

Abstract
Background
Pheochromocytomas are tumors arising from chromaffin tissue located in the adrenal medulla associated with typical symptoms and signs which may occasionally develop metastases, which are defined as the presence of tumor cells at sites where these cells are not found. This retrospective analysis was focused on clinical, genetic, and histopathologic characteristics of primary metastatic versus primary benign pheochromocytomas.
Materials and methods
We identified 41 subjects with metastatic pheochromocytoma and 108 subjects with apparently benign pheochromocytoma. We assessed dimension and biochemical profile of the primary tumor, age at presentation, and time to develop metastases.
Results
Subjects with metastatic pheochromocytoma presented at a significantly younger age (41.4±14.7 vs. 50.2±13.7 years; P<0.001), with larger primary tumors (8.38±3.27 cm vs. 6.18±2.75 cm; P<0.001) and secreted more frequently norepinephrine (95.1% vs. 83.3 %; P=0.046) compared to subjects with apparently benign pheochromocytomas. No significant differences were found in the incidence of genetic mutations in both groups of subjects (25.7 % in the metastatic group and 14.7 % in the benign group; P=0.13). From available histopathologic markers of potential malignancy, only necrosis occurred more frequently in subjects with metastatic pheochromocytoma (27.6 % vs. 0 %; P<0.001). The median time to develop metastases was 3.6 years with the longest interval 24 years.
Conclusions
In conclusion, regardless of a genetic background, the size of a primary pheochromocytoma and age of its first presentation are two independent risk factors associated with the development of metastatic disease.
Keywords: malignant pheochromocytoma, norepinephrine, epinephrine
Introduction
Pheochromocytomas and paragangliomas are tumors arising from chromaffin tissue located either in the adrenal medulla (pheochromocytomas) or at extra-adrenal sites along the sympathetic and/or the parasympathetic chain (paragangliomas) [1, 2]. These tumors typically occur in a benign form but may present in a malignant form and reach up to 36 % or even higher in subjects with extra-adrenal tumors, in particular when associated with the mutation of the gene encoding the B subunit of the mitochondrial complex II enzyme succinate dehydrogenase enzyme (SDHB) [3–8]. Metastatic pheochromocytoma or paraganglioma is defined as the presence of tumor cells at the sites where these cells are not found such as lymph nodes, bones, liver, or lungs [9].
To predict the likelihood of metastatic spread, several criteria and scoring systems have been developed on the basis of histopathology or clinical data, but no system has proven reliable [10]. For example, a multiparameter Pheochromocytoma of the Adrenal Gland Scaled Score (PASS), which includes several histopathologic features of the primary tumor such as mitotic activity and capsular or vascular invasion [11], has been shown unreliable due to its high interobserver and intraobserver variation [12]. From several clinical studies, the highest predictive value might include the extra-adrenal tumor location, presence of SDHB mutations, and tumor size [4, 13, 14]. Most of the studies focused on predictors of malignancy in pheochromocytomas or paragangliomas have not addressed adrenal or extra-adrenal tumors separately on a large scale due to their rarity [15, 16].
In the study of O’Riordain et al., subjects with paragangliomas that had tumors larger than 5 cm showed a worse prognosis compared to those that had smaller tumors [5]. In the study of Amar et al., the smallest tumor diameter of subjects with malignant pheochromocytoma and paraganglioma was 5 cm [3], and Kimura et al. showed that malignant adrenal pheochromocytoma was significantly larger than tumors of subjects with benign adrenal pheochromocytomas [17]. In contrast, Thompson found no significant difference in the tumor diameter between benign and metastatic pheochromocytoma [11].
In the present study, we retrospectively assessed clinical, genetic, and histopathologic characteristics of primary metastatic from primary benign adrenal pheochromocytomas. In particular, we focused on common histopathologic characteristics including the tumor dimension, the presence of necrosis, capsular, vascular, and lymphatic invasion and genetic background as possible risk factors of their metastatic behavior. Patients presenting with metastatic paragangliomas (extra-adrenal tumors) were not included.
Subjects and Methods
Patients
We identified 41 subjects with metastatic pheochromocytoma referred to either the National Institutes of Health (NIH), Bethesda, USA or to the 3rd Department of Medicine, General University Hospital, Prague, Czech Republic. Four additional subjects were not included in the analysis because the size of their primary tumors was not available. Metastasis was defined as the presence of tumor cells at sites where no chromaffin tissue is normally found and the metastatic lesion(s) were proven either by biopsy or the presence of persistently elevated catecholamine/metanephrine levels together with a positivity on pheochromocytoma specific imaging modalities and previous history of pheochromocytoma [9]. As the control group, we used 108 subjects with apparently benign pheochromocytoma with the average follow up of 5.8 ± 4.3 years. Study protocols were approved by the Institutional Review Board of the National Institute of Child Health and Human Development at NIH and by the Ethics Committee of the First Faculty of Medicine. All patients provided written informed consent. Reporting of the study conforms to STROBE along with references to STROBE and the broader EQUATOR guidelines [18].
Biochemical phenotype of tumors was characterized according to elevations of particular catecholamines (epinephrine, norepinephrine, and dopamine) or their metabolites (metanephrine and normetanephrine) either in urine or plasma. All elevations above the upper range of normal values were counted as positive and further detailed work-up was initiated. In most subjects, elevations of plasma or urinary catecholamines or metanephrines were many times above upper reference limits. Genetic testing for mutations in pheochromocytoma susceptible genes [RET (rearranged during transfection), VHL (von Hippel–Lindau gene), SDHB, and SDHD (succinate dehydrogenase subunit D gene) was performed at NIH, at Mayo Medical Laboratories, Rochester, MN or at Division of Molecular Diagnostics at the University of Pittsburgh Medical Center Department of Genetics of the Children’s Hospital of Philadelphia, PA as described elsewhere [6] or at Institute of Biology and Medical Genetics of the First Faculty of Medicine, Charles University in Prague. In total, 35 (85%) subjects with metastatic pheochromocytoma and 75 (69%) subjects with benign pheochromocytoma completed genetic testing. Diagnosis of neurofibromatosis type I was made based on a clinical presentation. Certain histopathologic parameters of potential tumor malignancy (vascular, lymphatic, and capsular invasion, increased mitotic figures, and necrosis) were obtained from available pathological reports [(29 (71%) subjects with metastatic pheochromocytoma and 108 (100%) subjects with benign pheochromocytoma].
Data are shown as mean ± standard deviation (SD) or median and interquartile ranges in case of non-parametric data distribution. Continuous variables were compared using the unpaired t-test and categorical variables by the Fisher exact test. Tumor dimension was assessed using the longest tumor diameter. Kaplan-Meier method was used to estimate the time from diagnosis of PHEO to diagnosis of malignancy and to estimate survival of subjects with metastatic PHEO, where survival was defined as the time from diagnosis of PHEO to the date of death or date of last follow-up. We used log-rank test to compare survival between subgroups of patients. P<0.05 values were considered as significant. Data were analyzed using the statistical software package Statistica version 9.1CZ (StatSoft, Tulsa, OK, USA).
Results
In total, we analyzed 41 subjects with metastatic adrenal pheochromocytoma and 108 subjects with benign adrenal pheochromocytoma. Primary tumors of subjects with malignant pheochromocytoma were diagnosed at a significantly younger age (41.4±14.7 vs. 50.2±13.7 years; P<0.001) compared to subjects with benign pheochromocytomas (P<0.001) (Table 1). No significant difference was found in gender distribution between subjects with malignant and benign adrenal pheochromocytomas, although male subjects tended to present more frequent metastases (Table 1). Among available histopathologic parameters, only necrosis occurred significantly more frequently in the metastatic pheochromocytoma group (P<0.001) (Table 2).
Table 1.
Baseline characteristics of subjects with metastatic and benign pheochromocytoma
| Parameter | Metastatic pheochromocytoma (n=41) |
Benign pheochromocytoma (n=108) |
P |
|---|---|---|---|
| Gender (male/female) | 19/22 | 44/62 | NS |
| Age (years) (min; max) | 41.4±14.7 (6;83) | 50.2±13.7 (21;78) | <0.001 |
| Age <40 years (%) | 19 (46.3%) | 26 (24.1%) | 0.008 |
| Tumor dimension (cm) (min; max) | 8.38±3.27 (2.4;17) | 6.18±2.75 (2;16) | <0.001 |
| Number of tumors ≥5cm | 37 (90%) | 72 (66.7%) | 0.002 |
| Right adrenal | 28 (68.3%) | 59 (54.6%) | 0.11 |
| Left adrenal | 12 (29.3%) | 44 (40.7%) | NS |
| Bilateral adrenal involvement | 4 (9.8%) | 5 (4.6%) | NS |
| Syndromic presentation | 9 (25.7%) | 11 (14.7%) | 0.13 |
| SDHB | 2 | 0 | |
| MEN 2 | 4 | 2 | |
| VHL | 3 | 5 | |
| NF1 | 0 | 4 |
SDHB, succinate dehydrogenase subunit B; MEN 2, multiple endocrine neoplasia type 2; VHL, von Hippel-Lindau; NF1, neurofibromatosis type 1; NS, non-significant.
Table 2.
Histopathologic markers of potential malignancy in subjects with benign and metastatic pheochromocytoma
| Parameter | Metastatic pheochromocytoma (n=29) |
Benign pheochromocytoma (n=108) |
P |
|---|---|---|---|
| Vascular invasion | 5 (17.2%) | 11 (10.2%) | NS |
| Capsular invasion | 4 (13.8) | 16 (16.7%) | NS |
| Increased mitotic figures | 2 (6.9%) | 8 (7.4) | NS |
| Necrosis | 8 (27.6%) | 0 (0%) | <0.001 |
| Invasion to adjacent adipose tissue | 2 (6.9%) | 5 (4.6%) | NS |
NS, non-significant.
Genetic testing for germ-line mutations or clinical assessment revealed 9 (25.7 %) carriers of germ-line mutations in patients with malignant pheochromocytoma (2× SDHB, 4× RET, and 3× VHL), compared to 11 (14.7 %) carriers in subjects with benign pheochromocytoma (2× RET, 5× VHL, and 4× neurofibromatosis type I) (Table 1). No mutation of the SDHD gene was found.
Primary pheochromocytomas in subjects with metastatic spread were significantly larger than in subjects with benign pheochromocytomas (P<0.001). The distribution of tumor dimensions was variable ranging from 2.4 cm to 17 cm (median 8 cm) in the malignant group and 2 cm to 16 cm (median 5.8 cm) in the benign group (Table 1). However, only four (10 %) subjects with metastatic pheochromocytoma presented initially with primary tumors smaller than 5 cm compared to 33.3 % subjects with benign pheochromocytoma (P<0.001) (Table 1). Malignant adrenal pheochromocytomas were diagnosed more frequently at the right side compared to the benign tumors, but this difference did not reach statistical significance. Incidence of bilateral tumors was similar in both groups of subjects (Table 1).
The analysis of catecholamine levels showed that subjects with malignant pheochromocytoma secreted significantly more often norepinephrine, thereby having a so called “noradrenergic phenotype” (P=0.04) (Table 3). On the contrary, significantly increased epinephrine levels, or so called “adrenergic phenotype”, were more common in subjects with benign pheochromocytomas, although not reaching statistical significance (P=0.06) (Table 3). Isolated norepinephrine secretion was the most common type of catecholamine secretion in subjects with malignant pheochromocytoma. In the benign group, co-secretion of norepinephrine and epinephrine was the most frequent type of catecholamine secretion (Table 2). Only one patient with malignant pheochromocytoma (4 cm primary pheochromocytoma diameter) secreted only epinephrine, whereas isolated epinephrine secretion was found in 11 (10.1 %) subjects with benign pheochromocytoma (Table 3). No significant difference was found in the frequency of dopamine secretion or in the number of subjects without catecholamine or metanephrine secretion, which are so called “biochemically silent tumors” (Table 3).
Table 3.
Biochemical patterns of subjects with metastatic and benign pheochromocytoma
| Parameter | Metastatic pheochromocytoma (n=41) |
Benign pheochromocytoma (n=108) |
P |
|---|---|---|---|
| Norepinephrine | 15 (36.6%) | 22 (20.4%) | 0.04 |
| Norepinephrine + epinephrine | 13 (31.7%) | 51 (47.7%) | 0.06 |
| Norepinephrine + epinephrine + dopamine | 8 (19.5%) | 12 (11.2%) | NS |
| Norepinephrine + dopamine | 3 (7.3%) | 5 (4.7%) | NS |
| Epinephrine | 1 (2.4%) | 11 (10.1%) | 0.11 |
| No catecholamine secretion | 1 (2.4%) | 6 (5.6%) | NS |
| Positive norepinephrine | 39 (95.1%) | 90 (83.3%) | 0.046 |
| Positive epinephrine | 22 (53.6%) | 74 (69.2%) | 0.06 |
| Positive dopamine or without catecholamine secretion | 12 (29.3%) | 23 (21.3%) | NS |
Norepinephrine, epinephrine, and dopamine denote elevated either catecholamines, or its metabolites either in urine or in plasma; NS, non-significant.
The median time from the diagnosis of the primary tumor to the diagnosis of metastatic spread was 3.6 years (in subjects younger 40 years, the median time was 7 years and in subjects older 40 years 3.1 years; P=0.24) (Table 4; Figure 1). In eleven subjects (27 %), malignant disease was diagnosed either at the time of diagnosis of a primary tumor or during the first year after diagnosis of pheochromocytoma. In almost half of the subjects (18 patients), metastatic involvement was discovered later than 5 years after the diagnosis of primary tumor (the longest interval between the diagnosis of primary tumor and metastases was 24 years) (Table 4). Sixteen subjects older than 40 years (76 %) developed metastases during the 5 years after diagnosis of pheochromocytoma compared to only 7 subjects younger than 40 years (32 %) (P=0.02). No other differences were found between subjects younger and older than 40 years regarding tumor size and type catecholamine secretion. Four subjects with malignant pheochromocytoma had a range of 17 to 27 years between the diagnosis of primary tumor and metastases, but they were not included in the analysis because of the inability to obtain information regarding the dimension of the primary tumor. The most common site of metastatic location were lymphatic nodes (66 %), liver (49 %), bones (49 %), and less frequently lungs (34 %) (Table 4).
Table 4.
Time to development of distant metastases and metastatic locations in subjects with metastatic pheochromocytoma
| Parameter | Metastatic pheochromocytoma (n=41) |
|---|---|
| Time to diagnosis of malignancy (years) | 3.6 (0.42; 9) (min 0, max 24) |
| Diagnosis of malignancy at presentation or until 1 year of diagnosis of pheochromocytoma | 11 (26.8%) |
| Diagnosis of malignancy 1 to 5 years after diagnosis of pheochromocytoma | 12 (29.3%) |
| Diagnosis of malignancy more than 5 years after diagnosis of pheochromocytoma | 18 (43.9%) |
| Metastatic locations | |
| Lymphatic nodes | 27 (65.9%) |
| Liver | 20 (48.8%) |
| Bone | 20 (48.8%) |
| Lung | 14 (34.1%) |
| Mediastinum | 4 (9.8%) |
Figure 1.
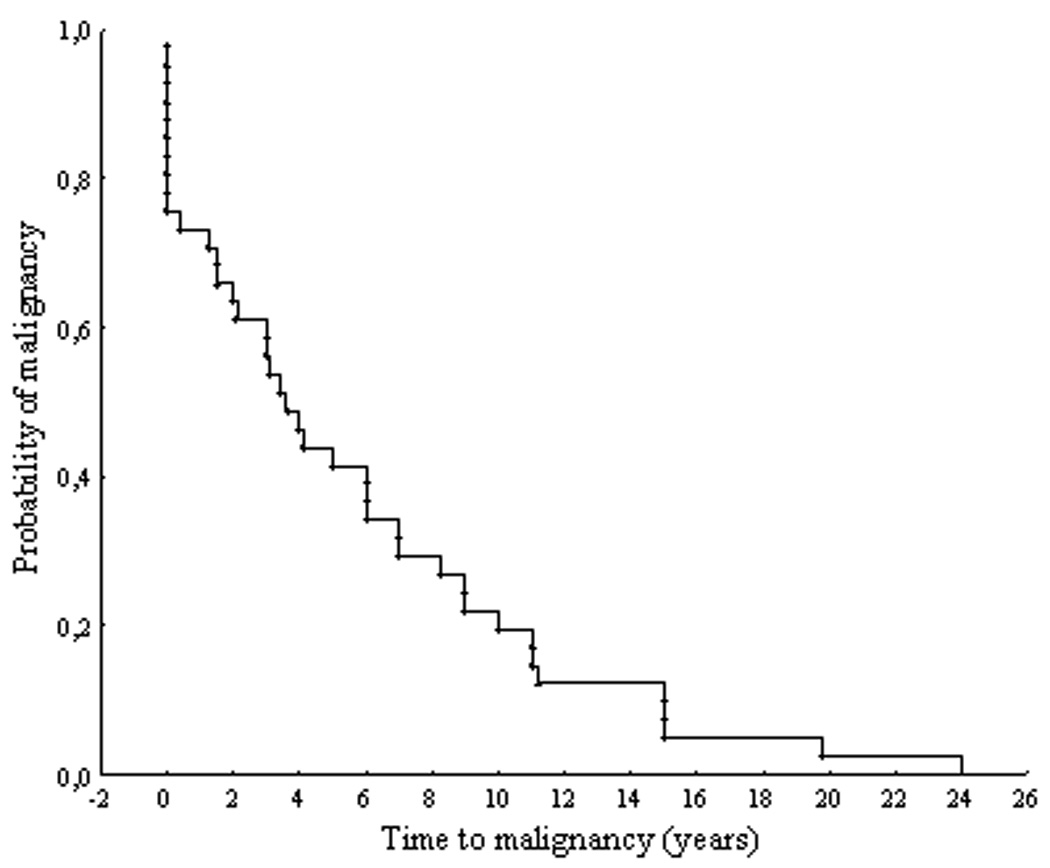
Distribution of time intervals from the diagnosis of primary tumor to diagnosis of metastases.
Survival analysis subjects with metastatic PHEO (median time for the censored observations was 9.0 years, range from 0.6 to 27.3 years) revealed significantly longer survival of subjects secreting epinephrine (p=0.02; Figure 2) and shorter survival of subjects who produced dopamine or presented with biochemically silent tumors (p=0.04; Figure 2). No survival differences were found according metastatic involvement (liver, lung, and bones).
Figure 2.
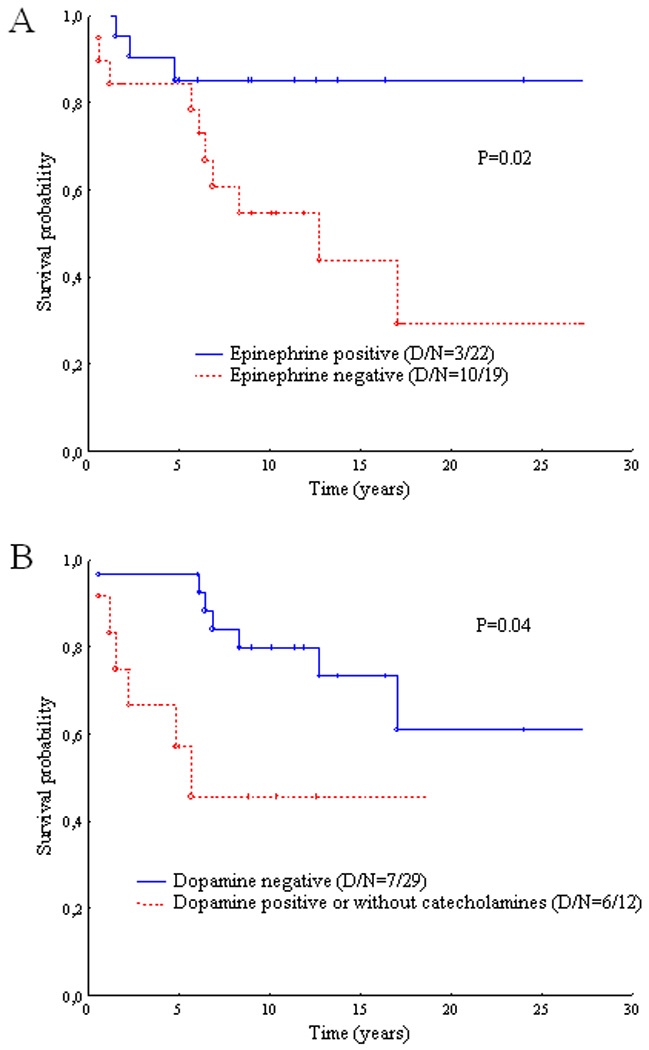
Survival of subjects with metastatic pheochromocytoma in respect to catecholamine secretion. Epinephrine or dopamine denote either maternal catecholamines or its metabolites in urine or in plasma; D, number of deaths; N, total number of subjects.
Discussion
In the present retrospective analysis that specifically focused on subjects with pheochromocytoma as the adrenal tumor, we have found that subjects with metastatic disease presented with larger primary tumors at significantly younger age, and produced more frequently norepinephrine compared to subjects with apparently benign tumors. There was no statistical difference in the gender distribution and in frequency of mutations of genes associated with the presence of pheochromocytoma.
Our findings that metastatic pheochromocytomas present with larger primary tumors than benign ones suggest that the size of a primary tumor is an important predictor of malignancy. Thus, as found in the present study, 90 % of subjects with metastatic pheochromocytomas presented with primary tumors larger than 5 cm. These findings that a tumor dimension plays an important role as a risk factor in the development of metastatic spread is further supported by previous reports [16, 17]. In the study of Kimura et al., the dimension and weight of primary adrenal pheochromocytomas were significantly higher in subjects malignant tumors compared to subjects with apparently benign disease [17]. Similar results were found in the study of Shen et al. where dimension of primary tumors was significantly larger in malignant pheochromocytomas compared to benign tumors [16]. In the study of Amar et al., the smallest diameter of the primary tumor (extra-adrenal or adrenal origin) which developed metastases was 5 cm [3]. In contrast, Linnoila et al. showed that weight of primary tumors of malignant pheochromocytomas or paragangliomas was significantly higher than benign tumors, but no correlation was found between tumor weight and development of metastases [19]. However, the number of tumors available for the analysis in this study was small and no attention was paid to discriminate between primary adrenal and extra-adrenal tumors. Similarly, Thompson found no significant difference in the primary tumor dimension between malignant and benign adrenal pheochromocytoma [11].
In contrast, only 10% of subjects with metastatic pheochromocytomas presented with tumors smaller than 5 cm. These results do not support findings of the study of Gupta et al., where 47% of subjects with metastatic pheochromocytoma presented with primary tumors <5 cm [20]. The smallest tumor diameter in our study was 2.4 cm which is in agreement with other authors who also found primary tumors <3 cm which presented in the later course with distant metastases [11, 21].
Until now, many authors have attempted to find predictors of malignancy in pheochromocytoma but no scoring system has been proven reliable [10]. The strongest predictors are extra-adrenal locations and the presence of mutation of the SDHB gene. Although the mostly used PASS score (includes vascular, capsular or adipose tissue invasion, large nests or diffuse growth, focal or confluent necrosis, high cellularity, tumor cell spindling, cellular monotony, increased mitotic figures, atypical mitotic figures, profound nuclear pleomorphism, and hyperchromasia) developed by Thompson [11] might be of some value [21, 22], other authors reported about its limitations due to high inter- and intraobserver variation [12]. According to our results, only necrosis occurred more frequently in malignant tumors but no difference was found in vascular and capsular invasion or invasion into adipose tissue and in the number of mitotic figures. Thus, present results of histopathologic findings support previous findings of Linnoila et al. and Kimura and al. who concluded that necrosis but not capsular or vascular invasion is indeed an important risk factor for the development of metastatic pheochromocytoma [17, 19].
Age
From the clinical point of view, age at time of a primary tumor associated with metastatic disease later on and time interval from the diagnosis of the primary tumor to the diagnosis of metastases are also important facts. First, pheochromocytomas associated with the development of metastatic disease were diagnosed at significantly younger age compared to subjects with benign pheochromocytoma, which is in the agreement with Kimura et al.[17]. Higher age in subjects with benign disease might be explained by the fact that the number of subjects with pheochromocytoma diagnosed incidentally during abdominal ultrasound, CT or MRI performed from other reasons is still increasing [23, 24]. Second, the number of subjects who developed metastases at least 5 years after the diagnosis of primary tumor was also higher in younger subjects compared to older subjects with malignant pheochromocytoma. One explanation could be that younger subjects may not be so complaint as older ones to undergo a regular follow-up which is mandatory in all subjects with diagnosed pheochromocytoma [25]. The age dependent function of immune system and behavior of tumor cells may be additional important factors that could contribute to the explanation of present findings.
Biochemistry
Analysis of biochemical phenotypes revealed that norepinephrine secretion in subjects with metastatic pheochromocytoma was more frequent than those with benign tumors. On the contrary, there was a tendency to have more frequent epinephrine secretion in subjects with benign tumors. Epinephrine production is the latest step in the catecholamine synthesis catalyzed by phenylethanolamine-N-methyltransferase. Thus, the failure of neuronal apoptosis during embryological development [26] in dopaminergic or noradrenergic chromaffin progenitors would be expected to occur earlier than in more fully mature adrenergic chromaffin cells and this may also explain younger age in subjects with malignant pheochromocytoma. Secretion of epinephrine or its metabolite metanephrine which may be regarded as a marker of cell differentiation was associated with better survival of subjects with metastatic PHEO whereas dopamine or its metabolite metanephrine was an indicator of a worse outcome of subjects with metastatic pheochromocytoma.
Both groups of subjects did not differ in the presence of tumors secretion either only dopamine or being biochemically silent. This could be explained by the relatively low number of subjects who have been tested positive for mutations in the SDHB-gene which has been found to be strongly associated not only with malignant disease but also with either dopamine secreting tumors or no catecholamine production at all [3, 27, 28].
Finally, it should be noted that a genetic background was not found to play any important role in the pathogenesis of malignant pheochromocytoma. However, only two patients with mutation of the SDHB-gene, which is strongly associated with a malignant behavior of extra-adrenal pheochromocytoma, were included in the present study. Therefore, larger samples of SDHB-related pheochromocytomas are needed to confirm these preliminary observations. This would most likely require multi-institutional studies since these tumors are quite rare.
Our study has several limitations. First, genetic testing could not be performed in all subjects and genetic testing for other pheochromocytoma susceptibility genes (isocitrate dehydrogenase 1 and -2, SDHAF2, TMEMI127, and SDHC [29–31]) was not carried out. Second, the interval for follow up in 49% of patients was less than 5 years and these patients will need to be followed for a longer time period.
In conclusion, subjects with metastatic adrenal pheochromocytoma presented with larger primary tumors that secreted more frequently norepinephrine and were significantly younger at the time of diagnosis of primary tumor than subjects with benign adrenal pheochromocytoma. According to our data, a minimal tumor diameter of 5 cm might be considered an important risk factor for the metastatic spread in subjects with pheochromocytoma. Almost half of the subjects with metastatic pheochromocytoma developed metastases at least 5 years after the diagnosis of the primary tumor which underlines the importance of very close lifelong follow-up of subjects with pheochromocytoma/paraganglioma. We propose that the optimal interval for subjects at higher risk of malignancy (younger subjects with tumors larger than 5 cm and secreting norepinephrine) would be for biochemical testing every 6 months and for morphological testing (CT or MRI scanning) once a year. On the other hand, testing on yearly basis would be sufficient for subjects with lower risk of malignancy (older subjects with tumors smaller than 5 cm and secreting epinephrine).
Acknowledgement
This research was supported by the Intramural Research Program of the NICHD/NIH and by Research projects of Czech Ministry of Education 0021620807 and 0021620808.
Footnotes
DISCLOSURE STATEMENT: The authors have nothing to disclose.
References
- 1.Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366:665–675. doi: 10.1016/S0140-6736(05)67139-5. [DOI] [PubMed] [Google Scholar]
- 2.Young WF., Jr Adrenal causes of hypertension: pheochromocytoma and primary aldosteronism. Rev Endocr Metab Disord. 2007;8:309–320. doi: 10.1007/s11154-007-9055-z. [DOI] [PubMed] [Google Scholar]
- 3.Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92:3822–3828. doi: 10.1210/jc.2007-0709. [DOI] [PubMed] [Google Scholar]
- 4.John H, Ziegler WH, Hauri D, Jaeger P. Pheochromocytomas: can malignant potential be predicted? Urology. 1999;53:679–683. doi: 10.1016/s0090-4295(98)00612-8. [DOI] [PubMed] [Google Scholar]
- 5.O'Riordain DS, Young WF, Jr, Grant CS, Carney JA, van Heerden JA. Clinical spectrum and outcome of functional extraadrenal paraganglioma. World J Surg. 1996;20:916–921. doi: 10.1007/s002689900139. [DOI] [PubMed] [Google Scholar]
- 6.Brouwers FM, Eisenhofer G, Tao JJ, Kant JA, Adams KT, Linehan WM, et al. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006;91:4505–4509. doi: 10.1210/jc.2006-0423. [DOI] [PubMed] [Google Scholar]
- 7.Burnichon N, Rohmer V, Amar L, Herman P, Leboulleux S, Darrouzet V, et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab. 2009;94:2817–2827. doi: 10.1210/jc.2008-2504. [DOI] [PubMed] [Google Scholar]
- 8.Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91:827–836. doi: 10.1210/jc.2005-1862. [DOI] [PubMed] [Google Scholar]
- 9.Tischler AS. Pheochromocytoma and extra-adrenal paraganglioma: updates. Arch Pathol Lab Med. 2008;132:1272–1284. doi: 10.5858/2008-132-1272-PAEPU. [DOI] [PubMed] [Google Scholar]
- 10.Tischler AS. Pheochromocytoma: time to stamp out "malignancy"? Endocr Pathol. 2008;19:207–208. doi: 10.1007/s12022-008-9047-x. [DOI] [PubMed] [Google Scholar]
- 11.Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551–566. doi: 10.1097/00000478-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Wu D, Tischler AS, Lloyd RV, DeLellis RA, de Krijger R, van Nederveen F, et al. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol. 2009;33:599–608. doi: 10.1097/PAS.0b013e318190d12e. [DOI] [PubMed] [Google Scholar]
- 13.Ahlman H. Malignant pheochromocytoma: state of the field with future projections. Ann N Y Acad Sci. 2006;1073:449–464. doi: 10.1196/annals.1353.049. [DOI] [PubMed] [Google Scholar]
- 14.Zelinka T, Timmers HJ, Kozupa A, Chen CC, Carrasquillo JA, Reynolds JC, et al. Role of positron emission tomography and bone scintigraphy in the evaluation of bone involvement in metastatic pheochromocytoma and paraganglioma: Specific implications for SDHB gene mutations. Endocr Relat Cancer. 2008;15:311–323. doi: 10.1677/ERC-07-0217. [DOI] [PubMed] [Google Scholar]
- 15.Glodny B, Winde G, Herwig R, Meier A, Kuhle C, Cromme S, et al. Clinical differences between benign and malignant pheochromocytomas. Endocr J. 2001;48:151–159. doi: 10.1507/endocrj.48.151. [DOI] [PubMed] [Google Scholar]
- 16.Shen WT, Sturgeon C, Clark OH, Duh QY, Kebebew E. Should pheochromocytoma size influence surgical approach? A comparison of 90 malignant and 60 benign pheochromocytomas. Surgery. 2004;136:1129–1137. doi: 10.1016/j.surg.2004.05.058. [DOI] [PubMed] [Google Scholar]
- 17.Kimura N, Watanabe T, Noshiro T, Shizawa S, Miura Y. Histological grading of adrenal and extra-adrenal pheochromocytomas and relationship to prognosis: a clinicopathological analysis of 116 adrenal pheochromocytomas and 30 extra-adrenal sympathetic paragangliomas including 38 malignant tumors. Endocr Pathol. 2005;16:23–32. doi: 10.1385/ep:16:1:023. [DOI] [PubMed] [Google Scholar]
- 18.Simera I, Moher D, Hoey J, Schulz KF, Altman DG. A catalogue of reporting guidelines for health research. Eur J Clin Invest. 2010;40:35–53. doi: 10.1111/j.1365-2362.2009.02234.x. [DOI] [PubMed] [Google Scholar]
- 19.Linnoila RI, Keiser HR, Steinberg SM, Lack EE. Histopathology of benign versus malignant sympathoadrenal paragangliomas: clinicopathologic study of 120 cases including unusual histologic features. Hum Pathol. 1990;21:1168–1180. doi: 10.1016/0046-8177(90)90155-x. [DOI] [PubMed] [Google Scholar]
- 20.Grogan RH, Mitmaker E, Vriens MR, Harari A, Gosnell JE, Shen WT, et al. Adrenal incidentaloma: Does an adequate workup rule out surprises? Surgery. 2010;148:392–397. doi: 10.1016/j.surg.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Strong VE, Kennedy T, Al-Ahmadie H, Tang L, Coleman J, Fong Y, et al. Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery. 2008;143:759–768. doi: 10.1016/j.surg.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 22.Gao B, Meng F, Bian W, Chen J, Zhao H, Ma G, et al. Development and validation of pheochromocytoma of the adrenal gland scaled score for predicting malignant pheochromocytomas. Urology. 2006;68:282–286. doi: 10.1016/j.urology.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 23.Amar L, Servais A, Gimenez-Roqueplo AP, Zinzindohoue F, Chatellier G, Plouin PF. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab. 2005;90:2110–2116. doi: 10.1210/jc.2004-1398. [DOI] [PubMed] [Google Scholar]
- 24.Noshiro T, Shimizu K, Watanabe T, Akama H, Shibukawa S, Miura W, et al. Changes in clinical features and long-term prognosis in patients with pheochromocytoma. Am J Hypertens. 2000;13:35–43. doi: 10.1016/s0895-7061(99)00139-9. [DOI] [PubMed] [Google Scholar]
- 25.Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. 1997;29:1133–1139. doi: 10.1161/01.hyp.29.5.1133. [DOI] [PubMed] [Google Scholar]
- 26.Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8:155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 27.Timmers HJ, Kozupa A, Eisenhofer G, Raygada M, Adams KT, Solis D, et al. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2007;92:779–786. doi: 10.1210/jc.2006-2315. [DOI] [PubMed] [Google Scholar]
- 28.Chen H, Sippel RS, O'Dorisio MS, Vinik AI, Lloyd RV, Pacak K. The north american neuroendocrine tumor society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775–783. doi: 10.1097/MPA.0b013e3181ebb4f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao L, Barontini M, Niederle B, Jech M, Pfragner R, Dahia PLM. Mutations of the metabolic genes IDH1, IDH2, and SDHAF2 are not major determinants of the pseudohypoxic phenotype of sporadic pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2010;95:1469–1472. doi: 10.1210/jc.2009-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mannelli M, Ercolino T, Giache V, Simi L, Cirami C, Parenti G. Genetic screening for pheochromocytoma: should SDHC gene analysis be included? J Med Genet. 2007;44:586–587. doi: 10.1136/jmg.2007.051045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229–233. doi: 10.1038/ng.533. [DOI] [PMC free article] [PubMed] [Google Scholar]