

Abstract
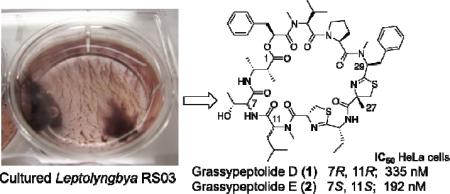
Two new grassypeptolides and a lyngbyastatin analogue, together with the known dolastatin 12, have been isolated from field collections and laboratory cultures of the marine cyanobacterium Leptolyngbya sp. collected from the SS Thistlegorm shipwreck in the Red Sea. The overall stereostructures of grassypeptolides D (1) and E (2) and Ibu-epidemethoxylyngbyastatin 3 (3) were determined by a combination of 1D and 2D NMR experiments, MS analysis, Marfey's methodology, and HPLC-MS. Compounds 1 and 2 contain 2-methyl-3-aminobutyric acid (Maba) and 2-aminobutyric acid (Aba), while biosynthetically distinct 3 contains 3-amino-2-methylhexanoic acid (Amha) and the β-keto amino acid 4-amino-2,-2-dimethyl-3-oxopentanoic acid (Ibu). Grassypeptolides D (1) and E (2) showed significant cytotoxicity to HeLa (IC50 = 335 and 192 nM, respectively) and mouse neuro-2a blastoma cells (IC50 = 599 and 407 nM, respectively), in contrast to Ibu-epidemethoxylyngbyastatin 3 (neuro-2a cells, IC50 > 10 μM) and dolastatin 12 (neuro-2a cells, IC50 > 1 μM).

Microbial metabolites appear to be characteristic of certain biotopes, both on an environmental and a species level, which has provided a diversity of chemical structures unparalleled by even the largest combinatorial libraries.1 Our research has recently focused on the isolation and structure elucidation of biologically active natural products from microorganisms inhabiting unique environments. The Red Sea represents an unexplored repository of diverse cyanobacteria, although in low abundance. This may result from the low annual rainfall, minimal freshwater input and high evaporation rate that make the Red Sea one of the most saline and pristine water bodies in the world.2 Despite these conditions, we have collected specimens from a range of cyanobacterial genera including Lyngbya, Phormidium, Symploca, and the Leptolyngbya that is the subject of this report. Thus, we are interested to compare the biosynthetic capabilities of these Red Sea organisms with those collected pantropically. As is the case for natural products in general, cyanobacterial metabolites often occur as sets of related analogues that possess varying biological selectivity, putatively for optimal adaptation to a range of environments.3 In addition, the capacity of any one cyanobacterium to produce several biosynthetically distinct metabolites,4 as well as the production of the same or biosynthetically related natural products by different genera of cyanobacteria is the subject of intense investigation.5 One proposal is that common heterotrophic bacteria associated with cyanobacteria may be the biosynthetic origin of the isolated products. Alternatively, horizontal gene transfer between different cyanobacteria or between heterotrophic bacteria may account for the presence of multiple biosynthetic gene clusters in cyanobacterial genomes.6 Remarkably, a Floridian Lyngbya confervoides has afforded lyngbyastatins 4–6,7,8 pompanopeptins A and B,9 largamides A–H,10 tiglicamides A–C,11 and grassypeptolides A–C.12 Here we report the isolation of grassypeptolides D (1) and E (2) and Ibu-epidemethoxylyngbyastatin 3 (3), as well as the known dolastatin 12,13 from a marine Leptolyngbya cyanobacterium collected from the Red Sea shipwreck SS Thistlegorm (46 – 98 ft). All four macrocyclic depsipeptides are also produced by the laboratory-cultured (monoclonal) Red Sea Leptolyngbya. While grassypeptolide D (1) is ~1.5-fold less cytotoxic to HeLa cervical carcinoma and neuro-2a mouse blastoma cells than grassypeptolide E (2), these threonine/N-methylleucine diastereomers do not show the dramatic natural structure-activity relationship observed between the N-methylphenylalanine epimers grassypeptolides A and C.12 Ibu-epidemethoxylyngbyastatin 3 (3, IC50 > 10 μM) was significantly less cytotoxic to neuro-2a cells than the grassypeptolides and related dolastatin 12 (IC50 > 1 μM).
Results and Discussion
A crude organic extract of the Red Sea Leptolyngbya was subjected to bioassay-guided fractionation via normal phase VLC using a stepped gradient of hexanes to EtOAC to MeOH. The fraction eluting with 25% MeOH-EtOAC was highly cytotoxic to mouse neuro-2a neurobastoma cells (30 μg/mL reduced cell viability by 99.6%). This VLC fraction was separated by C18 reversed-phase (RP18) solid phase extraction (SPE) and exhaustive RP-HPLC to yield three minor cytotoxic metabolites (1, 1.5 mg; 2, 0.5 mg and 3, 2.9 mg) and the known depsipeptide dolastatin 12 (5.0 mg) as the major component.
A molecular formula of C57H81N9O10S2 for both grassypeptolides D (1) and E (2) was provided by HR-MS ([M + Na]+ m/z 1138.5515 and 1138.5413, respectively) and supported by NMR spectroscopic (Table 1) data. The 1H and 13C NMR spectra for each compound were similar, and indicated peptidic metabolites due to the presence of three NH doublets (δH 6.45–7.50), three N-methyl substituents (δH 3.1–3.5), nine α-H multiplets (δH 3.4–5.9), numerous overlapped methyl doublets (δH 0.74–0.99) and 10 putative ester/amide carbonyl 13C signals (δC 168–175) in each case. These data suggested that 1 and 2 were structurally related to the Floridian Lyngbya confervoides metabolites grassypeptolides A–C, and similar to grassypeptolide C in particular.12 However, the 1D NMR spectra for both 1 and 2 contain a 3H singlet (δH-27 1.35 and 1.38, respectively) and a quaternary carbon (δC-25 84.0) not present in the spectra for grassypeptolide C. In addition, comparison of the α-CH chemical shifts for 1, 2, and grassypeptolide C revealed significant differences between compounds 1 and 2 (Table 1), whereas the α-CH chemical shifts for 1 more closely matched those for grassypeptolide C. These differences led us to investigate the structures of 1 and 2 more closely.
Table 1.
1H (700 MHz) and 13C (175 MHz) NMR Spectroscopic Data for Grassypeptolides D (1) and E (2) in CDCl3.
| unit | position | Grassypeptolide D (1) | Grassypeptolide E (2) | ||
|---|---|---|---|---|---|
| δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | δC, mult. | ||
| Maba | 1 | - | 172.4, C | - | 172.9, C |
| 2 | 2.50, dq (7.0, 6.8) | 45.6, CH | 2.51, dq (7.0, 4.5) | 45.5, CH | |
| 3 | 4.22, m | 48.5, CH | 4.23, m | 47.0, CH | |
| 4 | 1.19, d (6.7) | 19.9, CH3 | 1.10, d (7.0) | 19.3, CH3 | |
| 5 | 1.12, d (7.0) | 14.4, CH3 | 1.16, d (7.0) | 14.4, CH3 | |
| NH | 7.70, br | 6.45, d (9.9) | |||
| Thr | 6 | - | 169.3, C | - | 170.5, C |
| 7 | 4.45, (6.8, 6.1) | 59.1, CH | 3.36, dd (6.9, 3.3) | 57.0, CH | |
| 8 | 4.02, m | 69.1, CH | 3.30, m | 67.9, CH | |
| 9 | 1.22, d (6.4) | 19.7, CH3 | 0.87, d (6.5) | 19.4, CH3 | |
| OH | 4.00, br | - | |||
| NH | 6.90, d (7.3) | 6.90, d (6.9) | |||
| N-Me-Leu | 10 | - | 170.3, C | - | 170.0, C |
| 11 | 4.70, br | 57.5, CH | 5.15, t (7.7) | 54.6, CH | |
| 12a | 1.97, m | 37.2, CH2 | 1.78, m | 37.1, CH2 | |
| 12b | 1.67, m | ||||
| 13 | 1.58, m | 25.3, CH | 1.52, m | 25.3, CH | |
| 14 | 0.98, d (6.4) | 23.2, CH3 | 0.98, d (6.6) | 23.2, CH3 | |
| 15 | 0.94, d (6.5) | 22.8, CH3 | 0.93, d (6.7) | 22.4, CH3 | |
| 16 | 3.20, s | 33.2, CH3 | 3.49, s | 30.7, CH3 | |
| Aba-thn-ca | 17 | - | 170.4, C | - | 169.3, C |
| 18 | 5.29, m | 78.1, CH | 5.07, m | 75.9, CH | |
| 19a | 3.60, m | 33.3, CH2 | 4.15, br dd | 35.5, CH2 | |
| 19b | 3.26, m | 3.44, dd (−11.2, 8.5) | |||
| 20 | - | 178.0, C | - | 175.0, C | |
| 21 | 4.55, m | 54.2, CH | 4.71, ddd (9.0, 4.0, 2.7) | 53.2, CH | |
| 22a | 2.11, m | 25.2, CH2 | 1.95, dqd (−11.5, 7.4, 4.0) | 28.5, CH2 | |
| 22b | 1.86, m | 1.48, m | |||
| 23 | 0.96, t (6.9) | 11.6, CH3 | 0.74, t (7.4) | 10.0, CH3 | |
| NH | 7.10, d (7.7) | 7.50, d (9.0) | |||
| N-Me-Phe-thn-ca | 24 | - | 173.7, C | - | 168.9, C |
| 25 | - | 84.0, C | - | 84.0, C | |
| 26a | 3.74, d (−11.5) | 43.1, CH2 | 3.29, d (−11.6) | 41.5, CH2 | |
| 26b | 3.17, d(−11.3) | 3.18, d(−11.7) | |||
| 27 | 1.35, s | 24.3, CH3 | 1.38, s | 23.9, CH3 | |
| 28 | - | 173.9, C | - | 173.9, C | |
| 29 | 5.39, dd (10.7, 6.4) | 58.9, CH | 5.87, dd (12.2, 3.0) | 55.8, CH | |
| 30a | 3.24, m | 36.0, CH2 | 3.55, br | 36.1, CH2 | |
| 30b | 3.16, m | 3.28, m | |||
| 31 | - | 135.5, C | - | 137.7, C | |
| 32/36 | 7.33, m | 129.2, CH | 7.18, m | 129.6, CH | |
| 33/35 | 7.22, m | 128.5, CH | 7.03, br | 129.3, CH | |
| 34 | 7.20, m | 127.6, CH | 7.08, m | 127.3, CH | |
| 37 | 3.21, s | 30.7, CH3 | 3.17, s | 30.2, CH3 | |
| Pro | 38 | - | 172.7, C | - | 171.2, C |
| 39 | 4.84, dd (8.8,5.1) | 57.5, CH | 5.10, br | 58.8, CH | |
| 40a | 2.22, m | 27.5, CH2 | 2.30, m | 31.2, CH2 | |
| 40b | 1.93, m | 2.10, m | |||
| 41a | 1.99, m | 24.9, CH2 | 2.01, m | 21.7, CH2 | |
| 41b | 1.63, m | ||||
| 42a | 3.97, m | 47.8, CH2 | 3.80, m | 46.9, CH2 | |
| 42b | 3.53, m | 3.69, m | |||
| N-Me-Val | 43 | - | 168.6, C | - | 168.5, C |
| 44 | 4.98, d (11.0) | 60.0, CH | 5.01, d (10.8) | 58.1, CH | |
| 45 | 2.32, m | 27.4, CH | 2.35, m | 28.1, CH | |
| 46 | 0.98, d (6.4) | 19.6, CH3 | 0.95, d (6.7) | 19.5, CH3 | |
| 47 | 0.88, d (6.6) | 18.0, CH3 | 0.85, d (6.7) | 18.6, CH3 | |
| 48 | 3.12, s | 30.0, CH3 | 3.20, s | 30.3, CH3 | |
| Pla | 49 | - | 170.2, C | - | 171.4, C |
| 50 | 5.33, dd (9.9, 3.2) | 72.4, CH | 5.33, dd (9.7, 3.8) | 72.4, CH | |
| 51a | 3.09, dd (−14.5, 9.9) | 36.9, CH2 | 3.16, m | 36.9, CH2 | |
| 51b | 3.04, dd (−14.4, 3.2) | 3.01, m | |||
| 52 | - | 134.9, C | - | 136.1, C | |
| 53/57 | 7.22, m | 129.1, CH | 7.12, m | 129.1, CH | |
| 54/56 | 7.24, m | 129.5, CH | 7.31, m | 129.5, CH | |
| 55 | 7.27, m | 127.6, CH | 7.24, m | 127.4, CH | |
Analysis of the 2D NMR spectra in CDCl3 for 1 and 2 (HSQC, HSQC-TOCSY, HMBC, COSY, ROESY) confirmed that grassypeptolides D (1) and E (2) had the same connectivity of planar structure as grassypeptolide C. An HSQC-TOCSY experiment for 1 identified spin systems for Thr, Leu, Pro, and Val amino acid side chains, and key HMBC (supported by COSY and ROESY) correlations established that Leu and Val were N-methylated as in grassypeptolide C. A pair of methyl doublets (δH-4 1.19 and δH-5 1.12) correlated to two vicinally coupled methine multiplets (δH-2 2.50 and δH-3 4.22), distinguished the β-amino acid residue 2-methyl-3-aminobutyric acid (Maba). COSY correlations from an upfield methyl doublet (δH-23 0.96) to a relatively shielded methylene (δH-22 2.11/1.86), in turn coupled to a methine (δH-21 4.55), supported a 2-aminobutyric acid (Aba) residue. An HMBC correlation to the putative carbonyl 13C of Aba from a relatively deshielded methylene (δH-19 3.60/3.26), which also correlated to a second deshielded quaternary 13C (δC-17 170.4), was consistent with an Aba-derived thiazoline carboxylic acid (Aba-thn-ca). Chemical shift comparison with grassypeptolide C and HMBC analysis (Table S1) also confirmed the presence of two aromatic residues, phenyllactic acid (Pla) and N-methylphenylalanine (N-Me-Phe). The latter residue is incorporated into a thiazoline carboxylic acid in grassypeptolide C. However, no –CHCH2– motif consistent with this remaining thiazoline ring was apparent in the spectra for grassypeptolide D (1). Instead, an AB spin system of a fifth isolated methylene (δH-26 3.74/3.17) showed HMBC correlations to a methyl (δC-27 24.3), a midfield quaternary (δC-25 84.0) and two deshielded quaternary (δC-28 173.9 and δC-24 173.7) carbons. This indicated the presence of a 2–methylthiazoline carboxylic acid derived from N-methylphenylalanine (N-Me-Phe-4-Me-thn-ca). Although none of the previously reported grassypeptolides A–C contain a methylated thiazoline carboxylic acid, this unit has been reported in largazole14 and the hoiamides15 from marine cyanobacteria, as well as several terrestrial bacteria.
Analysis of the 2D NMR data for 2 revealed the same sequence of units as found in 1. However, the α-CH chemical shifts for Thr (δH/δC-7 3.36/ 57.0) and N-Me-Leu (δH/qC-11 5.15/54.6) in 2 were substantially different from those observed for 1 (Thr, δH/δC-7 4.45/59.1; N-Me-Leu, δH/δC-11 4.70/57.5) and grassypeptolide C (Thr, δH/δC-7 4.44/59.2; N-Me-Leu, δH/δC-11 4.70/57.6). Furthermore, the N-CH3-16 singlet (δH 3.49) for 2 was shifted slightly upfield compared to those for 1 and grassypeptolide C (δH 3.20 and 3.17, respectively). Overall, these chemical shift differences suggested different configurations for both Thr and N-Me-Leu in 2 relative to 1 and grassypeptolide C.
The absolute configurations of grassypeptolides D (1) and E (2) were determined by a combination of acid hydrolysis and oxidative ozonolysis followed by chiral LC-MS or Marfey's analysis. Chiral LC-MS of the acid hydrolysates of 1 and 2 established the presence of l-Pla, while analysis by RP18 HPLC of the ozonolysis and acid hydrolysate products of 1 and 2 derivatized with N-α-(5-fluoro-2,4-dinitrophenyl-)-l-leucinamide (Marfey's reagent) indicated the presence of N-Me-l-Val, l-Pro, N-Me-l-Phe, (2S)-MeCysA, d-Aba, l-Cya and (2R, 3R)-Maba. Consistent with the observed differences in the α-CH chemical shifts of Thr and N-Me-Leu described above, Marfey's analysis of the grassypeptolide D (1) ozonolysis and hydrolysis product matched the configurations observed for grassypeptolide C (d-allo-Thr and N-Me-d-Leu), while grassypeptolide E (2) hydrolysate retention times indicated the presence of l-Thr and N-Me-l-Leu.
Ibu-epidemethoxylyngbyastatin 3 (3) was isolated from the same first tier RP-HPLC fraction as the grassypeptolides. The molecular composition of 3 was established as C51H82N8O11 from HR-FTMS data ([M + Na]+ m/z 1005.5983). While the 1H NMR spectrum for compound 3 also exhibited resonances typical of a peptide, it was significantly different to those for grassypeptolides 1 and 2. In addition, inspection of the 13C NMR spectrum for compound 3 revealed the absence of a midfield quaternary (δC 84.0) and the presence of a downfield quaternary carbon (δC 208.4), which in combination with the reduced molecular mass, suggested a different planar structure for 3 relative to compounds 1 and 2. Instead, the 1D spectra for 3 were similar to those for dolastatin 12, isolated as the major component from the preceding first tier HPLC fraction. Examination of the 2D NMR data for 3 confirmed that an additional methyl triplet (δH 0.86) in the 1H NMR spectrum for 3 and increase of 14 mass units relative to dolastatin 12 were attributable to the presence of a 3-amino-2-methylhexanoic acid moiety (Amha) in 3, rather than the 3-amino-2-methylpentanoic acid (Ampa) in dolastatin 12. Thus, the planar structure of 3 could best be designated as demethoxylyngbyastatin 3, indicating replacement of the N,O-dimethylTyr in lyngbyastatin 316 with N-methylPhe in 3. The configurational assignment of the Ibu unit in the lyngbyastatin/dolastatin series has proven arduous given the propensity of this unit to decarboxylate and epimerize during acid hydrolysis to yield (2RS)-2-amino-4-methylpentan-3-one (Amp).4,17 The analysis and comparison of this series of depsipeptides containing Ibu is further confounded by whether or not the neighboring Ala unit is N-methylated. Williams and coworkers deduced that their cyanobacterial samples of dolastatin 12 and lyngbyastatins 1 and 3 each comprised Ibu epimeric mixtures following NaBH4 reduction of the natural products, acid hydrolysis and comparisons with the reduced Ibu standards, (3RS, 4S)-4-amino-2,2-dimethyl-3-hydroxy-pentanoic acid (Adhpa).16 It was shown that little enolization occurs during the NaBH4 reduction itself, which is performed to avoid extensive enolization of the β-keto moiety in Ibu during subsequent acid hydrolysis. To investigate further the timing of epimerization at C-15 in 3 and dolastatin 12 during acid hydrolysis, we first undertook an alternate strategy. Compound 3 and dolastatin 12 were subjected to Marfey's analysis following acid hydrolysis under two different conditions: (A) hydrolysis with 6N HCl for 50 seconds using a microwave and Ace high-pressure tube; and (B) treatment with 6N HCl at 110 °C for 18 h. Analysis of the reaction products of 3 by RP18 HPLC revealed the presence of both enantiomers of Amp in the S/R ratio of 4.1:1 and 1:2.2 for treatments A and B, respectively. A similar result was obtained in the analysis of dolastatin 12 under these conditions, suggesting that racemization of the Ibu unit likely occurs more slowly than peptide hydrolysis; a rapid acid hydrolysis reaction results in less enolization of the Ibu substrate. In tandem with the lack of extremely broad peaks (characteristic of the R-Ibu epimer lyngbyastatin 1,15) in the 1H NMR spectra for 3 and dolastatin 12, these data suggest that both compounds contain S-Ibu. Given the relatively sharp 1H NMR signals for the natural products, it is unlikely that epimerization from an R-Ibu to S-Ibu occurred prior to amide hydrolysis, despite that the S-Ibu configuration is thermodynamically favored.15 Noteworthy also is that the analysis of the S-Amp standard derivatized with Marfey's reagent under standard reaction conditions (40 °C, 1 h) showed the presence of both enantiomers in the S/R ratio of 6.7:1, whereas Marfey's derivatization for 24 h resulted in the ratio of 1.8:1. Thus, further enolization of the Amp unit in the natural product hydrolysate may occur during the derivatization process.
To corroborate our assignment of S-Ibu following the method of Williams et al.,16 compound 3 was reduced with NaBH4, subjected to microwave hydrolysis, and the Marfey's derivatives compared to synthetically prepared Adhpa standards (see Supporting Information). Analysis of the reaction products of 3 by RP18 HPLC-MS revealed the presence of enantiomers of Adhpa in the S/R ratio 13.8:1. The absolute configurations of the remaining stereogenic centers in Ibu-epidemethoxylyngbyastatin 3 were assigned by a combination of chiral LC-MS and Marfey's analysis using commercially available or synthetic (Amha) standards. Following acid hydrolysis of 3, chiral LC-MS analysis revealed the presence of (2S, 3S)-HMPA. Derivatization of the acid hydrolysate of 3 with Marfey's reagent, followed by RP18 HPLC or LC-MS established the presence of N-Me-l-Ala, N-Me-l-Val, N-Me-l-Phe and N-Me-l-Leu, and (2S, 3R)-Amha.
Diastereomeric grassypeptolides D (1) and E (2) both showed significant cytotoxicity to HeLa (IC50 335 and 192 nM, respectively) and neuro-2a (IC50 599 and 407 nM, respectively) cells. The small (~1.5-fold) difference in cytotoxicity between 1 and 2 indicates that the chirality of the N-Me-Leu and Thr α position is not critical for activity and that this region is not central to the pharmacophore of these structures. In contrast, Kwan et al.12 conclude that the N-Me-Phe region of grassypeptolides A–C is critical for their cytotoxicity given that grassypeptolide C (N-Me-l-Phe) showed 16–23– and 65–fold greater potency, respectively, than grassypeptolides A and B (both N-Me-d-Phe) against colorectal adenocarcinoma HT29 and cervical carcinoma HeLa cells. In addition the ethyl sidechain of the Aba-thn-ca in grassypeptolides A and C is associated with enhanced activity over the methyl of the Ala unit in grassypeptolide B.12 Like grassypeptolide C, grassypeptolides D (1) and E (2) also contain Aba and N-Me-l-Phe thiazoline carboxylic acid units. The only structural difference between 1 (IC50 335 nM, HeLa cells) and grassypeptolide C (IC50 45 nM, HeLa cells) is a methylated thiazoline of opposite chirality (25S). The apparent ~7.5–fold difference in cytotoxicity between grassypeptolides D (1) and C supports the hypothesis that the N-Me-Phe-thn-ca-Aba-thn-ca motif is central to the pharmacophore of the grassypeptolides. While lyngbyastatin 3 was reported previously to be potently cytotoxic to KB (epithelial carcinoma, subline of HeLa) and LoVo (colon carcinoma) cell lines (IC50 = 32 and 400 nM, respectively),16 we observed little effect of Ibu-epidemethoxylyngbyastatin 3 (3) on neuro-2a cells (IC50 > 10 μM). Similarly dolastatin 12 was only slightly more cytotoxic to these cells than 3 (IC50 > 1 μM).
A majority of the more than 300 known marine cyanobacterial metabolites have been reported from the genus Lyngbya.3 However, metabolites closely related to these “Lyngbya compounds” have also been reported from non-Lyngbya species, as in the case of the four compounds reported here. This has led to an inability to define strong chemotaxonomic trends for cyanobacterial genera. The taxonomic description of cyanobacteria has transitioned from the traditional phycological system to the modern bacteriological system, supported by use of the 16S rRNA gene for phylogenetic determinations. Phylogenetic revision of cyanobacterial taxonomy is in progress for several genera, including Lyngbya.5 In tandem with confirmation of the biogenetic source of compounds isolated from mixed field collections, this may clarify chemotaxonomic relationships. Although it belongs to a less commonly reported genus and is collected from a unique habitat, the Red Sea Leptolyngbya sp. RS03 reported here shows biosynthetic capabilities comparable to cyanobacteria collected pantropically. Bis-thiazoline-containing grassypeptolides D (1) and E (2) are closely related to grassypeptolide C, one of three structural analogues reported from a Florida Keys collection of Lyngbya confervoides,12 while Ibu-epidemethoxylyngbyastatin 3 (3) and dolastatin 12 are additional congeners of the large family of lyngbyastatins and micropeptins. A systematic classification of the cultured Red Sea Leptolyngbya sp. RS03 indicates that this organism is Leptolyngbya ectocarpi (Gomont) Anagnostidis et Komárek 1988, according to Komárek and Anagnostidis.18 A phylogenetic analysis of the partial 16S rRNA sequences from Leptolyngbya sp. RS03 (GenBank Acc. No. JF518829) and the marine Leptolyngbya reference strain Leptolyngbya ectocarpi ATCC 29409 revealed a distinct cluster of Leptolyngbya spp. from various marine habitats (Figure 1), with the exception of Phormidium persicinium SAG 80.79. However, the latter shares ~99.7% 16S rRNA gene sequence identity with the Leptolyngbya reference strains included in this analysis and may represent the same organism that has been maintained in different culture collections.19 Noteworthy is that the Floridian Lyngbya cf. confervoides VP0401 clusters with members of the Phormidium subgenus Geitlerinema Anagnostidis et Komárek 1988, which seemingly supports the notion that horizontal gene transfer plays an important role in the evolution of cyanobacteria.18 Also included in this analysis, is a separate cultured Red Sea Leptolyngbya sp. (RS02) that produces the brominated macrolide phormidolide,20 which was previously isolated from an Indonesian Phormidium sp. Further chemical and biological characterization of this second Red Sea Leptolyngbya is in progress. While it is well recognized that organisms isolated from their native environment may not produce the same secondary metabolites in laboratory culture,21 LC-MS analysis of extracts of monoclonal cultures of the Red Sea Leptolyngbya sp. RS03 confirmed its production of the four metabolites reported here. The stereoisomerism and natural SAR within the grassypeptolide series presents an intriguing example of the biosynthetic flexibility of cyanobacteria. Characterization and manipulation of the biosynthetic pathways for these natural products may provide exciting opportunities to produce new biologically active metabolites.
Figure 1.

The phylogenetic relationship of the Red Sea Leptolyngbya sp. RS03 with marine filamentous cyanobacteria of the order Oscillatoriales (III) based on 16S rRNA nucleotide sequences. Vibrio harveyi ATCC 14126 and V. parahaemolyticus ATCC 17802 (marine γ-proteobacteria) are included as outgroups. Labels on the terminal nodes indicate the species, strain, and GenBank accession numbers in parenthesis; an asterisk designates reference strains. Strains with 16S rRNA gene sequences determined in this study are indicated in blue. Support values < 70 are not indicated. The scale bar indicates 0.02 nucleotide substitutions per site.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a Jasco P-1010 polarimeter. UV spectra were measured on a SpectraMax190 (Molecular Devices). NMR data were acquired in CDCl3 referenced to residual CHCl3 chemical shifts (δC 77.2, δH 7.26) on a Bruker Avance III 700 MHz spectrometer equipped with a 5 mm 13C cryogenic probe for compound 2. NMR data for 1 and 3 were acquired in CDCl3 on a Bruker DPX 400 MHz spectrometer equipped with a 5mm BBI probe and also in methanol-d4 (residual CH3OH, δC 49.2, δH 3.31) on a Bruker DRX 600 MHz spectrometer equipped with a 5 mm TXI probe. HR-MS was performed in positive ion mode on Thermo Scientific LTQ FT Ultra Hybrid and AB SCIEX Triple TOF 5600 mass spectrometers. LC-ESIMS data were obtained on an AB SCIEX 3200 Q TRAP mass spectrometer. HPLC was performed using a Shimadzu dual LC-20AD solvent delivery system with a Shimadzu SPD-M20A UV/VIS photodiode array detector.
Collection, Isolation and Culture of the Red SeaLeptolyngbya
An apparent mixed assemblage of cyanobacteria was collected by hand using SCUBA from the SS Thistlegorm shipwreck (46 – 98 ft) in the Red Sea (N 27° 48.849' E 33° 55.222') on May 27, 2007. Morphological characterization was performed using a Zeiss phase contrast microscope (×100 objective) and the specimen was identified systematically according to Komárek and Anagnostidis.18 Live cyanobacteria were isolated microscopically (Olympus SZ40 stereomicroscope) and grown in triplicate in 24-well culture plates at 27 °C with a 12 hour light-dark cycle (~5 μmol photons s−1 m−2 provided by 40W cool white fluorescent lights). Four different enrichment mediums were used to investigate optimal conditions for survival in laboratory culture. These included 0.2 μm-filtered seawater from the Red Sea (RSW); RSW amended with soil extract; BG-11 medium containing DN vitamin mix,22 which had been modified to closely resemble conditions of the cyanobacterium's natural environment (RSM; pH 8.4 and salinity 41 ‰); and RSM + RSW (1:1). Monoclonal cultures of Leptolyngbya were isolated microscopically from serial dilutions of enrichment cultures in which exceptional growth was present (RSM:RSW) and maintained in RSM.
DNA Extraction and Amplification of Cyanobacterial 16S rRNA
Prior to DNA extraction, heterotrophic bacteria within the cyanobacterial cultures were reduced by sequential treatment through sonication, washing and addition of antibiotics as previously described.23 Genomic DNA was then extracted from approximately 40 mg of freeze-dried cyanobacterial tissue using the Wizard® Genomic DNA purification kit (Promega Inc., A1120) following the manufacturer's protocol. The isolated genomic DNA was subjected to further purification using an anion exchange column (Qiagen, Genomic-tip 20/G). DNA concentration and purity was measured on a Bio Rad SmartSpec™ 3000 spectrophotometer. Approximately 650 bp of the upstream cyanobacterial 16S rRNA sequence was amplified from these genomic extracts using the cyanobacteria-specific primers, CYA106F and CYA781Ra/b,24 while the cyanobacteria-specific primer (CYA359F) and general bacterial primer (1509R) were used to amplify 1,150 bp downstream.25 Sequences were amplified from approximately 50 ng of DNA using GoTaq® Hot Start polymerase (0.5 uL, Promega) according to manufacturer's specifications. Polymerase chain reactions (PCR) were performed in an Eppendorf Mastercycler® gradient thermal cycler (Eppendorf, Hauppauge, NY, USA) as follows: initial denaturation at 95 °C for 3 min; 15 cycles of Touch Down PCR: 95 °C for 30 sec, 65 °C for 45 sec (decreased by 1 °C per cycle), and 72 °C for 1 min; 15 additional cycles of amplification: 95 °C for 20 sec, 50 °C for 20 sec, and 72 °C for 1.5 min; and final elongation at 72 °C for 3 min. PCR products were gel-purified, cleaned with the QIAquick® Gel Extraction kit (cat. no. 28704, Promega) and directly sequenced on an ABI 3730 capillary sequencer by the Oregon State University Center for Genome Research and Biocomputing DNA Sequencing Core Facility. The amplification primers described above were used as sequencing primers with the addition of reverse complement primers CYA359R and CYA781Fa/b for additional sequence coverage at the end regions of the 16S rRNA gene amplification product. The 16S rRNA partial gene sequences were inspected visually and assembled using CAP3.26 The resulting contig was analyzed for chimeric sequences using Pintail27 and compared to sequences in the Ribosomal Database Project database (http://rdp.cme.msu.edu) and GenBank (http://www.ncbi.nlm.nih.gov). The consensus sequence was deposited in GenBank under accession number JF518829.
Phylogenetic Analysis
The 16S rRNA gene sequences of 29 marine cyanobacterial species within the Oscillatoriales (III) and two marine γ-proteobacteria strains Vibrio harveyi ATCC 14126 and Vibrio parahaemolyticus ATCC 17802 (included as outgroups) were collected from GenBank. The 16S rRNA gene library was screened for chimeric sequences using the computer program Mallard,28 aligned using ClustalX in MEGA 5,29 and the resulting alignment optimized with RASCAL30 for a total of 1250 positions (89.7 % identity) in the final dataset covering the V2 to V8 hypervariable regions within the 16S rRNA gene. Prior to phylogenetic predictions, a statistical selection of best fit models of nucleotide substitutions for the 16S rRNA data set was selected using Akaike and Bayesian information criteria (AIC and BIC) in jModelTest 0.1.1.31 Phylogenetic trees were calculated using the minimum-evolution (ME) algorithm in MEGA 5 as well as the Bayesian (MrBayes)32 and phylogenetic maximum likelihood (PhyML v3.0)33 algorithms in Geneious 5.3.34 The minimum evolutionary distances were determined using a maximum composite likelihood method performed with 1000 bootstrap replicates and the Close-Neighbor-Interchange (CNI) algorithm. All ambiguous positions were removed for each sequence pair (pairwise deletion option). The PhyML analysis was performed with 500 bootstrap replicates using the GTR+I+G model (selected by AIC and BIC in jModelTest; proportion of invariable sites (pINV) = 0.465, shape parameter (α) = 0.387, number of rate categories = 4). Bayesian analysis was performed with the GTR+I+G substitution model (pINV = 0.452, α = 0.372, number of rate categories = 4). The Markov chain length (one cold and three heated) was set to 3 million with sampling performed every 100 generations (25% burn in). The analysis was completed once convergence was achieved (~2.7 million generations), which was determined by an average standard deviation in split frequencies of < 0.01.
Extraction and Isolation of Compounds 1–3 and Dolastatin 12
The field collection of Leptolyngbya (500 mL, collection code EHu5-27-07-1) for chemical extraction was stored in 2-propanol at −20 °C until extraction to yield 1.26 g organic extract (CH2Cl2-MeOH, 2:1). The organic extract was subjected to bioassay-guided fractionation via NP VLC using a stepped gradient of hexanes to EtOAc to MeOH. The fraction eluting with 25% MeOH-EtOAc was further separated by RP-SPE using a stepped gradient of MeOH-H2O from 50% MeOH-H2O to 100% MeOH, followed by 100% CH2Cl2. Isocratic RP-HPLC (column: Synergi Fusion-RP, 10 × 250 mm, 70% MeCN-H2O, 3 mL/min) of the SPE fraction C eluting in 70% MeOH-H2O yielded two impure HPLC peaks targeted for further purification. RP-HPLC (Chirobiotic TAG, 4.6 × 250 mm, 98% EtOH-H2O, 0.5 mL/min) of the less polar of these fractions yielded Ibu-epidemethoxylyngbyastatin 3 (3, 2.9 mg) and a mixture of grassypeptolides D (1, 1.5 mg) and E (2, 0.5 mg), which were separated on the Chirobiotic TAG column using 75% MeOH-H2O (0.5 mL/min). Dolastatin 12 was also isolated from the 25% MeOH-EtOAc NP VLC fraction H. Repeated isocratic RP-HPLC (column: Synergi Fusion RP, 10 × 250 mm, 70% MeCN-H2O, 3 mL/min) of the SPE fraction (H2) eluting in 70% MeOH-H2O fraction yielded dolastatin 12 as the major component. Subsequent LC-MS profiling (Synergi Fusion-RP, 2 × 100 mm, 0.2 mL/min, linear gradient of 65 to 100% MeCN in 0.1% (v/v) aqueous TFA) of extracts from monoclonal Leptolyngbya cultures (2 × 1.5 L) yielded m/z 1138.6 (1 and 2, [M+Na]+), 1005 (3, [M+Na]+) at the same retention times as 1, 2 and 3 purified from the original field collection.
Grassypeptolide D (1)
colorless, amorphous solid; [α]21D +25.9 (c 0.15, CH2Cl2); UV (MeOH) λmax (log ε) 212 (3.82), 260 (3.56); 1H and 13C NMR data, see Table 1, Tables S1 and S4; HR-FTMS m/z [M + Na]+ 1138.5515 (calcd for C57H81N9O10S2Na, 1138.5445), m/z [M + H]+ 1116.5458 (calcd for C57H82N9O10S2, 1116.5620).
Grassypeptolide E (2)
colorless, amorphous solid; [α]21D +13.2 (c 0.15, CH2Cl2); UV (MeOH) λmax (log ε) 212 (3.73), 256 (3.48); 1H and 13C NMR data, see Table 1, Table S2; HR-TOFMS m/z [M + Na]+ 1138.5413 (calcd for C57H82N9O10S2Na, 1138.5445), m/z [M + H]+ 1116.5603 (calcd for C57H82N9O10S2, 1116.5620).
Ibu-epidemethoxylyngbyastatin 3 (3)
white, amorphous solid; [α]21D −48.6 (c 0.5, CHCl3); UV (MeOH) λmax (log ε) 212 (3.98), 256 (3.81); 1H and 13C NMR data, see Table S3; HR-FTMS m/z [M + Na]+ 1005.5983 (calcd for C51H83N8O11Na, 1005.6001), m/z [M + H]+ 983.6162 (calcd for C51H83N8O11, 983.6175).
Dolastatin 12
white, amorphous solid; [α]21d −79.8 (c 0.5, CHCl3); UV (MeOH) λmax (log ε) 212 (3.71), 256 (3.57); 1H and 13C NMR data, see Supporting Information;13 HR-TOFMS m/z [M + H]+ 969.6035 (calcd for C50H81N8O11, 969.6019).
Absolute Configuration of Grassypeptolides D (1) and E (2)
The amino acid standards relevant to compounds 1 and 2 were obtained commercially or as gifts and prepared as 50 mM solutions in H2O. Standards for (2R)-and (2S)-methylcysteine were kindly provided by Dr. W.H. Gerwick, Scripps Institution of Oceanography, University of California, San Diego. A portion of each standard (5.0 mg) was dissolved in 720 μL HCO2H at 0 °C. Next, 80 μL of H2O2 (30%) was added dropwise with continuous stirring and the reaction was carried out at 0 °C for 2 h to yield either (2R)- or (2S)-methylcysteic acid (MeCysA). The product mixture was dried under a steady stream of N2 gas and resuspended in H2O (50 mM). The N-benzoyl O-methyl ester of (2R, 3S)-2-methyl-3-aminobutyric acid (Maba) was gratefully received from Dr. Hendrik Luesch, Department of Medicinal Chemistry, University of Florida. Approximately 0.4 mg was deprotected with 500 μL 6 N HCl at 110 °C for 24 h, evaporated to dryness and resuspended in H2O (50 mM). Each standard was derivatized for Marfey's analysis by adding 10 μL of 1 M NaHCO3 and 50 μL of N-α-(5-fluoro-2,4-dinitrophenyl-)-l-leucinamide (l-FDLA or d-FDLA, 1% w/v in acetone) to 25 μL of each standard solution. The mixture was heated at 40 °C for 1 h with continuous stirring, cooled to room temperature, acidified with 5 μ,L 2N HCl, evaporated to dryness, and resuspended in 250 μL MeCN-H2O (1:1).
Approximately 0.1 mg of 1 and 0.2 mg of 2 were dissolved separately in 3 mL CH2Cl2 (−78 °C). Ozone was then bubbled through each solution for 15 min. The solution was dried under a stream of N2 gas, followed by an oxidative workup of the residue (0.6 mL of H2O2-HCOOH 1:2 at 70 °C for 20 min). The oxidation product was concentrated under vacuum and hydrolyzed with 1 mL of 6 N HCl at 110 °C for 18 h. The hydrolyzed products were resuspended in 25 μL H2O and derivatized for Marfey's analysis in a similar manner to the derivatized chromatographic standards. The Marfey's products of 1 and 2 were resuspended in 50 μL MeCN-H2O (1:1) and analyzed by RP-HPLC (Gemini C18 110 A, 4.6 × 150 mm, 5 μm, 1.0 mL/min, UV detection at 340 nm) using a linear gradient of 30 to 70% MeCN in 0.1% (v/v) aqueous TFA over 50 min. The retention time (tR min) of the residues in the hydrolysate of 1 matched standards for d-Aba (21.9; l-Aba, 17.1), l-Cya (7.1; d-Cya, 6.5), (2S)-MeCysA [6.2; (2R)-MeCysA, 7.8], N-Me-d-Leu (27.8; N-Me-l-Leu, 24.5), (2R, 3R)-Maba l-FDLA [18.8; (2R,3R)-Maba d-FDLA, 25.6; (2R,3S)-Maba l-FDLA, 18.6; (2R,3S)-Maba d-FDLA, 20.8], l-Pro (14.1; d-Pro, 17.0), N-Me-l-Phe (22.7; N-Me-d-Phe, 24.6), N-Me-l-Val (21.2; N-Me-d-Val, 25.6), d-allo-Thr (12.6; l-Thr, 10.2; l-allo-Thr, 11.1; d-Thr, 14.4). The retention times for the residues in the hydrolysate of 2 were consistent with the results for 1, with the exception of N-Me-l-Leu (24.5) and l-Thr (10.2) standards matching the corresponding residues in the hydrolysate of 2. The configuration of the Pla residue in the hydrolysates of both 1 and 2 was determined by chiral LC-MS. The retention time [Chirobiotic TAG, 4.6 × 250 mm; MeOH-10 mM NH4OAc (3:2, pH 5.50); flow rate, 0.4 mL/min; detection by ESIMS in negative ion mode] of the natural product hydrolysate matched that for l-Pla (7.4 min; d-Pla, 8.5).
Absolute Configuration of Ibu-epidemethoxylyngbyastatin (3)
Approximately 0.4 mg of 3 in 0.2 mL anhydrous MeOH was added to a solution of NaBH4 (2 mg) in anhydrous MeOH at 0 °C. After stirring for 30 min, the solution was acidified with 1N HCl until pH 6. The solution was then partitioned between EtOAc and H2O, and the organic layer concentrated to dryness for analysis by RP-HPLC (Synergi Fusion-RP, 10 × 250 mm, 62% MeCN-H2O, 3.5 mL/min). A single product consistent with the dihydro-form of 3 (tR 18.9 min) was present, while unreduced compound 3 (tR 21.3 min) was not detected. This reduction product and an additional ~0.4 mg of 3 were separately hydrolyzed with 6N HCl (method A: Ace high-pressure tube, 1200W microwave for 50 s and immediately cooled to 0 °C; or method B: 110 °C for 18 h), evaporated to dryness, and resuspended in H2O (50 mM). Standards for the 3-amino-2-methylhexanoic acid (Amha) unit were kindly provided by Dr. David Horgen, College of Natural Sciences, Hawaii Pacific University. The other amino acid standards relevant to compound 3 were available commercially or from synthesis (Amp and Adhpa, see Supporting Information) and also prepared as 50 mM solutions in H2O. Marfey's derivatization was performed by adding 10 μL of 1M NaHCO3 and 50 μL of N-α-(5-fluoro-2,4-dinitrophenyl)-l-leucinamide (l-FDLA, 1% w/v in acetone) to 25 μL of each 50 mM solution. The mixture was heated at 40 °C for 1 h with stirring, cooled to room temperature, acidified with 5 μL 2N HCl and evaporated to dryness. The derivatized product was resuspended in 250 μL MeCN-H2O (1:1) for each standard or 100 μL for the hydrolysate of 3 and analyzed by RP-HPLC (Gemini C18 110A, 4.6 × 150 mm, 5 μm, 1.0 mL/min, UV detection at 340 nm) using a linear gradient of 30 to 70% MeCN in 0.1% (v/v) aqueous TFA over 50 min. The retention times (tR min) of the derivatized residues in the hydrolysate of 3 matched N-Me-l-Ala (16.2; N-Me-d-Ala, 16.8), (2S,3R)-Amha [28.6; (2S,3S)-Amha, 23.2; (2R,3R)-Amha; (2R,3S)-Amha, 22.5] and N-Me-l-Val (21.2; N-Me-d-Val, 25.5). The retention times of N-Me-l-Phe (23.2) and N-Me-d-Phe (24.5) standards overlapped with (2S,3S)-Amha (23.2) and N-Me-l-Leu (24.5) standards. Thus, the derivatized hydrolysate was subjected to LC-MS analysis (Gemini C18, 2.0 × 150 mm, 3 μm, 0.2 mL/min, UV and ESIMS detection, 340 nm and negative ion mode, respectively) using a linear gradient of 30 to 70% of 0.1% (v/v) HCO2H in MeCN and 0.1% (v/v) HCO2H in H2O over 50 min. The retention times (tR min, base peak m/z) of the derivatized residues in the hydrolysate matched N-Me-l-Phe (23.1, 472.1) and N-Me-l-Leu (24.9, 439.1). For the assignment of the Ibu unit, both the decarboxylated product, (2S)-amino-4-methylpentan-3-one (Amp), and the reduced Ibu unit, (3RS, 4S)-amino-2,2-dimethyl-3-hydroxy-pentanoic acid (Adhpa) were prepared (see Supporting Information). Portions were separately derivatized with l-FDLA and d-FDLA reagents and analyzed by RP-HPLC (Kinetex XB-C18 110A, 4.6 × 100 mm, 2.6 μm, 1.8 mL/min, UV detection at 340 nm) using a linear gradient of 30 to 70% MeCN in 0.1% (v/v) aqueous TFA over 15 min or LC-MS (Gemini C18, 2.0 × 150 mm, 3 μm, 0.2 mL/min, UV and ESIMS detection, 340 nm and negative ion mode, respectively) using a linear gradient of 30 to 70% of 0.1% (v/v) HCO2H in MeCN and 0.1% (v/v) HCO2H in H2O over 50 min. For the hydrolysate of 3 for method A, both S- and R-Amp were detected by RP-HPLC at tR = 9.8 and 10.0 min, respectively, in the ratio of 4.1:1. For the hydrolysate of 3 for method B, both S- and R-Amp were detected by RP-HPLC at tR = 9.8 and 10.0 min, respectively, in the ratio of 1:2.2. The retention times (tR min; S/R ratio) of the Amp derivatized standards were as follows: l-FDLA-S-Amp (9.8, 6.7:1) and d-FDLA-S-Amp (10.0, 1:5.9). For the hydrolysate of the reduction product of 3 (method A), the assignment of the reduced Ibu unit was determined by LC-MS analysis due to overlap in the HPLC trace. The retention time (tR min, 4S/4R ratio, base peak m/z) of the reduced natural product hydrolysate matched l-FDLA-(3RS, 4S)-Adhpa (19.7, 13.8:1, 454.1), while d-FDLA-(3RS, 4S)-Adhpa eluted at 20.6 min.
Synthesis of (2S)-2-amino-4-methylpentan-3-one (Amp) and (3RS, 4S)-4-amino-2,2-dimethyl-3-hydroxy-pentanoic acid (Adhpa)
Preparation and Chiral LC-MS of 2-Hydroxy-3-methylpentanoic acid (HMPA) for (3)
Diazotization of l-Ile (100 mg, 0.75 mmol) dissolved in 50 mL of 0.2 N HClO4 (0 °C) was carried out by the dropwise addition of a cold (0 °C) 20 mL solution of NaNO2 (1.4 g, 20 mmol) with rapid stirring. The solution was stirred at room temperature until the evolution of N2 subsided (~60 min). The solution was then brought to boiling for 3 min, cooled to room temperature, saturated with NaCl, and extracted with 20 mL EtOAc. The extract was dried with anhydrous Na2SO4 and concentrated under vacuum to afford (2S, 3S)-HMPA as an oil. The three other stereoisomers (2S, 3R)-HMPA, (2R, 3R)-HMPA and (2R, 2S)-HMPA were synthesized in a similar manner from l-allo-Ile, d-Ile and d-allo-Ile, respectively. A portion of each standard was analyzed using chiral LC-MS. The retention time of the natural product hydrolysate matched that for (2S,3S)-HMPA (7.5 min; Chirobiotic TAG, 4.6 × 250 mm; MeOH-10 mM NH4OAc 3:2 at pH 5.50; flow rate, 0.4 mL/min; detection by ESIMS in negative ion mode). The retention times of the remaining HMPA standards were as follows: (2S, 3R)-HMPA (6.5 min), (2R,3R)-HMPA (9.2 min), (2R,2S)-HMPA (8.1 min).
Cell Viability Assays
Mouse neuroblastoma neuro-2a or HeLa cells (ATCC, Manassas, VA) were cultured in RPMI-1640 media with 2 mM L-glutamine, pH 7.4 (Mediatech Inc., Manassas, VA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 1 mM sodium pyruvate (Mediatech), and 1% penicillin/streptomycin (Mediatech) at 37 °C in a humidified chamber containing 5% CO2. Cells were seeded into 96-well plates (neuro-2a: 20,000 cells per well; HeLa: 3,000 cells per well) in 90 μL of medium 4 h before treatment. Purified compounds were added to cells at final concentrations ranging from 10 nM to 10 μM (neuro-2a cells) or 2 μM (HeLa cells), each added in a 10 μL aliquot generated by serial dilution in serum-free medium on the day of the experiment, from stock solutions of 200 μM (1 and 2, both cell lines) or 2 mM (3 and dolastatin 12, neuro-2a cells only) compound in 100% DMSO (neuro-2a cells) or 100% EtOH (HeLa cells). Each 96-well plate also contained untreated and vehicle-treated control cells. Neuro-2a cells were also treated with 30 μg/mL of the parent 25% MeOH-EtOAC fraction as a positive control. Cell viability was determined after 48 h treatment using a standard 3-(4,5-dimethylthiazol-2-yl)-2,5,diphenyly tetrazolium bromide (MTT) assay. Briefly, MTT reagent (0.5 mg/mL in PBS; Sigma, St. Louis, MO) was added to each well and incubated for 2 h at 37°C. The medium was then aspirated from all wells and the purple formazan product solubilized with DMSO. The optical density of each well was determined at 550 nm using a BioTek Synergy HT microplate reader with Gen5 software (Bio-Tek, Winooski, VT). The cytotoxicity of each purified compound was assessed in at least three independent cultures with the viability of vehicle-treated control cells defined as 100% in all experiments.
Supplementary Material
Acknowledgment
We thank the Red Sea Protectorate for permission to make collections of Red Sea cyanobacteria, and Brian Arbogast and Jeff Morre of the Environmental Health Sciences Center at OSU for MS data acquisition (NIEHS P30 ES00210). The National Science Foundation (CHE-0722319) and the Murdock Charitable Trust (2005265) are acknowledged for their support of the OSU Natural Products and Small Molecule Nuclear Magnetic Resonance. Funding was provided by the OSU College of Pharmacy and an undergraduate scholarship from the OSU Research Office (to J.M.M.).
Footnotes
Supporting Information Available. Experimental details for the syntheses of (2S)-2-amino-4-methylpentan-3-one (Amp) and (3RS, 4S)-4-amino-2,2-dimethyl-3-hydroxypentanoic acid (Adhpa). Tables of 1D and 2D NMR data for compounds 1, 2 and 3. Selected NMR spectra in CDCl3 and CD3OD for compounds 1, 2 and 3. These data may be accessed online at http://pubs.acs.org.
References and Notes
- (1).Clardy J, Walsh C. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- (2).Fenton M, Geiselhart S, Rohling EJ, Hemleben C. Mar. Micropaleontol. 2000;40:277–294. [Google Scholar]
- (3).Tan LT. J. Appl. Phycol. 2010;22:659–676. [Google Scholar]
- (4).Harrigan GG, Luesch H, Yoshida WY, Moore RE, Nagle DG, Paul VJ, Mooberry SL, Corbett TH, Valeriote FA. J. Nat. Prod. 1998;61:1075–1077. doi: 10.1021/np980321c. [DOI] [PubMed] [Google Scholar]
- (5).Engene N, Coates RC, Gerwick WH. J. Phycol. 2010;46:591–601. [Google Scholar]
- (6).Hess WR. In: Cyanobacteria. Herrero A, Flores E, editors. Caister Academic Press; Norwich, UK: 2008. pp. 89–116. [Google Scholar]
- (7).Matthew S, Ross C, Rocca JR, Paul VJ, Luesch H. J. Nat. Prod. 2007;70:124–127. doi: 10.1021/np060471k. [DOI] [PubMed] [Google Scholar]
- (8).Taori K, Matthew S, Rocca JR, Paul VJ, Luesch H. J. Nat. Prod. 2007;70:1593–1600. doi: 10.1021/np0702436. [DOI] [PubMed] [Google Scholar]
- (9).Matthew S, Ross C, Paul VJ, Luesch H. Tetrahedron. 2008;64:4081–4089. [Google Scholar]
- (10).Matthew S, Paul VJ, Luesch H. Planta Med. 2009;75:528–533. doi: 10.1055/s-0029-1185332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Matthew S, Paul VJ, Luesch H. Phytochemistry. 2009;70:2058–2063. doi: 10.1016/j.phytochem.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kwan JC, Ratnayake R, Abboud KA, Paul VJ, Luesch H. J. Org. Chem. 2010;75:8012–8023. doi: 10.1021/jo1013564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pettit GR, Kamano Y, Kizu H, Dufresne C, Herald CL, Bontems RJ, Schmidt JM, Boettner FE, Nieman RA. Heterocycles. 1989;28:553–558. [Google Scholar]
- (14).Taori K, Paul VJ, Luesch H. J. Amer. Chem. Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- (15).Bai R, Bates RB, Hamel E, Moore RE, Nakkiew P, Pettit GR, Sufi BA. J. Nat. Prod. 2002;65:1824–1829. doi: 10.1021/np020117w. [DOI] [PubMed] [Google Scholar]
- (16).Williams PG, Moore RE, Paul VJ. J. Nat. Prod. 2003;66:1356–1363. doi: 10.1021/np0302145. [DOI] [PubMed] [Google Scholar]
- (17).Carter DC, Moore RE, Mynderse JS, Niemczura WP, Todd JS. J. Org. Chem. 1984;49:236–242. [Google Scholar]
- (18).Komárek J, Anagnostidis K. In: Süßwasserflora von Mitteleuropa 19/2. Büdel B, Krienitz L, Gärtner G, Schagerl M, editors. Elsevier/Spektrum; Heidelberg: 2005. p. 759. [Google Scholar]
- (19).Marquardt J, Palinska KA. Arch. Microbiol. 2007;187:397–413. doi: 10.1007/s00203-006-0204-7. [DOI] [PubMed] [Google Scholar]
- (20).Williamson RT, Boulanger A, Vulpanovici A, Roberts MA, Gerwick WH. J. Org. Chem. 2002;67:7927–7936. doi: 10.1021/jo020240s. [DOI] [PubMed] [Google Scholar]
- (21).Scherlach K, Hertweck C. Org. Biomol. Chem. 2009;7:1753–1760. doi: 10.1039/b821578b. [DOI] [PubMed] [Google Scholar]
- (22).Castenholz RW, Lester Packer ANG. Methods in Enzymology. Volume 167. Academic Press; 1988. pp. 68–93. [Google Scholar]
- (23).Han AW, Oh KH, Jheong WH, Cho YC. J. Microbiol. Biotechnol. 2010;20:1152–1155. doi: 10.4014/jmb.1003.03028. [DOI] [PubMed] [Google Scholar]
- (24).Nubel U, Garcia-Pichel F, Muyzer G. Appl. Environ. Microbiol. 1997;63:3327–3332. doi: 10.1128/aem.63.8.3327-3332.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Martínez-Murcia AJ, Acinas SG, Rodriguez-Valera F. FEMS Microbiol. Ecol. 1995;17:247–255. [Google Scholar]
- (26).Huang X, Madan A. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. Appl. Environ. Microbiol. 2005;71:7724–7736. doi: 10.1128/AEM.71.12.7724-7736.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. Appl. Environ. Microbiol. 2006;72:5734–5741. doi: 10.1128/AEM.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tamura K, Dudley J, Nei M, Kumar S. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- (30).Thompson JD, Thierry JC, Poch O. Bioinformatics. 2003;19:1155–1161. doi: 10.1093/bioinformatics/btg133. [DOI] [PubMed] [Google Scholar]
- (31).Posada D. Mol. Biol. Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- (32).Huelsenbeck JP, Ronquist F. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- (33).Guindon S, Gascuel O. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- (34).Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A. Geneious. v5.3. http://www.geneious.com. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.