

Abstract
The Delta–Notch signal transduction pathway has widespread roles in animal development in which it appears to control cell fate. CBF1/RBP-Jκ, the mammalian homolog of Drosophila Suppressor of Hairless [Su(H)], switches from a transcriptional repressor to an activator upon Notch activation. The mechanism whereby Notch regulates this switch is not clear. In this report we show that prior to induction CBF1/RBP-Jκ interacts with a corepressor complex containing SMRT (silencing mediator of retinoid and thyroid hormone receptors) and the histone deacetylase HDAC-1. This complex binds via the CBF1 repression domain, and mutants defective in repression fail to interact with the complex. Activation by Notch disrupts the formation of the repressor complex, thus establishing a molecular basis for the Notch switch. Finally, ESR-1, a Xenopus gene activated by Notch and X-Su(H), is induced in animal caps treated with TSA, an inhibitor of HDAC-1. The functional role for the SMRT/HDAC-1 complex in CBF1/RBP-Jκ regulation reveals a novel genetic switch in which extracellular ligands control the status of critical nuclear cofactor complexes.
Keywords: Notch, transcriptional repressor, corepressor complex, SMRT, CBF1/RBP-Jκ
The Notch signaling pathway is believed to control cell fate decisions in multiple developmental programs (for review, see Artavanis-Tsakonas et al. 1995; Robey 1997; Weinmaster 1997). Notch proteins define a family of transmembrane receptors that contain multiple epidermal growth factor (EGF)-like repeats and a conserved cysteine-rich region in their extracellular domain, and cdc10/ankyrin repeats in their intracellular domain. During Drosophila sense organ development, activation of the Notch receptor in response to binding of the ligand Delta induces transcription of genes of the Enhancer of Split complex [E(spl)] (Jennings et al. 1994). The Drosophila Suppressor of Hairless [Su(H)] protein binds to the promoter regions of the E(Spl) genes and is a key component in Notch-mediated activation (Bailey and Posakony 1995; Lecourtois and Schweisguth 1995). Activation of Su(H) is thought to be dependent on a direct physical interaction with the Notch cytoplasmic domain. Homologs of these proteins have been described in Xenopus (Wettstein et al. 1997), mouse (Matsunami et al. 1989), and humans (Amakawa et al. 1993) suggesting a high degree of evolutionary conservation. For example, the mammalian homolog of Su(H), CBF1/RBP-Jκ, can bind and stimulate promoters of the mouse Hairy enhancer of split (HES-1) genes in the presence of vertebrate Notch (Jarriault et al. 1995).
CBF1/RBP-Jκ is a bifunctional protein. In the presence of Notch it functions as a transcriptional activator, whereas in the absence of Notch it represses transcription (Hsieh and Hayward 1995). The promoter of the adenovirus pIX polypeptide contains a consensus binding site for CBF1/RBP-Jκ that mediates repression both in vitro and in vivo (Dou et al. 1994). A similar repressive function has been ascribed to CBF1/RPB-Jκ binding sites in the IL-6 and NF-κB2 promoters (Kannabiran et al. 1997; Plaisance et al. 1997; Miyazawa et al. 1998). Direct evidence for CBF1/RPB-Jκ’s repressive activity comes from the finding that a GAL4–CBF1 fusion protein can repress activity of a thymidine kinase (TK) promoter linked to GAL4-binding sites (Hsieh and Hayward 1995). The repression domain of CBF1/RBP-Jκ has been mapped to the central portion of the protein that also mediates DNA binding and interaction with Notch. It has been proposed that Notch functions to mask the repression domain of CBF1/RBP-Jκ (Hsieh and Hayward 1995). A similar derepressing activity has been proposed for the Epstein–Barr virus nuclear antigen 2 (EBNA2) (Ling et al. 1993; Grossman et al. 1994; Henkel et al. 1994). Although these factors have been shown to bind to CBF1/RBP-Jκ and generate an activating complex, a molecular explanation for derepression has not been provided.
Recent advances have implicated the role of cofactors in mediating transcriptional repression. These include the related corepressors SMRT (silencing mediator of retinoic acid and thyroid hormone receptors) and N-CoR, which bind to nuclear hormone receptors in the absence of their ligands (Chen and Evans 1995; Horlein et al. 1995), mSin3A which binds to members of the Mad family of Max-interacting proteins (Ayer and Lawrence 1995), and the Krüppel-associated box (KRAB) domain containing zinc finger proteins (Friedman et al. 1996). Interestingly, SMRT/N-CoR and mSin3A can form a multiprotein complex that includes the histone deacetylase HDAC1 (Alland et al. 1997; Heinzel et al. 1997; Nagy et al. 1997). It is generally believed that transcriptional repression is mediated by the histone deacetylase.
The identification of these corepressors led us to explore the possibility that such factors may also underlie the ability of Notch to derepress CBF1/RBP-Jκ. Here we identify SMRT as a transcriptional corepressor for CBF1/RBP-Jκ and reveal its critical role in Notch signaling.
Results
The repression activity of CBF1 is mediated by a soluble cofactor
Although the presence of a soluble corepressor for CBF1/RBP-Jκ has been proposed, its identity and biochemical properties remain unknown (Grossman et al. 1994; Henkel et al. 1994; Ling et al. 1994; Hsieh and Hayward 1995; Waltzer et al. 1995). To provide evidence for such a factor, we examined repression by a GAL4–CBF1 fusion protein in the presence of increasing amounts of CBF1/RBP-Jκ. Consistent with previous reports (Hsieh and Hayward 1995), GAL4–CBF1 effectively repressed activity of a luciferase reporter containing four copies of a GAL4-binding sequence upstream of the TK promoter TK–MH100x4–Luc (Fig. 1, lanes 1,2 ). Coexpression of CBF1/RBP-Jκ effectively relieved repression by GAL4–CBF1 in a dose-dependent manner, whereas a repression-defective mutant, CBF1 (EEF233AAA), failed to do so. This supports suggestions that CBF1/RBP-Jκ associates with a soluble factor(s) required for repression by the GAL4–CBF1 fusion protein.
Figure 1.


Wild-type CBF1, but not mutant CBF1 (EEF233AAA), derepresses GAL4–CBF1. GAL4–CBF1 was transfected along with increasing amounts of either wild-type CBF1 (lanes 2–6) or mutant CBF1 (EEF233AAA) (lanes 7–11) expression plasmids as indicated. Luciferase activity is normalized by β-galactosidase activity (described in Materials and Methods). The diagram illustrates how CBF1 overexpression may result in derepression of GAL4–CBF1.
CBF1 interacts with SMRT in yeast two-hybrid assays and in vitro
Previously, we have described a nuclear receptor corepressor termed SMRT (Chen and Evans 1995), which mediates repression by unliganded receptors. To test whether SMRT could also interact with CBF1/RBP-Jκ, yeast two-hybrid assays were carried out using a β-galactosidase reporter under the control of a GAL4 UAS. As expected, CBF1/RBP-Jκ fused to the GAL4 activation domain (AD) gave low background levels of reporter activity (data not shown). However, cotransfection of a fusion protein containing the GAL4 DNA-binding domain (DBD) fused to intact SMRT (GAL–SMRT) enhanced reporter activity, indicating an association between CBF1/RBP-Jκ and SMRT (Fig. 2A,B). By use of a series of deletion mutants, a CBF1/RBP-Jκ interaction domain (CID) was mapped to amino acids 649–811 of SMRT. This interaction is specific because a 3-amino-acid substitution mutant of CBF1 that does not repress [EEF233AAA (Hsieh and Hayward 1995)] failed to interact with SMRT (Fig. 2C). A GST pull-down assay was used to determine whether the interaction between SMRT and CBF1/RBP-Jκ is likely to be direct (Fig. 2D). Although GST–SMRT(649–811) showed significant retention of in vitro-translated CBF1/RBP-Jκ, GST alone and GST–SMRT(336–448) gave no binding. Furthermore, GST–SMRT(649–811) bound strongly to wild-type GAL–CBF1 but not to mutant GAL–CBF1 (EEF233AAA) (Fig. 2E). Barring the presence of a bridging protein in both yeast and reticulocyte lysates, these results argue that the interaction between CBF1/RBP-Jκ and SMRT is specific and direct.
Figure 2.




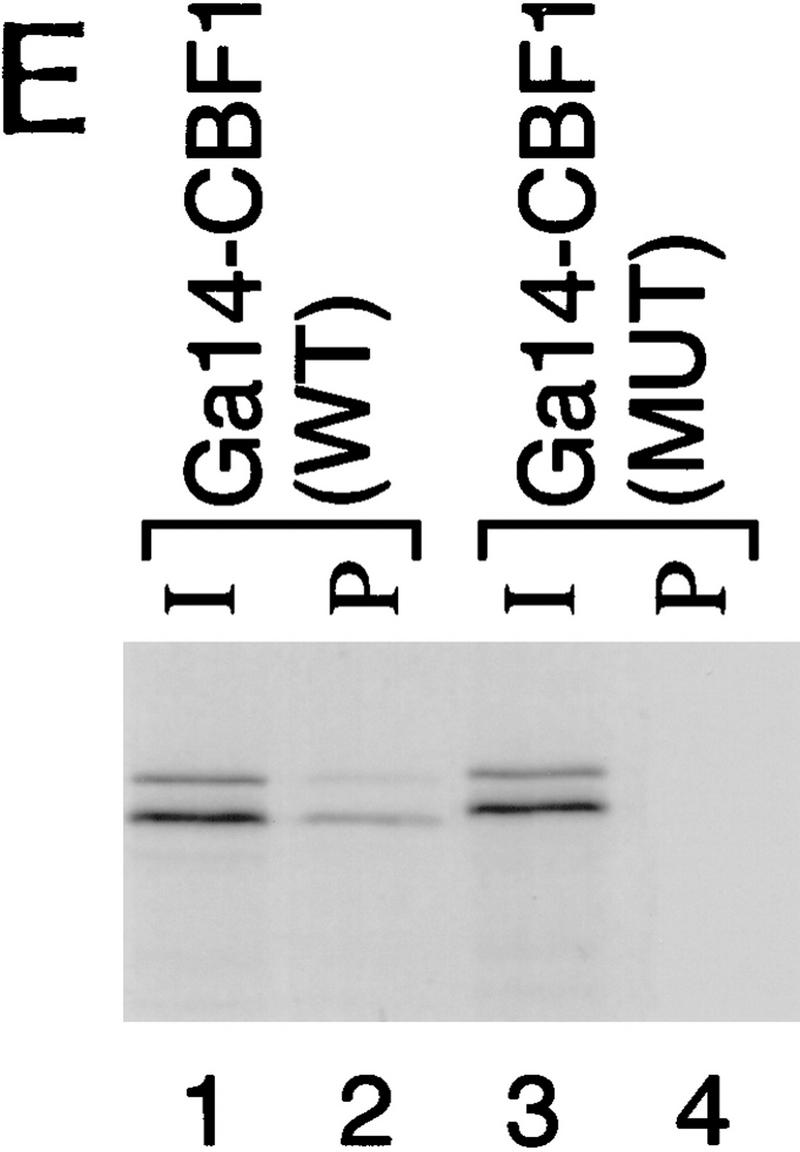
CBF1 interacts with SMRT in yeast and in GST pull-down assays. (A) Mapping of CBF1 interaction domain of SMRT in yeast. SMRT fragments fused to GAL4 DBD (GAL–SMRT) were cotransformed with GAL4 activation domain fused to CBF1(179–500) into yeast strain Y190. (SRD) SMRT repression domain; (RID) receptor interaction domain. (B) Quantitation of CBF1 and SMRT interaction in yeast. (C) Mutant CBF1 (EEF233AAA) fails to interact with SMRT in a yeast two-hybrid assay. Wild-type (lanes 1–3) or mutant GAL4–CBF1 (lanes 4–6) were cotransformed with or without AD–SMRT. (D) CBF1 interacts with GST–SMRT in vitro as shown in lanes 4 and 5. (E) Mutant GAL–CBF1 (EEF233AAA) (cf. lanes 2 and 4) does not interact with GST–SMRT(548–811). (I) Loaded in this experiment. (P) Pellet.
CBF1 interacts with SMRT in mammalian cells
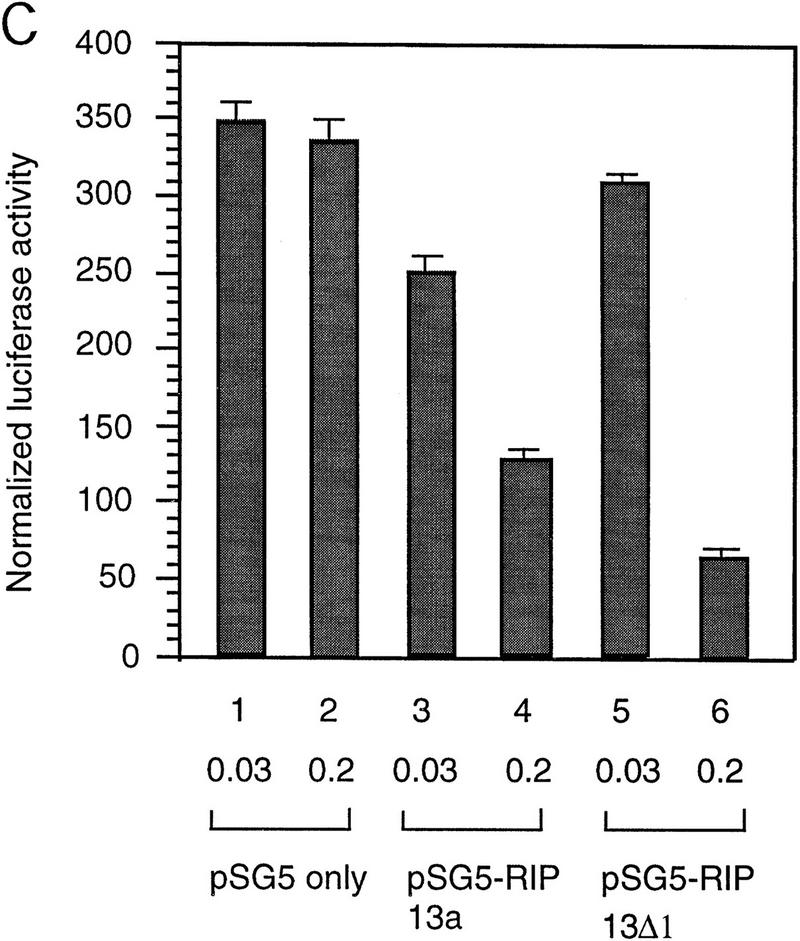
We assessed the interaction between CBF1/RBP-Jκ and SMRT in mammalian cells in two ways. First, we measured the interaction by coimmunoprecipitation. NIH-3T3 cells were transfected with a Flag epitope-tagged CBF1/RBP-Jκ protein (Flag–CBF1) with or without full-length SMRT. Whole-cell extracts were prepared, immunoprecipitated with an anti-Flag antibody, and coimmunoprecipitated proteins were detected by Western blot using a SMRT antibody (Fig. 3A). SMRT was detected only in the presence of Flag–CBF1, indicating that SMRT and CBF1/RBP-Jκ interact in transfected mammalian cell extracts. Second, we assessed the abilities of three GAL4–CBF1 fusion proteins to interact with VP16–SMRT (VP-F–SMRT) in a mammalian two-hybrid assay. GAL4–CBF1(179–361) contains an intact repression domain, whereas GAL4–CBF1(179–327) does not (Fig. 3B). As shown in Figure 3C, GAL4–CBF1(179–361) interacts strongly with SMRT but GAL4–CBF1(179–327) fails to interact with SMRT. GAL4–CBF1 (EEF233AAA) is another version of the protein that lacks repression activity (Hsieh and Hayward 1995). Only wild-type GAL4–CBF1 supported good transcriptional activation in the presence of VP16–SMRT (Fig. 3D, lanes 1–4), demonstrating that SMRT interacts with CBF1/RBP-Jκ in mammalian cells only when the latter contains a functional repression domain. To determine whether the SMRT-related protein N-CoR was also able to interact with CBF1/RBP-Jκ, we tested both N-CoR (amino acids 1681–2442) and RIP13Δ1 (amino acids 627–1277), an isoform of N-CoR (Seol et al. 1996). Both N-CoR and RIP13Δ1 show robust interaction with wild-type but not mutant CBF1 (see Fig. 3D, lanes 5–10). These results suggest that both members of this corepressor family may regulate CBF1/RBP-Jκ-mediated repression. It remains to be determined whether there is a physiological significance to the apparent difference in the affinity of N-CoR and SMRT for CBF1/RBP-Jκ.
Figure 3.



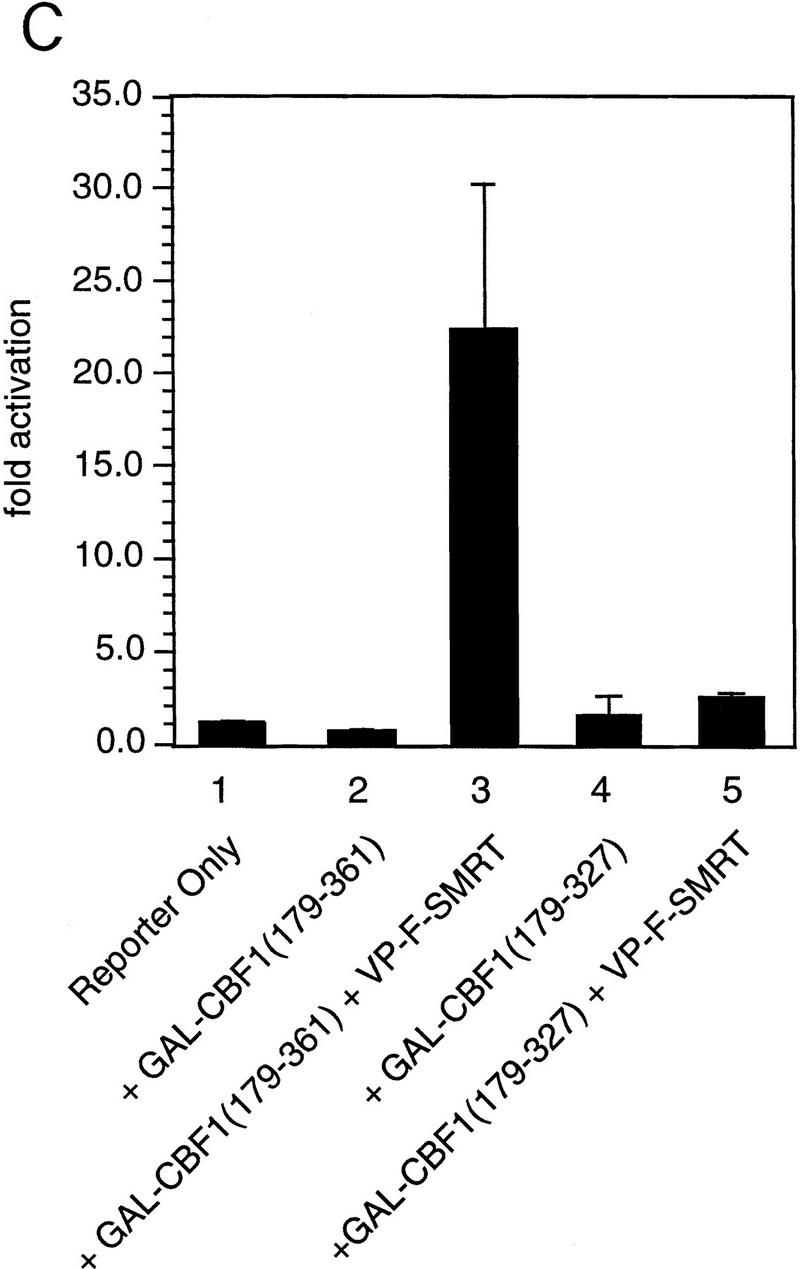

Functional interaction of CBF1 and SMRT. (A) SMRT interacts with CBF1 in vivo. NIH–3T3 cells were cotransfected with pBluescript (lanes 1,5), CBF1–Flag (lanes 2,6), SMRT (lanes 3,7), and CBF1–Flag + SMRT (lanes 4,8). Cells were harvested and lysed in RIPA buffer and extracts were analyzed by Western blotting. For immunoprecipitation, lysates were incubated with anti-Flag antibody and protein A/G–agarose and analyzed by Western blotting. (Left) Whole-cell extract probed with anti-SMRT antibody; (right) immunoprecipitated protein probed with anti-SMRT antibody. (B) NIH–3T3 cells were transfected with GAL5–TK–CAT reporter (1 μg) and the indicated GAL4–CBF1 fusion proteins (1 μg). Full-length CBF1 and CBF1(179–361) repress reporter; CBF1(179–327) does not repress (cf. lanes 2–4). The schematic highlights the minimal repression domain and the mutations that abolish repression activity of CBF1. (C) SMRT interaction with CBF1 requires an intact CBF1 repression domain. NIH–3T3 cells were cotransfected with a GAL4–E1B–luc reporter (0.3 μg), GAL4–CBF1 (1 μg) plasmids as indicated, and VP16–SMRT (1 μg). (D) Interaction with SMRT/N-CoR/RIP13Δ is impaired in a mutant CBF1 (EEF233AAA). CV-1 cells were transfected with a GAL4–E1B–luc reporter (0.1 μg), pCMX–lacZ (0.1 μg), and expression vectors as indicated.
SMRT antagonizes Notch (TAN-1)-mediated activation of CBF1
Next, we asked whether SMRT and Notch are antagonists. Specifically we asked whether SMRT could inhibit the activity of Notch signaling through a CBF1/RBP-Jκ-dependent reporter. Consistent with previous reports, GAL4–CBF1/RBP-Jκ is a strong activator in the presence of TAN-1 (translocation-associated Notch), a truncated form of Notch1 that contains only the cytoplasmic domain (Hsieh et al. 1996) (Fig. 4A, lanes 1,6). GAL4 DBD alone gave a low activation of the reporter; however, overexpression of SMRT led to strong inhibition of TAN-1 activity (Fig. 4A). Expression of TAN-1 and/or SMRT did not affect a reporter containing the TK promoter alone (data not shown). To assess the ability of SMRT to antagonize Notch activation through endogenous CBF1/RBP-Jκ, we monitored the activity of a luciferase reporter containing multiple CBF1/RBP-Jκ-binding sites in its promoter. This promoter was inactive in the absence of Notch and highly stimulated in the presence of TAN-1 (Fig. 4B). The addition of SMRT inhibited this activity in a dose-dependent manner. Similarly, N-CoR (data not shown) and two isoforms, RIP13a and RIP13Δ1, can inhibit TAN-1 activity through CBF1 (Fig. 4C). These findings were extended to determine whether SMRT could inhibit the activity of a natural promoter, the mouse HES-1 promoter. The HES-1 promoter has been shown previously to be activated by a TAN-1-CBF1/RBP-Jκ complex (Jarriault et al. 1995). Coexpression of SMRT inhibited TAN-1 activation of this promoter (Fig. 4D).
Figure 4.

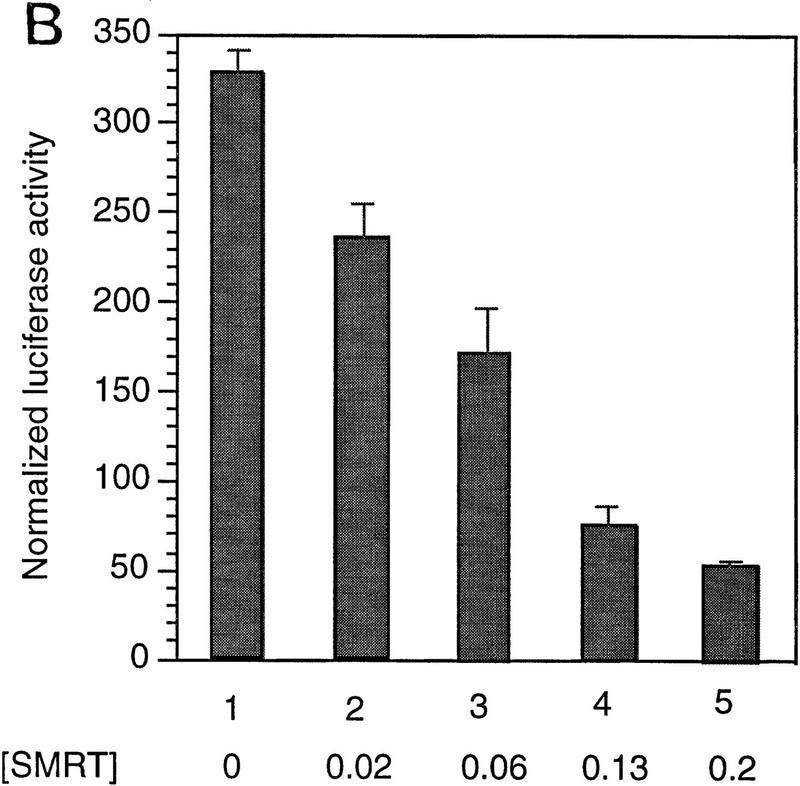


SMRT antagonizes TAN-1 activity. (A) SMRT inhibits TAN-1 activation of GAL4–CBF1. CV-1 cells were transfected with 0.1 μg of TK–MH100x4–Luc, pCMX–lacZ, 0.02 μg of pTAN-1, and pCMX–Gal4 DBD (lanes 1–5) or pCMX–Gal4 DBD–CBF1(179–500) (lanes 6–10). (B) SMRT inhibits Notch-mediated activation of endogenous CBF1. CV-1 cells were transfected with 0.1 μg of a CBF1–luc reporter, pCMX–lacZ, and pTAN-1 (0.02 μg). (C) RIP13a and RIP13Δ1 antagonize TAN-1-mediated activation of endogenous CBF1. Experiments were carried out similar to B and D. SMRT inhibits Notch activation of the HES-1 promoter. Experiments were carried out similar to B using 0.1 μg of a HES-1 promoter construct. (E) SMRT inhibits Notch interaction with CBF1 in vivo. 293T cells were transfected with the indicated plasmids. Flag–CBF1 (4 μg) was immunoprecipitated from whole-cell lysates in RIPA buffer. Immunoprecipitates were washed four times in RIPA buffer and loaded onto an 6% SDS–polyacrylamide gel. (Top) IP reactions probed with TAN-1 antibody; (middle and bottom) Western blots of whole cell extract. Blot was first probed with TAN-1 antibody and subsequently probed with Flag antibody to detect CBF1. Increasing amounts of SMRT were cotransfected in lanes 4 (1 μg) and 5 (2 μg).
To address the mechanism of inhibition, 293T cells were transfected with Flag–CBF1 and TAN-1 in the absence or presence of SMRT. Immunoprecipitations of whole-cell extracts by an anti-Flag antibody followed by Western analyses using a Notch1 antibody demonstrated that CBF1/RBP-Jκ interacts with TAN-1 in transfected cell extracts (Fig. 4E). However, the addition of increasing amounts of SMRT disrupts this interaction suggesting that SMRT and TAN-1 compete for binding to CBF1/RBP-Jκ. Western blots of the whole-cell extracts showed that SMRT did not effect the expression of either TAN-1 or Flag–CBF1 (Fig. 4E, bottom panels). Our results indicate that the interaction between SMRT and CBF1/RBP-Jκ occurs both in vitro and in vivo and that this interaction antagonizes the formation of a TAN-1-CBF1/RBP-Jκ complex.
TSA potentiates expression of a CBF1 target gene
SMRT has been shown previously to associate with mSin3A and the histone deacetylase HDAC1 as part of a large corepressor complex (Alland et al. 1997; Heinzel et al. 1997; Nagy et al. 1997). We reasoned that CBF1-mediated repression should be compromised in the presence of an inhibitor of histone deacetylase such as trichostatin A (TSA). We chose to test this in Xenopus animal caps, which are known to respond to the Notch signaling pathway. Specifically, we assessed the effects of TSA on the expression of ESR-1, an E(spl)-related gene from Xenopus. Expression of ESR-1, like similar genes in Drosophila, is induced in neural tissue by activated (cytoplasmic) forms of Xenopus Notch or by the Notch ligand, X-Delta-1 (Wettstein et al. 1997). Because induction of ESR-1 expression by Notch appears to be mediated by Xenopus Su(H) [X-Su(H)], we asked whether induction was enhanced by TSA. Such a result would support the idea that X-Su(H) might inhibit the expression of Notch target genes such as ESR-1 via the SMRT deacetylase complex. As shown in Figure 5, B and C, expression of ESR-1 in neuralized animal caps was induced by X-Delta-1 in a dose-dependent manner, with the effects of X-Delta-1 on ESR-1 expression saturating at 1 ng of injected RNA. In the presence of TSA, the induction of ESR-1 transcripts in response to X-Delta-1 was enhanced two- to threefold at each dose. In the presence of TSA the lowest dose of X-Delta-1 RNA (0.25 ng) induced levels of ESR-1 RNA comparable to the saturating level induced in the absence of TSA (cf. lanes 2, 3, and 10). These results are consistent with the prediction that Notch target genes are derepressed by treatment with TSA, possibly through the inhibition of SMRT-associated histone deacetylases. To assay whether HDAC-1 associated with CBF1 in vivo, coimmunoprecipitation experiments were performed. Cells were transfected with Flag–CBF1 in the presence or absence of HDAC-1 and immunoprecipitated with Flag antibody. The HDAC-1 was coimmunoprecipiated only in the presence of CBF1 (Fig. 5D). An interaction between CBF1 and HDAC-1 was also assessed through GST pull-down assays. HDAC-1 was shown to interact with GST–CBF1 (Fig. 5E) but not GST. As with SMRT, HDAC-1 did not interact with a repression-defective mutant, CBF1 (Fig. 5E, lane 3).
Figure 5.


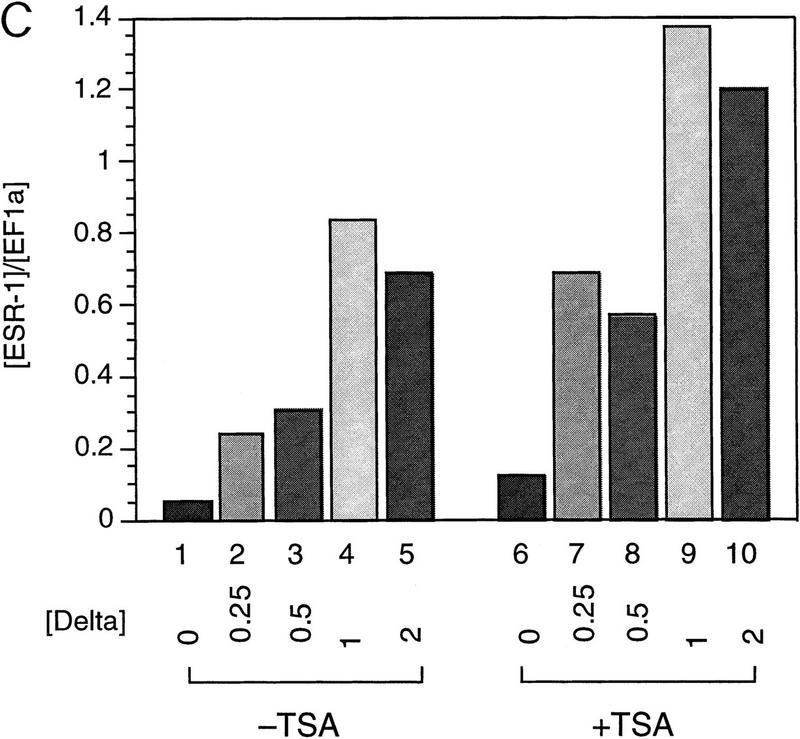
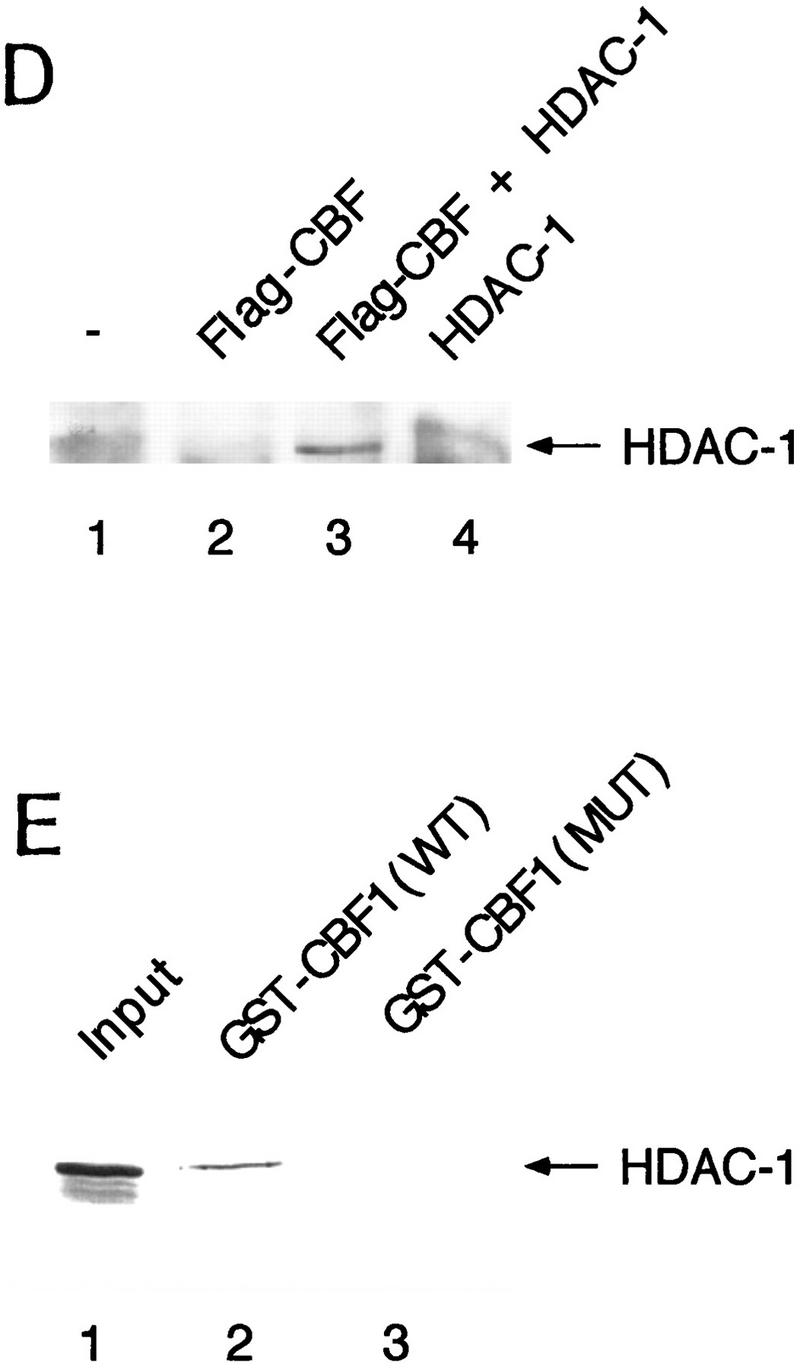
TSA derepresses expression of the Notch target gene ESR-1. (A) Diagram of the animal cap assay. Each blastomere of a two-cell Xenopus embryo was injected with varying amounts of X-Delta-1 RNA along with RNA (1.0 ng) encoding the neural inducer Noggin. For each assay, eight animal caps were explanted at late blastula stages (st. 9). From stage 12 to 17, treated animal caps were incubated with 400 nm TSA, after which total RNA was extracted. (B) RNase protection assay. RNA isolated from animal caps was assayed simultaneously for the levels of ESR-1, X-Delta-1, and EF-1α (an internal measure of total RNA). Arrows or bracket indicate the position of the protected probe for each RNA. The Xenopus Delta-1 probe detects both injected as well as endogenous RNA. (C) Quantitation of the assay. Protected band intensities were quantitated and normalized for total RNA using a PhosphorImager (Molecular Dynamics). (D) Association of CBF1 and HDAC-1 in vivo. CV-1 cells were cotransfected with CBF1–Flag (lane 3). Cells were harvested and lysed in RIPA buffer, and extracts were analyzed by Western blotting. For immunoprecipitation, lysates were incubated with anti-Flag antibody and protein A/G–agarose and analyzed by Western blotting with anti-HDAC-1 antibody. (E) HDAC-1 interacts with wild-type GST–CBF1 but not mutant GST–CBF1 (EEF233AAA) in vitro.
On the basis of these results, we propose a model (Fig. 6, top) to explain this switch in which CBF1/RBP-Jκ mediates repression of genes through the recruitment of a corepressor complex containing SMRT and histone deacetylase activity. In the absence of any positive acting factor, that is, TAN-1, the CBF1/RBP-Jκ exists as a corepressor complex. In the presence of activated Notch signaling, the intracellular domain of Notch would translocate to the nucleus and displace the corepressor complex (Fig. 6, bottom). It remains to be tested whether a Notch/CBF1 complex recruits a coactivating complex.
Figure 6.

A model for transcriptional regulation of Notch-activated genes. (Top) In the absence of activated Notch, gene transcription is repressed by binding of CBF1/RBP-Jκ and the recruitment of a corepressor complex containing histone deacetylase activity. (Bottom) Upon activation of Notch signaling by binding of the Notch ligand Delta, an intracellular form of Notch is released by proteolytic cleavage and translocates to the nucleus where it interacts with CBF1/RBP-Jκ. This interaction displaces the corepressor complex and results in activation of transcription presumably by removal of the histone deacetylase activity. It remains to be determined whether transcriptional activation by a Notch : CBF1/RBP-Jκ complex involves the recruitment of histone acetylases.
Discussion
In this work we identify a SMRT/HDAC-1 complex as a corepressor for the transcription factor CBF1/RBP-Jκ. A novel domain has been mapped in SMRT that directs interaction with CBF1/RBP-Jκ both in vitro and in vivo. In addition, we demonstrate a role for SMRT in Notch signaling through its ability to antagonize TAN-1 activation of CBF1/RBP-Jκ in a dosage-dependent manner. We show that expression of ESR-1, a Notch target gene in Xenopus, was induced in animal caps treated with TSA, an inhibitor of histone deacetylase HDAC-1. Furthermore, coimmunoprecipitation and GST pull-down assays indicate association of HDAC-1 and CBF1.
CBF1/RBP-Jκ has been described as both a transcriptional activator and transcriptional repressor. The switch from repressor to activator is mediated by interactions with proteins such as TAN-1, an activated form of the Notch receptor, and the Epstein–Barr virus protein EBNA2. Our studies were initiated to determine whether the mechanism of CBF1/RBP-Jκ-mediated repression was direct (e.g., through direct interactions with the basal machinery) or a consequence of one or more corepressors. The ability of overexpressed CBF1/RBP-Jκ to relieve repression by GAL4–CBF1 supported the latter possibility. The corepressors SMRT and N-CoR have been shown to be responsible for transcriptional repression by unliganded nuclear receptors. The binding of ligand leads to the dissociation of the corepressors and recruitment of a complex of transcriptional coactivators. On the basis of the possibility that a similar transition in bound proteins may regulate the switch of CBF1/RBP-Jκ from repressor to activator, we chose to examine whether the SMRT/N-CoR family of corepressors could mediate repression by CBF1/RBP-Jκ. Our initial results from yeast two-hybrid assays and GST pull-down experiments demonstrated that SMRT can bind to CBF1/RBP-Jκ. This binding localized to amino acids 649–811 in SMRT, defining a novel protein interaction region, as it does not overlap with those regions required for interactions with the nuclear receptors. The interaction was also detected in mammalian cells using both an in vivo two-hybrid analysis and coimmunoprecipitation from extracts of transfected cells. Importantly, SMRT did not bind a mutant CBF1 (EEF233AAA) that is defective in repression, a result that is expected if SMRT is a bona fide, physiological corepressor for CBF1/RBP-Jκ. Given that the CID is conserved in the SMRT-related protein N-CoR, we considered it likely that N-CoR also functions as a corepressor for CBF1/RBP-Jκ. The N-CoR isoforms RIP13a and RIP13Δ, which are most similar to SMRT, inhibited Notch-ediated activation of CBF1/RBP-Jκ, suggesting that repression may be mediated by the SMRT/ N-CoR family of proteins.
It has been proposed that Notch masks the repression domain of CBF1/RBP-Jκ. We reasoned that this might occur in one of two ways: Either Notch neutralizes SMRT associated corepressor complex in the context of a ternary complex with CBF1/RBP-Jκ or Notch simply displaces SMRT from CBF1/RBP-Jκ. Our observation that overexpressed SMRT antagonized Notch-mediated activation of CBF1/RBP-Jκ in vivo argued in favor of the latter possibility, and this was confirmed by our observation that SMRT antagonized the Notch–CBF1/RBP-Jκ interaction in vitro. Although these results relied on overexpressed SMRT to dampen Notch activity, they raise the possibility that the Notch signal may be actively regulated by SMRT/N-CoR. Recent results of Lassar and coworkers (M. Lazar, pers. comm.) indicate that levels of N-CoR may be subject to control by cell-specific proteolysis.
SMRT has been shown to exist in a multiprotein complex that includes the histone deacetylase HDAC1. The importance of histone deacetylase activity in transcriptional repression and other biological processes can be probed with TSA, an inhibitor of HDAC-1. To test the role of histone deacetylase activity on Notch signaling, we examined the effect of TSA on the response of Xenopus ESR-1 to X-Delta-1. TSA treatment led to increased ESR-1 expression, even in the absence of added Delta. This result supports a physiological role for histone deacetylase in repressing ESR-1 transcription in general and is consistent with our model that this is a specific consequence of SMRT/N-CoR interacting with CBF1/RBP-Jκ. In agreement with this result, we detect association of CBF1/RBP-Jκ and HDAC-1 by coimmunoprecipitation and GST pull-down assays. It is not clear from these experiments whether the interaction of HDAC-1 with CBF1/RBP-Jκ is direct or indirect. We cannot exclude the possibility of endogenous bridging proteins in the lysate mediating CBF1 and HDAC-1 interaction. Nevertheless, these results are particularly interesting, as the transcriptional repressor promyelocytic leukemia zinc finger (PLZF) also interacts with all known components of the corepressor complex such as SMRT/N-CoR, mSin3A, and HDAC-1 (Grignani et al. 1998; Lin et al. 1998).
Recent work by Honjo and coworkers (Taniguchi et al. 1998) has identified a LIM protein, KyoT2, as a possible negative regulator of CBF1/RBP-Jκ-mediated transcription. KyoT2 inhibits CBF1/RBP-Jκ by interfering with its DNA binding. These results do not address the mechanism by which CBF1/RBP-Jκ functions as a transcriptional repressor once bound to DNA. In various cell extracts there is a significant amount of endogenous CBF1/RBP-Jκ DNA-binding activity (data not shown). Although it is likely that multiple factors may regulate CBF1 activity, SMRT fulfills all of the criteria for a bona fide corepressor for CBF1, including the lack of binding of SMRT to a repression-defective mutant of CBF1. Taken together, our data provide evidence that the switch of CBF1/RBP-Jκ from a repressor to an activator is mediated by a direct effect of Notch on binding by the nuclear receptor cofactor SMRT. The parallels between Notch-mediated activation of CBF1/RBP-Jκ and ligand-mediated activation of hormone suggest this unusual type of regulation may be even more widely conserved and that corepressors may function as integrators of multiple cell growth and signal transduction pathways.
Materials and methods
Yeast two-hybrid
Yeast two-hybrid assays were performed in strain Y190 as described by the manufacturer’s protocol (Clontech). β-Galactosidase activity from three independent transformants was measured according to the manufacturer’s protocol (Clontech). Mean values of six independent measurements are presented.
GST pull-down assays
GST–SMRT fragments were constructed by standard techniques, expressed, and purified according to the manufacturer (Pharmacia). 35S-Labeled CBF1 was translated in vitro (Promega). Purified GST–SMRT peptides were incubated with 5 μl of in vitro-translated CBF1 in G buffer (20 mm Tris-HCl at pH 7.9, 150 mm KCl, 1 mm EDTA, 4 mm MgCl2, 0.2% NP-40, 10% glycerol) for 1 hr at 4°C on a nutator and washed five times with G buffer. Bound proteins were loaded onto a 10% SDS–polyacrylamide gel and visualized by autoradiography.
Transient transfection assays
Transient transfections in CV1 cells were carried out as described previously (Nagy et al. 1997) with 0.1 μg of TK–MH100x4–Luc as a reporter gene and pCMX–lacZ as an internal control. For NIH–3T3 and 293T cells, transfections were carried out using calcium phosphate coimmunoprecipitation with the amounts of plasmids indicated in the legends to Figures 3 and 4. Luciferase and chloramphenicol acetyltransferase (CAT) activities of each sample were normalized to β-galactosidase activity. CAT assays were done according to Gorman et al. (1982). Each transfection was carried out in triplicate, and experiments were repeated at least three times.
Coimmunoprecipitation
Immunoprecipitation (IP) was carried out with cell extracts prepared from NIH–3T3 or 293T cells 48 hr after transfection. Cells were lysed in RIPA buffer, scraped, and passed several times through a 21-gauge needle. Extract (150 μl) was diluted to 500 μl in RIPA buffer and incubated with 1–2 μg of antibody for 1 hr at 4°C. To the IP reaction was added 20 μl of protein A/G–agarose (Santa Cruz), and the mix was incubated for 1 hr to overnight at 4°C. The beads were washed four times in RIPA buffer: 1× PBS, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS, resuspended in SDS–polyacrylamide gel loading buffer, and proteins were analyzed by Western blotting.
Animal cap assays
Synthesis and injection of RNA was carried out as described previously (Wettstein et al. 1997). To generate neuralized animal caps, 1.0 ng of RNA encoding Noggin (Lamb et al. 1993) was injected into each side of a two-cell-stage embryo. Different amounts of RNA encoding X-Delta-1 were injected along with the Noggin RNA (see Fig. 5 legend). Animal caps were isolated and cultured for ∼8 hr (equivalent to neural plate stage 12). They were then cultured until neuralae (stage 17) in the presence or absence of 400 nm TSA, after which RNA was extracted using the Tri Reagent (Molecular Research Center, Inc.). ESR-1 expression was analyzed using RNase protection assay as described previously. Quantitation was carried out on a PhosphorImager (Molecular Dynamics). Protected band intensities (ESR-1 and EF-1α) were integrated and background corrected.
Acknowledgments
We thank Ester Banayo for technical assistance and Richard Lin for providing constructs. We also thank Spyros Artavanis-Tsakonas for the TAN-1 expression plasmid and Diane Hayward for the Flag–CBF1 expression vector and CBF1–luciferase reporter. We acknowledge Daniel A. Wettstein for assistance in animal cap assays. We thank Lita Ong and Elaine Stevens for administrative assistance and members of the Evans laboratory for providing reagents and comments on the manuscript. We acknowledge Henry Juguilon for help in tissue culture. N. K.-Nakagawa is supported by The Naito Foundation and by the Human Frontier Science Program. M.D. is a C.J. Martin Research fellow from the National Health and Medical Research Foundation of Australia. P.O. was supported by National Institutes of Health (NIH) grant AI36878. R.M.E. is an investigator of the Howard Hughes Medical Institute (HHMI) and March of Dimes Chair of Molecular Developmental Biology at the Salk Institute for Biological Studies. This work was supported by the HHMI (to R.M.E. and T.K.) and NIH grants to R.M.E. (GM26444 and HD27183) and C.R.K.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL evans@salk.edu; FAX (619) 455-1349.
References
- Alland L, Muhle R, Hou H, Potes JJ, Chin L, Schreiber-Agus N, DePinho RA. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387:49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- Amakawa R, Jing W, Ozawa K. Human Jk recombination signal binding protein gene (IGKJRB): Comparison with its mouse homologue. Genomics. 1993;17:306–315. doi: 10.1006/geno.1993.1326. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- Ayer DE, Lawrence QA. Mad-Max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell. 1995;80:767–776. doi: 10.1016/0092-8674(95)90355-0. [DOI] [PubMed] [Google Scholar]
- Bailey AM, Posakony JW. Suppressor of hairless directly activates transcription of enhancer of split complex genes in response to Notch receptor activity. Genes & Dev. 1995;9:2609–2622. doi: 10.1101/gad.9.21.2609. [DOI] [PubMed] [Google Scholar]
- Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- Dou S, Zeng X, Cortes P. The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol Cell Biol. 1994;14:3310–3319. doi: 10.1128/mcb.14.5.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Fredericks W, Jensen D, Speicher D, Huang X, Neilson E, Rauscher FR. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes & Dev. 1996;10:2067–2078. doi: 10.1101/gad.10.16.2067. [DOI] [PubMed] [Google Scholar]
- Gorman CM, Moffat LF, Howard BH. Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol Cell Biol. 1982;2:1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grignani F, De Matteis S, Clara N, Tomasson L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara FF, Zamir I, Seiser C, Grignani F, Lazar MA, Minucci S, Pelicci PG. Fusion proteins of the retinoic acid receptor-a recruit histone deacetylase in promyelocytic leukemia. Nature. 1998;391:815–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- Grossman SR, Johansen E, Tong X, Yalamanchili R, Kieff E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the Jκ recombination signal binding protein. Proc Natl Acad Sci. 1994;91:7568–7572. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel T, Lavinsky RM, Mullen T-M, Soderstrom M, Laherty CD, Torchia J, Yang W-M, Brad G, Ngo SG, Davie JR, Seto E, Eisenman R, Rose DW, Glass CK, Rosenfeld MG. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- Henkel T, Ling PD, Hayward SD, Peterson MG. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein Jκ. Science. 1994;265:92–95. doi: 10.1126/science.8016657. [DOI] [PubMed] [Google Scholar]
- Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, Rosenfeld MG. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- Hsieh JJ-D, Hayward SD. Masking of the CBF1/RBPJκ transcriptional repression domain by Epstein-Barr virus EBNA2. Science. 1995;268:560–563. doi: 10.1126/science.7725102. [DOI] [PubMed] [Google Scholar]
- Hsieh JJ-D, Henkel T, Salmon P, Robey E, Peterson MG, Hayward D. Truncated mammalian Notch 1 activated CBF1/RBPJκ-repressed genes by a mechanism resembling that of Epstein-Barr Virus EBNA2. Mol Cell Biol. 1996;16:952–959. doi: 10.1128/mcb.16.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signaling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- Jennings B, Preiss A, Deildakis C, Bray S. The Notch signalling pathway is required for Enhancer of split bHLH protein expression during neurogenesis in the Drosophila embryo. Development. 1994;120:3537–3548. doi: 10.1242/dev.120.12.3537. [DOI] [PubMed] [Google Scholar]
- Kannabiran C, Zeng XY, Vales LD. The mammalian transcriptional repressor RBP (CBF1) regulates interleukin-6 gene expression. Mol Cell Biol. 1997;17:1–9. doi: 10.1128/mcb.17.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb TM, Knecht AK, Smith WC, Stachel SE, Economides AN, Stahl N, Yancopolous GD, Harland RM. Neural induction by the secreted polypeptide noggin. Science. 1993;262:713–718. doi: 10.1126/science.8235591. [DOI] [PubMed] [Google Scholar]
- Lecourtois M, Schweisguth F. The neurogenic suppressor of hairless DNA-binding protein mediates the transcriptional activation of the enhancer of split complex genes triggered by Notch signaling. Genes & Dev. 1995;9:2598–2608. doi: 10.1101/gad.9.21.2598. [DOI] [PubMed] [Google Scholar]
- Lin RJ, Nagy L, Inoue S, Shao W, Miller WH, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukemia. Nature. 1998;391:811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- Ling PD, Rawlins DR, Hayward SD. The Epstein-Barr virus immortalizing protein EBNA-2 is targeted to DNA by cellular enhancer-binding protein. Proc Natl Acad Sci. 1993;90:9237–9241. doi: 10.1073/pnas.90.20.9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling PD, Hsieh JJ-D, Ruf IK, Rawlins DR, Hayward SD. EBNA-2 upregulation of Epstein-Barr virus latency promotes and the cellular CD23 promoter utilizes a common targeting intermediate. J Virol. 1994;68:5375–5383. doi: 10.1128/jvi.68.9.5375-5383.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunami N, Hamaguchi Y, Yamamoto Y. A protein binding to the J Kappa recombination sequence of immunoglobin genes contains a sequence related to the integrase motif. Nature. 1989;342:934–937. doi: 10.1038/342934a0. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Mori A, Yamamoto K, Okudaira H. Transcriptional roles of CCAAT/Enhancer binding protein-β, nuclear factor-κB, and C-promoter binding factor 1 in interleukin (IL)-1β-induced IL-6 synthesis by human rheumatoid fibroblast-like synoviocytes. J Biol Chem. 1998;273:7620–7627. doi: 10.1074/jbc.273.13.7620. [DOI] [PubMed] [Google Scholar]
- Nagy L, Kao HY, Chakravarti D, Lin R, Hassig CA, Ayer DE, Schreiber SL, Evans RM. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373–380. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- Plaisance S, Vanden Berghe W, Boone E, Fiers W, Haegeman G. Recombination signal sequence binding protein κ is constitutively bound to the NFκ-B site of the Interleukin-6 promoter and acts as a negative regulatory factor. Mol Cell Biol. 1997;17:3733–3743. doi: 10.1128/mcb.17.7.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey E. Notch in vertebrates. Curr Opin Genes & Dev. 1997;7:551–557. doi: 10.1016/s0959-437x(97)80085-8. [DOI] [PubMed] [Google Scholar]
- Seol W, Mahon MJ, Lee YK, Moore DD. Two receptor interacting domains in the nuclear hormone receptor corepressor RIP13/N-CoR. Mol Endocrinol. 1996;10:1646–1655. doi: 10.1210/mend.10.12.8961273. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Furukawa T, Tun T, Han H, Honjo T. LIM protein KYOT2 negatively regulates transcription by association with the RBP-J DNA-binding protein. Mol Cell Biol. 1998;18:644–654. doi: 10.1128/mcb.18.1.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltzer L, Bourillot PY, Sergeant A, Manet E. RBP-J κ repression activity is mediated by a co-repressor and antagonized by the Epstein-Barr virus transfection factor EBNA2. Nucleic Acids Res. 1995;23:4939–4945. doi: 10.1093/nar/23.24.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinmaster G. The ins and outs of notch signaling. Mol Cell Neurosci. 1997;9:91–102. doi: 10.1006/mcne.1997.0612. [DOI] [PubMed] [Google Scholar]
- Wettstein DA, Turner DL, Kintner CR. The Xenopus homolog of Drosophila Suppressor of Hairless mediates Notch signaling during primary neurogenesis. Development. 1997;124:693–702. doi: 10.1242/dev.124.3.693. [DOI] [PubMed] [Google Scholar]