

Abstract

We have developed a ligand that reversibly binds to aniline substrates allowing for the control of regioselectivity and enantioselectivity in hydroformylation. In this paper we address how the electronics of the aniline ring affect both binding of the substrate to the ligand and the enantioselectivity in this reaction.
Asymmetric hydroformylation (AHF) is an efficient and practical means of producing chiral aldehyde products. The challenges in AHF are controlling both the enantioselectivity and the regioselectivity of the reaction.1 Hydroformylation of terminal olefins generally has the preference to form the achiral linear isomers, whereas 1,2-disubstituted olefins show poor regiocontrol. Therefore the majority of AHF has been performed on symmetrical substrates or olefins with an electronic preference to form the branched isomers (such as styrene). These classes of substrates have been found to be receptive to AHF, and a variety of ligands have been developed that promote the reaction with high enantioselectivity.2–14 Most recently, Zhang15 and Landis16 have also demonstrated success in the AHF of substrates that contain an internal directing group. Our research group has tried to diversify the substrate scope of AHF by expanding to substrates that are not electronically activated and do not have a directing group as part of the molecule. This is achieved by having a phosphorous-based ligand that reversibly and covalently binds to common organic functionalities.17–29 Upon binding the substrate, the phosphine ligand serves as the directing group controlling the regioselectivity. Importantly, the reversibility of the bonding between the substrate and ligand allows the ligand to be employed catalytically.30–34 We recently applied this concept to the AHF of p-methoxyphenyl (PMP) protected amines towards the synthesis of β-amino alcohols.35 Although the PMP group is a useful protecting group, aniline derivatives are an important class of molecules found broadly in biologically active compounds. In this paper we explore how the electronics of the nitrogen affects both binding of the substrate to the ligand as well as the enantioselectivity of the hydroformylation reaction.
We initiated our investigation by studying the exchange reaction of a variety of electronically modified anilines with ligand 1.36 The electronics of the aniline affect the affinity of the substrate for the ligand, but do not prevent binding in any of the tested substrates. Electron poor anilines have a lower binding affinity than electron rich rings, with all of the Keq values being within one order of magnitude of each other (table 1). To gain further insight into the factors that affect binding, the equilibrium data was plotted versus both σp and σ+ hammett parameters. The data correlate slightly better to the σ+ values than the σp, consistent with there being a minor resonance component to the binding affinity (Figure 1). The ρ value is small and negative (−0.47), suggesting only a modest favourability for binding of electron rich rings. One interpretation of these data is that the nitrogen lone pair is conjugated to the aromatic ring in the secondary anilines, but upon binding to the ligand the steric repulsion between the aromatic ring and ligand rotates the lone pair out of conjugation. Since resonance stabilizing electron-withdrawing groups are in direct conjugation with the nitrogen lone pair, the equilibrium shifts towards the starting materials as compared to donating groups. Alternatively, when the aniline is bound to the ligand, the aniline nitrogen lone pair can participate in donation into the σ* of either the C-N or C-P bonds of the heterocyclic ring.37–38 In this case donating groups would enhance this interaction while withdrawing groups would mitigate it.
Table 1.
Electronic effects on binding to (±)−1
| entry | R | Keqa |
|---|---|---|
| 1 | OMe (2a) | 2.9 ± 0.7 |
| 2 | Me (2b) | 1.3 ± 0.5 |
| 3 | H (2c) | 1.2 ± 0.3 |
| 4 | Cl (2d) | 1.2 ± 0.2 |
| 5 | CN (2e) | 0.64 ± 0.03 |
| 6 | NO2 (2f) | 0.43 ± 0.07 |
Keq values were determined by 1H NMR in triplicate.
Figure 1.

Hammett plots

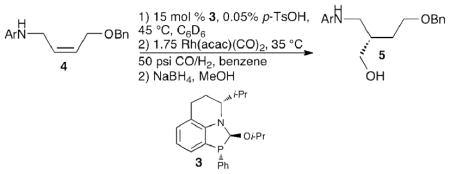
Having investigated the effects of electronics on binding affinity to the ligand, we probed the consequences on hydroformylation. For the hydroformylation studies the benzyl ethers were used as substrates, because these compounds are readily accessible in large quantities as geometrically pure compounds from commercially available cis,1,4-butenediol (note: hydroformylation of (Z)-N-(but-2-en-1-yl)aniline (2c) afforded the desired product in 74% 1H NMR yield and 90% ee). As previously reported using ligand 3, p-methoxy phenyl (PMP) substrates yield the product in high enantioselectivity (92% ee) and good yield (70%, table 2, entry 1).39 Unsubstituted and electronically neutral aromatics form the product in comparable levels of enantioselectivity and slightly elevated isolated yields (table 2, entries 2–4). When an electron-withdrawing substituent is in the para position of the aromatic ring both the yield and enantioselectivities decrease (table 2, entries 5 and 6). Based on gas uptake curves, these reactions stall before reaching 100% conversion, consistent with the active catalyst decomposing during the reaction. The exchange data shows that the ligand bound to electron deficient aniline derivatives are thermodynamically less stable, which may be leading to a more rapid decomposition of the ligand. Analysis of the pre-exchange reaction prior to hydroformylation by 31P NMR indicates considerable decomposition of ligand 3 to a variety of unidentifiable phosphorous compounds in the presence of compounds 4e and 4f. The Hammett data suggests that electron-withdrawing groups not directly in conjugation with the aniline lone pair would be thermodynamically more stable. Using a substrate with a m-NO2 yields the alcohol product in improved enantioselectivity and yield (table 2, entry 8), consistent with this prediction. Similarly, a substrate with an electron-deficient pyridine ring affords the desired product in moderate yield (64%) and good enantioselectivity (87% ee). We also attempted to perform hydroformylations with substitution, such as Cl and CH3, at the ortho position of the aromatic ring. These provided low conversion to product; we believe this is a result of difficulties binding sterically large substrates to the ligand.
Table 2.
Electronic effects on hydroformylation of aniline substrates
| entry | Ar | product | yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1d |
 (4a) |
5a | 70 | 92 |
| 2 |
 (4b) |
5b | 79 | 92 |
| 3 |
 (4c) |
5c | 77 | 91 |
| 4 |
 (4d) |
5d | 72 | 88 |
| 5 |
 (4e) |
5e | 49 | 65 |
| 6 |
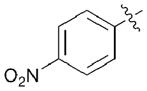 (4f) |
5f | 41 | 52 |
| 7 |
 (4g) |
5g | 60 | 89 |
| 8 |
 (4h) |
5h | 61 | 84 |
| 9 |
 (4i) |
5i | 64 | 87 |
Unless otherwise noted, the following reaction conditions were used: 1) 15 mol % 3, 0.05 mol % p-TsOH, 45 °C, C6D6; 2) 1.75 mol % Rh(acac)(CO)2, 35 °C, 50 psi H2/CO, benzene; 3) NaBH4, MeOH.
Isolated yield.
Enantiomeric excess was determined by HPLC analysis.
Standard conditions, except 0.03 mol % p-TsOH was used for the pre-exchange.
Conclusions
The asymmetric hydroformylation of aniline derivatives has been achieved using a chiral scaffolding ligand. By studying the substrate/ligand exchange and hydroformylation reactions, we found a correlation between binding affinity and the yield and enantioselectivity of the hydroformylation reaction. Substrates with high affinity for the ligand generally afford improved enantioselectivity. Substrates with a low affinity for ligand yield lower enantioselecitvity and yield, which is potentially a result of decomposition of the ligand during the reaction. With this information, we are currently developing ligands with improved stability with the aim of improving yields and selectivities.
Experimental
All general considerations are the same as previously reported.35 The following compounds were made according to literature procedures and matched reported spectra: Ligand 1,26 ligand 3(OMe),35 and ligand 3(OiPr),35 N-(but-2-yn-1-yl)-4-methoxyaniline,35 (Z)-N-(but-2-en-1-yl)-4-methoxyaniline (2a),35 2-(but-2-yn-1-yl)isoindoline-1,3-dione,40 but-2-yn-1-amine,41 (Z)-4-(benzyloxy)but-2-en-1-ol, (Z)-N-(4-(benzyloxy)but-2-en-1-yl)-4-methoxyaniline (4a),35 (Z)-(7((4-chlorobut-2-en-1-yl)oxy)methyl)benzene,35 2-isobutyrylcyclohexanone,42 1-iodo-3-nitrobenzene,43 3-iodopyridine,44 (S)-4-(benzyloxy)-2-(((4-methoxyphenyl)amino)methyl)butan-1-ol (5a).35
Equilibrium Substrates
General Procedure 1
To a flame-dried flask was added p-toluidine (3.36 g, 31.4 mmol), CH3CN (30 mL), and 1-bromo-2-butyne (0.55 mL, 6.3 mmol). The reaction was stirred overnight, diluted with Et2O (100 mL), washed with water (2 × 40 mL), and washed with saturated NH4Cl (3 × 25 mL). The organic were dried over MgSO4, filtered, and concentrated.
General Procedure 2
A flame-dried flask was charged with Lindlar’s catalyst (172 mg) and purged with nitrogen. N-(but-2-yn-1-yl)-4-methylaniline (1.23 g, 7.74 mmol) in EtOH (15 mL) was added, followed by quinoline (82 μL, 0.70 mmol). The flask was purged with H2 (4x), fitted with a H2 balloon, and stirred at room temperature for 40 minutes. The reaction was filtered through a plug of silica and concentrated. Column chromatography (10% EtOAc/Hex) afforded the title compound.
N-(but-2-yn-1-yl)-4-methylaniline
General Procedure 1. Column chromatography (5% EtOAc/Hex) yielded an orange oil (781 mg, 78%). 1H NMR (CDCl3, 500 MHz) δ 7.01 – 7.04 (m, 2H), 6.60 – 6.62 (m, 2H), 3.85 (s, 2H), 3.71 (br s, 1H), 2.26 (app d, 3H, J = 4.2), 1.80 – 1.81 (m, 3H); 13C NMR (CDCl3, 125 MHz) δ 145.2, 129.9, 127.7, 113.8, 79.1, 76.5, 34.5, 20.6, 3.5; IR: 1616, 1517, 1249, 806, 502 cm−1; HRMS (DART-TOF) calcd. for C11H14N1 [M+H]+: 160.1126, found: 160.1119.
(Z)-N-(but-2-en-1-yl)-4-methylaniline (2b)
General Procedure 2, yielding an orange oil (1.02 g, 82%). 1H NMR (CDCl3, 500 MHz) δ 6.99 (d, 2H, J = 7.8), 6.55 – 6.58 (m, 2H), 5.63 – 5.67 (m, 1H), 5.55 – 5.59 (m, 1H), 3.74 (d, 2H, J = 6.6), 3.50 (br s, 1H), 2.23 (s, 3H), 1.71 – 1.73 (m, 3H); 13C NMR (CDCl3, 125 MHz) δ 146.3, 129.9, 128.1, 127.2, 127.0, 113.3, 41.4, 20.6, 13.3; IR: 1616, 1517, 1313, 1256, 805 cm−1; HRMS (DART-TOF) calcd. for C11H16N1 [M+H]+: 162.1283, found: 162.1277.
N-(but-2-yn-1-yl)aniline
General Procedure 1 using aniline (7.73 mL, 84.8 mmol). Column chromatography (5% EtOAc/Hex) yielded a yellow oil (1.9 g, 78%). 1H NMR (CDCl3, 500 MHz) δ 7.20 – 7.24 (m, 2H), 6.77 – 6.80 (m, 1H), 6.67 – 6.70 (m, 2H), 3.88 (s, 2H), 3.85 (br s, 1H), 1.81 – 1.82 (m, 3H); 13C NMR (CDCl3, 125 MHz) δ 147.5, 129.4, 118.4, 113.6, 79.2, 76.3, 34.2, 3.7; IR: 1601, 1502, 1313, 747, 690 cm−1; HRMS (DART-TOF) calcd. for C10H12N1 [M+H]+: 146.0970, found: 146.0967.
(Z)-N-(but-2-en-1-yl)aniline (2c, 7% (E)-isomer)
General Procedure 2 using N-(but-2-yn-1-yl)aniline (1.01 g, 7.02 mmol), affording an orange oil (720 mg, 73%). 1H NMR (CDCl3, 500 MHz) δ 7.17 – 7.21 (m, 2H), 6.71 – 6.74 (t, 1H, J = 7.3), 6.62 – 6.64 (m, 2H), 5.65 – 5.68 (m, 1H), 5.54 – 5.59 (m, 1H), 3.78 (d, 2H, J = 6.6), 3.69 (d, 2H(E)-isomer, J = 6.1), 3.63 (br s, 1H), 1.71 – 1.72 (m, 3H); 13C NMR (CDCl3, 125 MHz) δ 148.5, 129.4, 127.8, 127.4, 117.6, 113.1, 41.0, 13.3; IR: 1601, 1503, 1312, 1260, 747, 690, 507 cm−1; HRMS (DART-TOF) calcd. for C10H14N1 [M+H]+: 148.1126, found: 148.1119.
N-(but-2-yn-1-yl)-4-chloroaniline
General Procedure 1 using 4-chloroaniline (1.88 g, 14.7 mmol). Column chromatography (20% EtOAc/Hex) yielded a light red oil (331 mg, 66%). 1H NMR (CDCl3, 500 MHz) δ 7.15 – 7.17 (m, 2H), 6.59 – 6.61 (m, 2H), 3.86 (s, 2H), 1.81 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 146.0, 129.2, 123.1, 114.7, 79.5, 75.9, 34.3, 3.7; IR: 1599, 1496, 1312, 814, 500 cm−1; HRMS (DART-TOF) calcd. for C10H11Cl1N1 [M+H]+: 180.0580, found: 180.0581.
(Z)-N-(but-2-en-1-yl)-4-chloroaniline (2d)
General Procedure 2 using N-(but-2-yn-1-yl)-4-chloroaniline (1.02 g, 5.68 mmol), affording an orange oil (814 mg, 81%). 1H NMR (CDCl3, 500 MHz) δ 7.11 – 7.13 (m, 2H), 6.52 – 6.55 (app dd, 2H, J = 6.6, 2.2), 5.65 – 5.68 (m, 1H), 5.51 – 5.54 (m, 1H), 3.73 (d, 2H, J = 6.6), 3.64 (br s, 1H), 1.70 – 1.72 (m, 3H); 13C NMR (CDCl3, 125 MHz) δ 147.0, 129.2, 127.7, 127.4, 122.1, 114.1, 41.1, 13.3; IR: 1599, 1496, 1311, 1176, 812, 503 cm−1; HRMS (DART-TOF) calcd. for C10H13Cl1N1 [M+H]+: 182.0737, found: 182.0739.
4-(but-2-yn-1-ylamino)benzonitrile
A flame-dried flask was charged with K2CO3 (6.00 g, 45.4 mmol), 4-aminobenzonitrile (7.00 g, 56.7 mmol), DMF (140 mL), and 1-bromo-2-butyne (2.51 g, 18.9 mmol). The reaction was stirred overnight at 80 °C. The reaction was diluted with EtOAc (200 mL) and washed with water (2 × 100 mL). The aqueous layer was washed with EtOAc (200 mL) and the organics were dried over MgSO4, filtered, and concentrated. Column chromatography (20 % EtOAc/Hex) afforded an orange oil (1.32 g, 16%). 1H NMR (CDCl3, 500 MHz) δ 7.42 – 7.44 (m, 2H), 6.60 – 6.62 (m, 2H), 4.47 (br s, 1H), 3.87 – 3.90 (m, 2H), 1.78 – 1.79 (app t, 3H, J = 2.4); 13C NMR (CDCl3, 125 MHz) δ 150.6, 133.7, 120.4, 112.9, 99.7, 80.0, 74.8, 33.4, 3.6; IR: 3371, 2212, 1604, 1522, 1324, 1174, 824, 543 cm−1; HRMS (DART-TOF) calcd. for C11H11N2 [M+H]+: 171.0922, found: 171.0923.
(Z)-4-(but-2-en-1-ylamino)benzonitrile (2e, 5% (E)-isomer)
General Procedure 2 using 4-(but-2-yn-1-ylamino)benzonitrile (800 mg, 4.70 mmol), afforded an orange oil (622 mg, 77%). 1H NMR (CDCl3, 500 MHz) δ 7.38 – 7.42 (m, 2H), 6.54 – 6.56 (m, 2H)), 5.67 – 5.74 (m, 1H), 5.47 – 5.53 (m, 1H), 4.27 (br s, 1H) 3.78 – 3.81 (t, 2H, J = 5.6), 3.70 – 3.72 (m, 2H(E)-isomer), 1.71 – 1.73 (m, 3H), 1.69 – 1.70 (m, 2H(E)-isomer); 13C NMR (CDCl3, 125 MHz) δ 151.4, 133.8, 128.5, 126.3, 120.7, 112.4, 98.8, 40.2, 13.3; IR: 3367, 2209, 1602, 1521, 1171, 820, 542 cm−1; HRMS (DART-TOF) calcd. for C11H13N2 [M+H]+: 173.1079, found: 173.1080.
N-(but-2-yn-1-yl)-4-nitroaniline
To a flame-dried flask was added KF (426 mg, 6.17 mmol), K2CO3 (853 mg, 6.17 mmol), and 1-fluoro-4-nitrobenzene (871 mg, 6.17 mmol). But-2-yn-1-amine (426 mg, 6.17 mmol) was added to the flask as a solution in DMSO (20 mL). The reaction was stirred at room temperature overnight. Water (5 mL) was added and the mixture was extracted with Et2O (150 mL). The organic layer was dried over MgSO4, filtered, and concentrated. Column chromatography (15% EtOAc/Hex) afforded a yellow oil (660 mg, 56%). 1H NMR (CDCl3, 500 MHz) δ 8.11 (d, 2H, J = 9.3), 6.59 – 6.62 (m, 2H), 4.69 (br s, 1H), 3.93 – 3.95 (m, 2H), 1.79 – 1.80 (m, 3H); 13C NMR (CDCl3, 125 MHz) δ 152.5, 138.9, 126.4, 111.9, 80.5, 74.4, 33.7, 3.7; IR: 1616, 1515, 1231, 805, 512 cm−1; HRMS (DART-TOF) calcd. for C10H11N2O2 [M+H]+: 191.0821, found: 191.0819.
(Z)-N-(but-2-en-1-yl)-4-nitroaniline (2f, 14% (E)-isomer)
General Procedure 2 using N-(but-2-yn-1-yl)-4-nitroaniline (66 mg, 0.35 mmol) afforded a yellow oil (60 mg, 90%). 1H NMR (CDCl3, 500 MHz) δ 8.07 – 8.11 (m, 2H), 6.52 – 6.55 (m, 2H), 5.71 – 5.77 (m, 1H), 5.46 – 5.49 (m, 1H), 3.85 – 3.87 (m, 2H), 3.77 – 3.79 (m, 2H(E)-isomer), 1.73 – 1.75 (m, 3H), 1.71 – 1.72 (m, 3H(E)-isomer); 13C NMR (CDCl3, 125 MHz) δ 153.4, 138.3, 129.0, 126.6, 125.9, 111.4, 40.5, 13.4; IR: 3378, 1600, 1503, 1471, 1318, 1302, 1283, 1111 cm−1; HRMS (DART-TOF) calcd. for C10H13N2O2 [M+H]+: 193.0977, found: 193.0985.
Equilibrium Experiments
In a dry box, a solution of isopropanol (100 μL, 1.31 mmol) in benzene-d6 (1.63 M) was prepared. The solution was dispensed into three NMR tubes. A second solution of (Z)-N-(but-2-enyl)-4-methoxyaniline (70 mg, 0.43 mmol), ligand (25 mg, 0.086 mmol), and p-TsOH (298 μL, 7.2 × 10−4M in C6H6; note C6H6 was removed prior to mixing with substrate and ligand) in C6D6 (1.5 mL) was made. The solution was dispensed into three NMR tubes. The total volume of each tube was brought to 0.7 mL. Each reaction was allowed to equilibrate overnight at 45 °C.
Hydroformylation Substrates
General Procedure 3:45
To a flame-dried flask was added L-proline (325 mg, 2.82 mmol), CuI (537 mg, 2.82 mmol), and K2CO3 (1.17 g, 8.46 mmol). (Z)-4-(benzyloxy)but-2-en-1-amine (500 mg, 2.82 mmol) was added to the flask as a solution in DMSO (5 mL), followed by iodobenzene (314 μL, 2.82 mmol). The reaction was heated to 60 °C for 8 hours. The mixture was diluted with EtOAc(100 mL) and washed with water (70 mL). The organic layer was dried over anhydrous MgSO4, filtered, and concentrated.
General Procedure 4
To a flame-dried flask was added K2CO3 (420 mg, 3.04 mmol), 4-fluorobenzonitrile (368 mg, 3.04 mmol), (Z)-4-(benzyloxy)but-2-en-1-amine (700 mg, 3.94 mmol), and DMSO (10 mL), which was heated to 90 °C overnight. The reaction was quenched by the addition of water (25 mL) and was diluted with Et2O (70 mL). The organic layer was dried over anhydrous MgSO4, filtered and concentrated.
General Procedure 5:42
To an oven-dried flask was added CuI (232 mg, 1.22 mmol) and Cs2CO3 (2.00 g, 6.10 mmol). (Z)-4-(benzyloxy)but-2-en-1-amine (600 mg, 3.05 mmol), 1-iodo-3-methylbenzene (392 μL, 3.05 mmol), DMF (1.5 mL), and 2-isobutyrylcyclohexanone (410 mg, 2.44 mmol) were added. The reaction was stirred for 12 hours. The reaction was filtered through a pad of Celite, washed with EtOAc (100 mL), and extracted with water (2 × 25 mL). The organic layer was dried over MgSO4, filtered and concentrated.
(Z)-N-(4-(benzyloxy)but-2-en-1-yl)-4-methylaniline (4b)
To a flame-dried flask was added p-toluidine (3.26 g, 30.5 mmol), (Z)-(((4-chlorobut-2-en-1-yl)oxy)methyl)benzene (1.20 g, 6.10 mmol), and acetonitrile (30 mL). The reaction was stirred for 12 hours. The reaction was diluted with Et2O (100 mL) and was washed with H2O (3 × 50 mL) and saturated aqueous NH4Cl (3 × 50 mL). The organics were dried over anhydrous MgSO4, filtered, and concentrated. Column chromatography (15% EtOAc/Hex) afforded an orange oil (748 mg, 46%). 1H NMR (CDCl3, 500 MHz) δ 7.28 – 7.36 (m, 5H), 7.00 (d, 2H, J = 8.1), 6.54 (d, 2H, J = 8.6), 5.74 – 5.82 (m, 2H), 4.54 (s, 2H), 4.15 (d, 2H, J = 4.9), 3.75 (d, 2H, J = 4.5), 3.55 (br s, 1H), 2.25 (s, 3H) 13C NMR (CDCl3, 125 MHz) δ 145.9, 138.3, 131.0, 129.9, 129.0, 128.6, 128.0, 127.9, 127.2, 113.4, 72.7, 65.9, 41.9, 20.6; IR: 1616, 1518, 1252, 1070, 806, 734, 696 cm−1; HRMS (DART-TOF) calcd. for C18H22N1O1 [M+H]+: 268.1701, found: 268.1698.
(Z)-2-(4-(benzyloxy)but-2-en-1-yl)isoindoline-1,3-dione
A flame-dried flask was charged with (Z)-4-(benzyloxy)but-2-en-1-ol (3.74 g, 21.0 mmol), triphenylphosphine (3.74 g, 21.0 mmol), phthalimide (3.09 g, 21.0 mmol), tetrahydrofuran (105 mL), and diisopropyl azodicarboxylate (4.13 mL, 21.0 mmol). The reaction was stirred at room temperature overnight. The mixture was concentrated. Column chromatography (20% EtOAc/Hex) gave a colorless oil (4.23 g, 65%). 1H NMR (CDCl3, 400 MHz) δ 7.81 – 7.85 (m, 2H), 7.69 – 7.72 (m, 2H), 7.28 – 7.39 (m, 5H), 5.78 – 5.84 (m, 1H), 5.62 – 5.68 (m, 1H), 4.58 (s, 2H), 4.32 – 4.34 (m, 2H), 4.30 – 4.32 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 168.1, 138.4, 134.2, 132.4, 131.0, 128.6, 128.1, 127.9, 126.4, 123.5, 72.7, 65.9, 35.2; IR: 1709, 1390, 1088, 1072, 735, 715, 698 cm−1; HRMS (DART-TOF) calcd. for C19H18N1O3 [M+H]+: 308.1287, found: 308.1284.
(Z)-4-(benzyloxy)but-2-en-1-amine
A 100-mL flask was charged with (Z)-2-(4-(benzyloxy)but-2-en-1-yl)isoindoline-1,3-dione (4.23 g, 13.7 mmol), hydrazine hydrate (1.6 mL, 25.7 mmol), and ethanol (10 mL). The mixture was heated to 70 °C overnight. The solid was filtered and washed with water (100 mL). The aqueous solution was acidified to pH ≈ 2 with concentrated HCl and was washed with Et2O (120 mL). The aqueous layer was then basified to pH ≈ 13 by solid KOH pellets. The solution was washed with Et2O (200 mL). The organic layer was dried over anhydrous MgSO4, filtered, and concentrated to obtain the title compound as a pale yellow oil (1.92 g, 79%). 1H NMR (CDCl3, 500 MHz) δ 7.27 – 7.36 (m, 5H), 5.61 – 5.71 (m, 2H), 4.51 (s, 2H), 4.07 (d, 2H, J = 6.0), 3.31 (d, 2H, J = 6.1), 1.13 (br s, 2H); 13C NMR (CDCl3, 125 MHz) δ 138.3, 134.9, 128.6, 128.0, 127.8, 126.7, 72.5, 65.7, 39.3; IR: 3067, 2854, 1453, 1088, 734, 696 cm−1; HRMS (DART-TOF) calcd. for C11H16N1O1 [M+H]+: 178.1232, found: 178.1240.
(Z)-N-(4-(benzyloxy)but-2-en-1-yl)aniline (4c)
General Procedure 3. Column chromatography (20% EtOAc/Hex) yielded a pale orange oil (301 mg, 42%). 1H NMR (CDCl3, 500 MHz) δ 7.28 – 7.37 (m, 5H), 7.16 – 7.21 (m, 2H), 6.75 (app t, 1H, J = 7.3), 6.61 (d, 2H, J = 7.6), 5.74 – 5.83 (m, 2H), 4.55 (s, 2H), 4.15 (d, 2H, J = 5.4), 3.78 (d, 2H, J = 5.6), 3.68 (br s, 1H); 13C NMR (CDCl3, 125 MHz) δ 148.2, 138.3, 130.8, 129.5, 129.1, 128.6, 128.0, 127.9, 117.9, 113.2, 72.7, 65.9, 41.6; IR: 3027, 2856, 1603, 1504, 1094, 1071, 749, 695 cm−1; HRMS (DART-TOF) calcd. for C17H20N1O1 [M+H]+: 254.1545, found: 254.1555.
(Z)-N-(4-(benzyloxy)but-2-en-1-yl)-4-chloroaniline (4d)
General Procedure 3 using 1-bromo-4-chlorobenzene (540 mg, 2.82 mmol). Column chromatography (20% EtOAc/Hex) yielded an orange oil (358 mg, 44%). 1H NMR (CDCl3, 500 MHz) δ 7.28 – 7.37 (m, 5H), 7.10 – 7.13 (m, 2H), 6.49 – 6.51 (m, 2H), 5.79 – 5.84 (m, 1H), 5.70 – 5.75 (m, 1H), 4.54 (s, 2H), 4.14 (d, 2H, J = 6.4), 3.74 (d, 2H, J = 6.6), 3.71 (br s, 1H); 13C NMR (CDCl3, 125 MHz) δ 146.7, 138.2, 130.4, 129.4, 129.3, 128.7, 128.0, 127.9, 122.5, 114.2, 72.8, 65.8, 41.7; IR: 2854, 1598, 1496, 1357, 1070, 814, 735, 696, 504 cm−1; HRMS (DART-TOF) calcd. for C17H19Cl1N1O1 [M+H]+: 288.1155, found: 288.1145.
(Z)-4-((4-(benzyloxy)but-2-en-1-yl)amino)benzonitrile (4e)
General Procedure 4. Column chromatography (20% EtOAc/Hex) afforded an orange oil (201 mg, 37%). 1H NMR (CDCl3, 500 MHz) δ 7.39 – 7.42 (m, 2H), 7.29 – 7.37 (m, 5H), 6.51 – 6.53 (m, 2H), 5.82 – 5.87 (m, 1H), 5.67 – 5.71 (m, 1H), 4.55 (s, 2H), 4.26 (br s, 1H), 4.12 – 4.13 (m, 2H), 3.81 (br m, 2H); 13C NMR (CDCl3, 125 MHz) δ 151.2, 138.0, 133.9, 130.0, 129.4, 128.7, 128.1, 128.0, 120.6, 112.5, 99.2, 72.9, 65.8, 40.8; IR: 3367, 1606, 1525, 1207 cm−1; HRMS (DART-TOF) calcd. for C18H19N2O1 [M+H]+: 279.1497, found: 279.1498.
(Z)-N-(4-(benzyloxy)but-2-en-1-yl)-4-nitroaniline (4f)
General Procedure 4 using 1-fluoro-4-nitrobenzene (429 mg, 3.04 mmol). Column chromatography (20% EtOAc/Hex) yielded a bright yellow oil (457 mg, 50%). 1H NMR (CDCl3, 500 MHz) δ 8.07 (d, 2H, J = 9.3), 7.31 – 7.37 (m, 5H), 6.49 (d, 2H, J = 9.3), 5.86 – 5.90 (m, 1H), 5.69 – 5.74 (m, 1H), 4.63 (br s, 1H), 4.56 (s, 2H), 4.15 (d, 2H, J = 6.4), 3.86– 3.89 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 153.2, 138.4, 138.0, 130.3, 128.9, 128.7, 128.1, 128.0, 126.5, 111.4, 72.9, 65.8, 40.9; IR: 3372, 1595, 1299, 1278, 1107, 830, 694 cm−1; HRMS (DART-TOF) calcd. for C17H19N2O3 [M+H]+: 299.1396, found: 299.1407.
(Z)-N-(4-(benzyloxy)but-2-en-1-yl)-3-methylaniline (4g)
General Procedure 5. Column chromatography (10% EtOAc/Hex) afforded an orange oil (653 mg, 80%). 1H NMR (CDCl3, 500 MHz) δ 7.30 – 7.37 (m, 5H), 7.08 (t, 1H, J = 7.3), 6.56 (d, 1H, J = 7.1), 6.43 – 6.42 (m, 2H), 5.76 – 5.81 (m, 2H), 4.55 (s, 2H), 4.16 (d, 2H, J = 5.4), 3.76 (d, 2H, J = 5.6), 3.63 (br s, 1H), 2.29 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 148.2, 139.2, 138.3, 130.9, 129.3, 129.0, 128.6, 128.0, 127.9, 118.8, 114.0, 110.4, 72.7, 65.9, 41.6, 21.8; IR: 1604, 1491, 1089, 1070, 769, 735, 692 cm−1; HRMS (DART-TOF) calcd. for C18H22N1O1 [M+H]+: 268.1701, found: 268.1702.
(Z)-N-(4-(benzyloxy)but-2-en-1-yl)-3-nitroaniline (4h)
General Procedure 3 using 1-iodo-3-nitrobenzene (886 mg, 3.56 mmol). Column chromatography (15% EtOAc/Hex) afforded a bright orange oil (561 mg, 53%). 1H NMR (DMSO, 500 MHz) δ 7.31 – 7.36 (m, 5H), 7.27 – 7.29 (m, 2H), 6.95 – 6.97 (m, 1H), 6.52 – 6.55 (m, 1H), 5.69 – 5.74 (m, 1H), 5.58 – 5.62 (m, 1H), 4.50 (s, 2H), 4.16 – 4.17 (m, 2H), 3.76 – 3.78 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 149.6, 148.9, 138.1, 130.1, 129.9, 129.5, 128.7, 128.1, 128.0, 119.1, 112.4, 106.6, 72.9, 65.9, 41.3; IR: 2857, 1621, 1581, 1344, 1089, 1070, 734, 689, 672 cm−1; HRMS (DART-TOF) calcd. for C17H19N2O3 [M+H]+: 299.1396, found: 299.1400.
(Z)-N-(4-(benzyloxy)but-2-en-1-yl)pyridin-3-amine (4i)
General Procedure 5 using 3-iodopyridine (833 mg, 4.06 mmol). Column chromatography (50% EtOAc/Hex) afforded an orange oil (366 mg, 37%). 1H NMR (CDCl3, 500 MHz) δ 8.00 – 8.01 (d, 1H, J = 3.0), 7.97 – 7.99 (dd, 1H, J = 4.6, 1.0), 7.29 – 7.35 (m, 5H), 7.06 – 7.08 (m, 1H), 6.83 – 6.86 (m, 1H), 5.81 – 5.86 (m, 1H), 5.72 – 5.76 (m, 1H), 4.54 (s, 2H), 4.13 (d, 2H, J = 6.1), 3.78 (d, 2H, J = 5.1), 3.78 (br s, 1H); 13C NMR (CDCl3, 125 MHz) δ 144.1, 139.3, 128.1, 136.5, 130.0, 129.7, 128.7, 128.1, 128.0, 123.8, 118.8, 72.8, 65.8, 41.1; IR: 2861, 1519, 1092, 808, 736, 697 cm−1; HRMS (DART-TOF) calcd. for C16H19N2O1 [M+H]+: 255.1497, found: 255.1484.
Hydroformylation Products
General Hydroformylation Procedure
(Z)-N-(4-(benzyloxy)but-2-en-1-yl)-4-methylaniline (53.4 mg, 0.200 mmol), (iPrO)-2 (205 μL, 0.146 M solution in C6D6), 0.05% p-toluenesulfonic acid in benzene (175 μL, 1 × 10−4 mmol), and C6D6 (0.4 mL) were mixed and heated to 45 °C in a sealed NMR tube. The solution was concentrated in a dry glove box, and then was redissolved in C6D6. The solution was heated to 45 °C for 4 h before being concentrated again in a glove box. The resulting residue was dissolved in benzene (1.5 mL), mixed with 1.75% Rh(acac)(CO)2 (0.9 mg, 0.0035 mmol), and injected into the Endeavor, followed by 0.5 mL benzene to wash the injection port. The Endeavor was purged with nitrogen (4 × 100 psi). The temperature was held at 35 °C for 10 minutes and the Endeavor was pressurized to 50 psi with H2/CO. The hydroformylation was carried out at 35 °C and 50 psi H2/CO for 14 hours with stirring at 700 rpm. The Endeavor was depressurized and cooled to ambient temperature. A solution of 1,3,5-trimethoxybenzene in CHCl3 (100 μL, 0.1863 M) was added, and the sample was concentrated. The resulting residue was added, as a solution in MeOH (3 mL), to a flame-dried flask containing NaBH4 (23.0 mg, 0.600 mmol). The reaction was stirred at room temperature for 1.5 hours. The reaction was quenched with water (5 mL) and extracted with CH2Cl2 (3 × 15 mL). The organics were dried over Na2SO4, filtered, and concentrated. The crude reaction was chromatographed (1% MeOH/DCM) to yield the title compound.
(S)-4-(benzyloxy)-2-((p-tolylamino)methyl)butan-1-ol (5b)
Yellow oil (47.4 mg, 79%). HPLC (OD-H, 1.0 mL/min, 23% iPrOH: 77% Hexanes, 240 nm) trmajor = 11.9 min and trminor = 20.6 min, 92% ee; 1H NMR (CDCl3, 500 MHz) δ 7.29 – 7.37 (m, 5H), 6.98 (d, 2H, J = 8.6), 6.54 (d, 2H, J = 8.3), 4.54 (s, 2H), 3.69 (d, 2H, J = 5.1), 3.55 – 3.64 (m, 2H), 3.11 – 3.18 (m, 2H), 2.24 (s, 3H), 2.00 – 2.02 (m, 1H), 1.72 – 1.76 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 146.1, 138.1, 129.9, 128.7, 128.0, 127.9, 127.1, 113.6, 73.5, 68.7, 65.4, 47.5, 38.8, 30.3, 20.6; IR: 3380, 2923, 2854, 1521, 1260, 1095, 807 cm−1; HRMS (DART-TOF) calcd. for C19H26N1O2 [M+H]+: 300.1964, found: 300.1977. [α]D20 = +21.8 (c = 0.330, CHCl3, l = 50 mm).
(S)-4-(benzyloxy)-2-((phenylamino)methyl)butan-1-ol (5c)
Yellow oil (43.9 mg, 77%). HPLC (OD-H, 1.0 mL/min, 15% iPrOH, 85% Hexanes, 240 nm) trmajor = 15.5 min and trminor = 20.4 min, 91% ee; 1H NMR (CDCl3, 500 MHz) δ 7.29 – 7.38 (m, 5H), 7.15 – 7.18 (m, 2H), 6.69 – 6.72 (app t, 1H, J = 7.3), 6.60 (d, 2H, J = 7.8), 4.54 (s, 2H), 3.70 (d, 2H, J = 5.1), 3.55 – 3.65 (m, 2H), 3.13 – 3.21 (m, 2H), 1.99 – 2.02 (m, 1H), 1.74 – 1.78 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 148.6, 138.0, 129.4, 128.7, 128.0, 117.6, 113.2, 73.5, 68.8, 65.2, 46.8 38.9, 30.3; IR: 2924, 2862, 1602, 1092, 1025, 748, 610 cm−1; HRMS (DART-TOF) calcd. for C18H24N1O2 [M+H]+: 286.1807, found: 286.1806. [α]D20 = +19.6 (c = 0.310, CHCl3, l = 50 mm).
(S)-4-(benzyloxy)-2-(((4-chlorophenyl)amino)methyl)butan-1-ol (5d)
Orange oil (45.9 mg, 72%). HPLC (OD-H, 1.0 mL/min, 15% iPrOH: 85% Hexanes, 240 nm) trmajor = 13.5 min and trminor = 18.5 min, 88% ee; 1H NMR (CDCl3, 500 MHz) δ 7.29 – 7.38 (m, 5H), 7.07 – 7.10 (m, 2H), 6.47 – 6.50 (m, 2H), 4.53 (s, 2H), 3.68 (d, 2H, J = 4.9), 3.55 – 3.62 (m, 2H), 3.07 – 3.17 (m, 2H), 1.96 – 2.01 (m, 1H), 1.72 –1.76 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 147.2, 138.0, 129.2, 128.7, 128.1, 128.0, 122.0, 114.1, 73.5, 68.7, 65.0, 46.8, 38.8, 30.2; IR: 3378, 2924, 2860, 1600, 1500, 1093, 816 cm−1; HRMS (DART-TOF) calcd. for C18H23Cl1N1O2 [M+H]: 320.1417, found: 320.1425. [α]D20 = +20.3 (c = 0.325, CHCl3, l = 50 mm).
(S)-4-((4-(benzyloxy)-2-(hydroxymethyl)butyl)amino)benzonitrile (5e)
Orange oil (30.3 mg, 49%). HPLC (OD-H, 1.0 mL/min, 15% iPrOH: 85% Hexanes, 220 nm) trmajor = 17.3 min and trminor = 20.3 min, 65% ee; 1H NMR (CDCl3, 500 MHz) δ 7.30 – 7.38 (m, 7H), 6.44 – 6.47 (m, 2H), 4.90 (br s, 1H), 4.53 (s, 2H), 3.70 (d, 2H, J = 4.7), 3.56 – 3.63 (m, 2H), 3.12 – 3.25 (m, 2H), 2.25 (br s, 1H), 1.98 – 2.02 (m, 1H), 1.70 – 1.81 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 151.8, 137.9, 133.7, 128.8, 128.2, 128.1, 120.8, 112.3, 98.5, 73.7, 68.7, 64.8, 45.9, 38.8, 30.1; IR: 3373, 2922, 2867, 2211, 1606, 1528, 1173 cm−1; HRMS (DART-TOF) calcd. for C19H23N2O2 [M+H]: 311.1760, found: 311.1756; [α]D20 = + 34.2 (c = 0.090, CHCl3, l = 50 mm).
(S)-4-(benzyloxy)-2-(((4-nitrophenyl)amino)methyl)butan-1-ol (5f)
Yellow oil (27.2 mg, 41%). HPLC (AS-H, 1.0 mL/min, 15% iPrOH: 85% Hexanes, 220 nm) trmajor = 53.9 min and trminor = 46.6 min, 52% ee; 1H NMR (CDCl3, 500 MHz) δ 8.01 – 8.04 (m, 2H), 7.32 – 7.38 (5H, m), 6.37 – 6.39 (m, 2H), 5.36 (br s, 1H), 4.54 (s, 2H), 3.71 (br s, 2H), 3.60 – 3.62 (t, 2H, J = 5.5), 3.17 – 3.31 (m, 2H), 2.28 (br s, 1H), 2.00 –2.05 (m, 1H), 1.71 – 1.81 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 153.9, 137.8, 137.7, 128.8, 128.3, 128.2, 126.6, 111.0, 73.7, 68.8, 64.9, 46.2, 38.8, 30.0; IR: 3370, 2922, 2855, 1598, 1308, 1279, 1108, 697 cm−1; HRMS (DART-TOF) calcd. for C18H23N2O4 [M+H]: 331.1658, found: 331.1657; [α]D20 = +28.7 (c = 0.060, CHCl3, l = 50 mm).
(S)-4-(benzyloxy)-2-((m-tolylamino)methyl)butan-1-ol (5g)
Yellow oil (35.8 mg, 60%). HPLC (OD-H, 1.0 mL/min, 23% iPrOH: 77% Hexanes, 240 nm) trmajor = 10.3 min and trminor = 12.3 min, 89% ee; 1H NMR (CDCl3, 500 MHz) δ 7.29 – 7.38 (m, 5H), 7.04 – 7.07 (m, 1H), 6.53 (d, 1H, J = 7.3), 6.41 – 6.42 (m, 2H), 4.54 (s, 2H), 3.69 (d, 2H, J = 5.3), 3.55 – 3.63 (m, 2H), 3.11 – 3.19 (m, 2H), 2.27 (s, 3H), 2.00 – 2.02 (m, 1H), 1.73 – 1.77 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 148.6, 139.2, 138.1, 129.3, 128.7, 128.0, 127.8, 118.6, 114.0, 110.3, 73.5, 68.8, 65.3, 46.9, 39.0, 30.3, 21.8; IR: 3378, 2919, 2858, 1604, 1092, 1028, 769, 737, 695; HRMS (DART-TOF) calcd. for C19H26NO2 [M+H]: 300.1964, found: 300.1958; [α]D20 = +28.2 (c = 0.160, CHCl3, l = 50 mm).
(S)-4-(benzyloxy)-2-(((3-nitrophenyl)amino)methyl)butan-1-ol (5h)
Orange oil (40.4 mg, 61%). HPLC (OD-H, 1.0 mL/min, 7% iPrOH: 93% Hexanes, 240 nm) trmajor = 55.9 min and trminor = 37.8 min, 84% ee; 1H NMR (CDCl3, 500 MHz) δ 7.45 – 7.47 (app dd, 1H, J = 8.1, 1.5), 7.28 – 7.37 (m, 6H), 7.19 – 7.22 (t, 1H, J = 8.1), 6.75 – 6.77 (m, 1H), 4.65 (br s, 1H), 4.53 (s, 2H), 3.70 (d, 2H, J = 4.4), 3.56 – 3.64 (m, 2H), 3.14 – 3.24 (m, 2H), 2.48 (br s, 1H), 1.99 – 2.04 (m, 1H), 1.71 – 1.82 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 149.7, 149.5, 137.9, 129.8, 128.8, 128.2, 128.1, 119.0, 111.8, 106.2, 73.6, 68.7, 65.0, 46.5, 38.8, 30.1; IR: 3386, 2923, 2859, 1527, 1454, 1092, 733, 698 cm−1; HRMS (DART-TOF) calcd. for C18H23N2O4 [M+H]: 331.1658, found: 331.1662; [α]D20 = +19.8 (c = 0.200, CHCl3, l = 50 mm).
(S)-4-(benzyloxy)-2-((pyridin-3-ylamino)methyl)butan-1-ol (5i)
Orange oil (36.5 mg, 64%). HPLC (OD-H, 1.0 mL/min, 21% iPrOH: 79% Hexanes, 240 nm) trmajor = 17.2 min and trminor = 27.6 min, 87% ee; 1H NMR (CDCl3, 500 MHz) δ 7.93 (br s, 2H), 7.28 – 7.36 (m, 5H), 7.05 (br s, 1H), 6.82 (d, 1H, J = 5.0), 4.52 (s, 2H), 4.26 (br s, 1H), 3.70 (d, 2H, J = 4.7), 3.55 – 3.63 (m, 2H), 3.11 – 3.21 (m, 2H), 1.98 – 2.02 (m, 1H), 1.73 – 1.77 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 144.8, 138.4, 138.0, 136.0, 128.7, 128.1, 128.0, 123.9, 118.7, 73.5, 68.7, 64.8, 46.3, 38.7, 30.1; IR: 2858, 1589, 1091, 794, 734, 632 cm−1; HRMS (DART-TOF) calcd. for C17H23N2O2 [M+H]: 287.1760, found: 287.1759; [α]D20 = +3.5 (c = 0.465, CHCl3, l = 50 mm).
Supplementary Material
Acknowledgments
We thank the ACS-PRF (DNI-5001400) and NIGMS (RO1GM087581) for funding this project. Mass spectrometry instrumentation at Boston College is supported by funding from the NSF (DBI-0619576).
Footnotes
Supporting Information. NMR spectra for all compounds, HPLC traces, and equilibrium data. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Breit B, Seiche W. Synthesis. 2001;1:1–36. [Google Scholar]
- 2.Gual A, Godard C, Castillon S, Claver C. Tetrahedron: Asymmetry. 2010;21:1135–1146. [Google Scholar]
- 3.Claver C, Dieguez M, Pamies O, Castillon S. Top Organometal Chem. 2006;18:35–64. [Google Scholar]
- 4.Klosin J, Landis CR. Acc Chem Res. 2007;40:1251–1259. doi: 10.1021/ar7001039. [DOI] [PubMed] [Google Scholar]
- 5.For recent examples of enantioselective hydroformylation, see and refs 6–14: Watkins AL, Landis CR. J Am Chem Soc. 2010;132:10306–10317. doi: 10.1021/ja909619a.
- 6.Noonan GM, Newton D, Cobley CJ, Suarez A, Pizzano A, Clarke ML. Adv Synth Catal. 2010;352:1047–1054. [Google Scholar]
- 7.Zhao B, Peng X, Wang Z, Xia C, Ding K. Chem-Eur J. 2008;14:7847–7857. doi: 10.1002/chem.200800388. [DOI] [PubMed] [Google Scholar]
- 8.Robert T, Abiri Z, Wassenaar J, Sandee AJ, Romanski S, Neudorfl JM, Schmalz HG, Reek JNH. Organometallics. 2010;29:478–483. [Google Scholar]
- 9.Chikkali SH, Bellini R, Berthon-Gelloz G, van der Vlugt JI, de Bruin B, Reek JNH. Chem Commun. 2010;46:1244–1246. doi: 10.1039/b924046b. [DOI] [PubMed] [Google Scholar]
- 10.Doro F, Reek JNH, van Leeuwen PWNM. Organometallics. 2010;29:4440–4447. [Google Scholar]
- 11.Wassenaar J, de Bruin B, Reek JNH. Organometallics. 2010;29:2767–2776. [Google Scholar]
- 12.Chercheja S, Nadakudity SK, Eilbracht P. Adv Synth Catal. 2010;352:637–643. [Google Scholar]
- 13.Gual A, Godard C, Castillon S, Claver C. Adv Synth Catal. 2010;352:463–477. [Google Scholar]
- 14.Zhang X, Cao B, Yan Y, Yu S, Ji B, Zhang X. Chem-Eur J. 2010;16:871–877. doi: 10.1002/chem.200902238. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Coo B, Yu S, Zhang X. Angew Chem Int Ed. 2010;49:4047–4050. doi: 10.1002/anie.201000955. [DOI] [PubMed] [Google Scholar]
- 16.McDonald RI, Wong GW, Neupane RP, Stahl SS, Landis CR. J Am Chem Soc. 2010;132:14027–14029. doi: 10.1021/ja106674n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.For early examples of reversible covalent bonding with ligands for metal catalysts, see refs 17–20: Lewis LN, Smith JF. J Am Chem Soc. 1986;108:2728–2735.
- 18.Lewis LN. Inorg Chem. 1985;24:4433–4435. [Google Scholar]
- 19.Preston SA, Cupertino DC, Palma-Ramirez P, Cole-Hamilton DJ. J Chem Soc, Chem Commun. 1986:977–978. [Google Scholar]
- 20.Iraqi A, Fairfax NR, Preston SA, Cupertino DC, Irvine DJ, Cole-Hamilton DJ. J Chem Soc, Dalton Trans. 1991:1929–1935. [Google Scholar]
- 21.For reviews on reversible covalent bonding with ligands for metal catalysts see ref 21 and 22: Park YJ, Park J, Jun C. Acc Chem Res. 2008;41:222–234. doi: 10.1021/ar700133y.
- 22.Rousseau G, Breit B. Angew Chem Int Ed. 2011;50:2450–2494. doi: 10.1002/anie.201006139. [DOI] [PubMed] [Google Scholar]
- 23.For examples of reversible covalent bonding between substrates and ligands in hydroformylation see refs 23–29: Lightburn TE, de Paolis OA, Cheng KH, Tan KL. Org Lett. 2011;13:2686–2689. doi: 10.1021/ol200782d.
- 24.Sun X, Frimpong K, Tan KL. J Am Chem Soc. 2010;132:11841–11843. doi: 10.1021/ja1036226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Worthy AD, Gagnon MM, Dombrowski MT, Tan KL. Org Lett. 2009;11:2764–2767. doi: 10.1021/ol900921e. [DOI] [PubMed] [Google Scholar]
- 26.Lightburn TE, Dombrowski MT, Tan KL. J Am Chem Soc. 2008;130:9210–9211. doi: 10.1021/ja803011d. [DOI] [PubMed] [Google Scholar]
- 27.Grünanger CU, Breit B. Angew Chem Int Ed. 2010;49:967–970. doi: 10.1002/anie.200905949. [DOI] [PubMed] [Google Scholar]
- 28.Grünanger CU, Breit B. Angew Chem Int Ed. 2008;47:7346–7349. doi: 10.1002/anie.200802296. [DOI] [PubMed] [Google Scholar]
- 29.Usui I, Nomura K, Breit B. Org Lett. 2011;13:612–615. doi: 10.1021/ol1028546. [DOI] [PubMed] [Google Scholar]
- 30.For examples of supramolecular catalysis controlling regioselectivity see refs 30–34: Slagt VF, Kamer PCJ, van Leeuwen PWNM, Reek JNH. J Am Chem Soc. 2004;126:1526–1536. doi: 10.1021/ja0386795.
- 31.Kuil M, Soltner T, van Leeuwen PWNM, Reek JNH. J Am Chem Soc. 2006;128:11344–11345. doi: 10.1021/ja063294i. [DOI] [PubMed] [Google Scholar]
- 32.Dydio P, Wojciech ID, Lutyz M, de Bruin B, Reek JNH. Angew Chem Int Ed. 2011;50:396–400. doi: 10.1002/anie.201005173. [DOI] [PubMed] [Google Scholar]
- 33.Smejkal T, Breit B. Angew Chem Int Ed. 2008;47:311–315. doi: 10.1002/anie.200703192. [DOI] [PubMed] [Google Scholar]
- 34.Smejkal T, Gribkov D, Geier J, Keller M, Breit B. Chem-Eur J. 2010;16:2470–2478. doi: 10.1002/chem.200902553. [DOI] [PubMed] [Google Scholar]
- 35.Worthy AD, Joe CL, Lightburn TE, Tan KL. J Am Chem Soc. 2010;132:14757–14759. doi: 10.1021/ja107433h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ligand 1 was used in the exchange experiments instead of 2, because it is more easily accessible in large quantities.
- 37.Juaristi E, Cuevas G. Tetrahedron. 1992;48:5019–5087. [Google Scholar]
- 38.Juaristi E, Cuevas G. The Anomeric Effect. CRC Press; Boca Raton: 1995. [Google Scholar]
- 39.The regioselectivities cannot be determined, because the product of hydroformylation of the carbon distal from the aniline functionality is not stable under the reaction conditions. This product likely forms a cyclic hemi-aminal that decomposes under the acidic conditions. Crude 1H NMR analysis of the p-chloro substrate shows clean formation of a single compound consistent with high regioselectivity, see supporting information for details.
- 40.Campi EM, Fallon GD, Jackon WR, Nilsson Y. Aust J Chem. 1992;45:1167–1178. [Google Scholar]
- 41.MacInnes I, Walton JC. J Chem Soc Perkin Trans II. 1987;8:1077–1082. [Google Scholar]
- 42.Shafir A, Buchwald SL. J Am Chem Soc. 2006;128:8742–8743. doi: 10.1021/ja063063b. [DOI] [PubMed] [Google Scholar]
- 43.Kraszkiewicz L, Sosnowski M, Skulski L. Synthesis. 2006;7:1195–1199. [Google Scholar]
- 44.Trécourt F, Breton G, Bonnet V, Mongin F, Marsais F, Quéguiner G. Tetrahedron. 2000;56:1349–1360. [Google Scholar]
- 45.Ma D, Cai Q, Zhang H. Org Lett. 2003;5(14):2453–2455. doi: 10.1021/ol0346584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.