MN. TOR2 is required for organization of the actin cytoskeleton in yeast. Proc Natl Acad Sci. 1996;93:13780–13785. doi: 10.1073/pnas.93.24.13780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence JC., Jr Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc Natl Acad Sci. 1998;95:7772–7777. doi: 10.1073/pnas.95.13.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60:3504–3513. [PubMed] [Google Scholar]
- Shioi T, Kang PM, Douglas PS, Hampe J, Yballe CM, Lawitts J, Cantley LC, Izumo S. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 2000;19:2537–2548. doi: 10.1093/emboj/19.11.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MJ, Berry CO, Zilberman F, Thomas G, Kozma SC. The Drosophila p70s6k homolog exhibits conserved regulatory elements and rapamycin sensitivity. Proc Natl Acad Sci. 1996;93:10791–10796. doi: 10.1073/pnas.93.20.10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svanberg E, Jefferson LS, Lundholm K, Kimball SR. Postprandial stimulation of muscle protein synthesis is independent of changes in insulin. Am J Physiol. 1997;272:E841–E847. doi: 10.1152/ajpendo.1997.272.5.E841. [DOI] [PubMed] [Google Scholar]
- Toker A. Protein kinases as mediators of phosphoinositide 3-kinase signaling. Mol Pharmacol. 2000;57:652–658. [PubMed] [Google Scholar]
- Verdu J, Buratovich MA, Wilder EL, Birnbaum MJ. Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nat Cell Biol. 1999;1:500–506. doi: 10.1038/70293. [DOI] [PubMed] [Google Scholar]
- Vilella-Bach M, Nuzzi P, Fang Y, Chen J. The FKBP12–rapamycin-binding domain is required for FKBP12-rapamycin-associated protein kinase activity and G1 progression. J Biol Chem. 1999;274:4266–4272. doi: 10.1074/jbc.274.7.4266. [DOI] [PubMed] [Google Scholar]
- Wang X, Campbell LE, Miller CM, Proud CG. Amino acid availability regulates p70 S6 kinase and multiple translation factors. Biochem J. 1998;334:261–267. doi: 10.1042/bj3340261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinkove D, Neufeld TP, Twardzik T, Waterfield MD, Leevers SJ. Regulation of imaginal disc cell size, cell number and organ size by drosophila class I(A) phosphoinositide 3-kinase and its adaptor. Curr Biol. 1999;9:1019–1029. doi: 10.1016/s0960-9822(99)80450-3. [DOI] [PubMed] [Google Scholar]
- Weng QP, Andrabi K, Kozlowski MT, Grove JR, Avruch J. Multiple independent inputs are required for activation of the p70 S6 kinase. Mol Cell Biol. 1995;15:2333–2340. doi: 10.1128/mcb.15.5.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Marshall CA, Lin TA, Kwon G, Munivenkatappa RB, Hill JR, Lawrence JC, Jr, McDaniel ML. Insulin mediates glucose-stimulated phosphorylation of PHAS-I by pancreatic beta cells. An insulin-receptor mechanism for autoregulation of protein synthesis by translation. J Biol Chem. 1998;273:4485–4491. doi: 10.1074/jbc.273.8.4485. [DOI] [PubMed] [Google Scholar]
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Yokogami K, Wakisaka S, Avruch J, Reeves SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol. 2000;10:47–50. doi: 10.1016/s0960-9822(99)00268-7. [DOI] [PubMed] [Google Scholar]
- Zaragoza D, Ghavidel A, Heitman J, Schultz MC. Rapamycin induces the G0 program of transcriptional repression in yeast by interfering with the TOR signaling pathway. Mol Cell Biol. 1998;18:4463–4470. doi: 10.1128/mcb.18.8.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XF, Florentino D, Chen J, Crabtree GR, Schreiber SL. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell. 1995;82:121–130. doi: 10.1016/0092-8674(95)90058-6. [DOI] [PubMed] [Google Scholar]
- Zinke I, Kirchner C, Chao LC, Tetzlaff MT, Pankratz MJ. Suppression of food intake and growth by amino acids in Drosophila: The role of pumpless, a fat body expressed gene with homology to vertebrate glycine cleavage system. Development. 1999;126:5275–5284. doi: 10.1242/dev.126.23.5275. [DOI] [PubMed] [Google Scholar]