

Abstract
Establishment of primary mouse embryo fibroblasts (MEFs) as continuously growing cell lines is normally accompanied by loss of the p53 or p19ARF tumor suppressors, which act in a common biochemical pathway. myc rapidly activates ARF and p53 gene expression in primary MEFs and triggers replicative crisis by inducing apoptosis. MEFs that survive myc overexpression sustain p53 mutation or ARF loss during the process of establishment and become immortal. MEFs lacking ARF or p53 exhibit an attenuated apoptotic response to myc ab initio and rapidly give rise to cell lines that proliferate in chemically defined medium lacking serum. Therefore, ARF regulates a p53-dependent checkpoint that safeguards cells against hyperproliferative, oncogenic signals.
Keywords: Myc signaling, ARF tumor suppressor, p53, apoptosis, immortalization
The INK4a–ARF locus is a common target of deletion and mutation in human cancers, possibly second in frequency only to p53. The product of the INK4a gene, p16INK4a, acts as an inhibitor of cyclin D-dependent kinases, preventing them from phosphorylating the retinoblastoma (pRb) protein and thus inhibiting S-phase entry during the cell division cycle (Serrano et al. 1993). A second product of this locus, p19ARF, encoded in part by an alternative reading frame of INK4a exon 2, is completely unrelated in its primary structure to p16INK4a and induces both G1- and G2-phase arrest in rodent fibroblasts (Quelle et al. 1995b) in a p53-dependent manner (Kamijo et al. 1997). Thus, both p16INK4a and p19ARF act as potent tumor suppressors by targeting pRb and p53 function, respectively.
Establishment of mouse embryo fibroblasts (MEFs) as continuously growing cell lines is usually accompanied by either ARF or p53 loss of function, implying that the two proteins act epistatically in a single pathway (Kamijo et al. 1997; Zindy et al. 1997). The p53 protein is a transcription factor (Kern et al. 1992) that induces several known target genes, including the cyclin-dependent kinase inhibitor p21/Cip1/Waf1 (El-Deiry et al. 1993; Harper et al. 1993; Xiong et al. 1993) and mdm2 (Barak et al. 1993; Wu et al. 1993). In turn, Mdm2 acts in a feedback loop to catalyze p53 ubiquitination and degradation, limiting the p53 response (Haupt et al. 1997; Honda et al. 1997; Kubbutat et al. 1997). The ARF protein can physically interact in binary or ternary complexes with p53 and Mdm2, and its overexpression induces p53 stabilization and activates p53-dependent transcription (Kamijo et al. 1998; Pomerantz et al. 1998; Zhang et al. 1998). Although the levels of p19ARF expressed in normal MEFs are relatively low, an unexplained feature is that p19ARF expression is significantly elevated in p53-null fibroblasts (Quelle et al. 1995b). Conversely, reintroduction of p53 into p53-null cells returns the level of p19ARF to normal levels (Kamijo et al. 1998). Together, these data suggest that a feedback loop also acts to limit p19ARF expression once p53 is activated, and the ability of Mdm2 to bind both p53 and p19ARF raises the possibility that Mdm2 might be responsible for their joint down-regulation.
The physiologic signals that induce ARF remain unknown. ARF is dispensable for p53 activation in response to ionizing or UV radiation (Kamijo et al. 1997), suggesting that it does not function in a DNA damage-signaling pathway. Observations that ARF-null MEFs are immortal and can be transformed by oncogenic ras alleles without a requirement for collaborating oncogenes such as myc and adenovirus E1A (Kamijo et al. 1997) led us to consider the possibility that myc and E1A might regulate p19ARF function. Either of these oncogenes are capable of immortalizing primary rodent fibroblasts (Land et al. 1983; Ruley 1983). Whether induced by enforced myc or E1A expression, chemical carcinogens, or by loss of p53 or ARF function, establishment and immortalization enable MEFs to be transformed into tumor cells by oncogenic ras genes alone (Land et al. 1983; Newbold and Overell 1983; Ruley 1983, 1990; Hicks et al. 1991; Lin et al. 1995; Serrano et al. 1996; Kamijo et al. 1997). myc and E1A seem to inactivate cellular responses that are normally required for ras-mediated inhibition of cell proliferation, thereby converting ras into a growth-promoting gene (Franza et al. 1986; Hicks et al. 1991; Hirakawa and Ruley 1991).
Given their apparent immortalizing functions, it seems paradoxical that myc and E1A are also potent inducers of apoptosis (Askew et al. 1991; White et al. 1991; Evan et al. 1992; Rao et al. 1992). The sensitivity of rodent fibroblasts to myc- or E1A-induced apoptosis correlates directly with the levels of oncoprotein expression and is greatly potentiated by depriving cells of extracellular survival factors (Evan et al. 1992; Lowe and Ruley 1993). Both Myc and E1A can induce p53 stabilization and trigger p53-dependent transcription (Lowe and Ruley 1993; Hermeking and Eick 1994; Wagner et al. 1994). Several lines of evidence indicate that p53 mediates apoptosis by myc and E1A in primary fibroblasts, with p53 loss rendering cells highly resistant to their deleterious effects (Debbas and White 1993; Lowe and Ruley 1993; Hermeking and Eick 1994; Wagner et al. 1994). For cells overexpressing myc to grow, programmed cell death must be actively suppressed (Askew et al. 1991; Evan et al. 1992; Hermeking and Eick 1994; Wagner et al. 1994). Therefore, myc overexpression should provide a strong selective pressure for events that dismantle apoptotic signaling pathways. Here, we show that ARF is a target of myc activation and that loss of ARF, like loss of p53, can attenuate myc-induced cell death. We suggest that ARF’s normal role is to respond to hyperproliferative signals, thereby facilitating p53 activation through a signaling pathway that differs from those induced by DNA damage.
Results
ARF is induced by explanting MEFs into culture
ARF is not detectably expressed during mouse embryogenesis, and disruption of the gene has no effect on development (Kamijo et al. 1997; Zindy et al. 1997). However, when MEFs were explanted into culture and cells were serially transferred on a 3-day schedule (3T9 protocol), p19ARF was induced at early passages and increased steadily thereafter (Fig. 1, wild-type MEFs). Its accumulation inversely correlates with the rate of MEF cell proliferation, which gradually slows and eventually ceases as cells reach replicative “crisis” (passages 17–20 on this protocol) (Kamijo et al. 1997). Expression of p19ARF in p53-null MEFs was elevated and temporally advanced as compared to that in wild-type cells (Fig. 1), consistent with the ability of ARF and p53 to regulate each other’s levels and activities (see introductory section). In contrast, the loss of p21Cip1, a p53-responsive gene product that negatively regulates progression through the cell cycle (El-Deiry et al. 1993; Harper et al. 1993; Xiong et al. 1993), did not affect ARF levels (Fig. 1).
Figure 1.

Expression of p19ARF in early-passage, primary MEF strains. MEFs of the indicated genotypes (left) propagated on a 3T9 protocol were harvested at passage numbers given at the top, lysed, and immunoblotted for p19ARF protein expression. Equal quantities of protein (200 μg) were loaded per lane.
Although expression of p19ARF in MEFs could connote a role in replicative senescence, the basis for its accumulation was puzzling. One clue was provided by observations that Rb-null MEFs greatly overexpressed p19ARF (Fig. 1), possibly reflecting a propensity of Rb-regulated E2F transcription factors to influence ARF gene expression. A previous survey of INK4a responses to various E2F family members noted that the level of ARF mRNA rose in response to infection of REF52 cells by adenoviruses encoding E2F-1 and, to a lesser extent, E2F-2, but not those specifying E2F3, E2F4, or E2F5 (DeGregori et al. 1997). Second, the ability of ARF-null MEFs to grow continuously and to be transformed by ras alone mimics the effects of myc and E1A on normal MEFs (Land et al. 1983; Ruley 1983). This led us to the idea that some immortalizing function of myc might be dispensable in ARF-null cells. The underlying hypothesis is that ARF normally functions to safeguard cells against sustained and potentially oncogenic hyperproliferative signals (as opposed to DNA damage), thereby explaining why its loss strongly predisposes to tumor development. (For supporting data involving E1A, see de Stanchina et al. 1998).
Induction of ARF by myc
We examined the effects of ectopic myc, Ha-ras (Val-12), and E2F-1 on ARF gene expression by infecting early passage (p5) MEFs with retrovirus vectors expressing these genes, or with a control vector expressing the T cell coreceptor CD8. Wild-type, ARF-null, and p53-null cells were infected three times at 4 hr intervals with high titer replication-defective viruses. By 48 hr after infection, >95% of MEFs infected with the control virus expressed cell-surface CD8, as determined by fluorescence-activated flow cytometry (FACS) using a cognate antibody (data not shown), indicating that virtually all cells were productively infected.
Patterns of various RNAs and proteins expressed 48 hr after infection are illustrated in Figure 2, A and B, respectively. When wild-type MEFs were infected with myc virus, we observed induction of ARF mRNA without significant changes in the levels of INK4a transcripts (Fig. 2A, lanes 1,2). This correlated with increased expression of p19ARF protein without an observable change in p16INK4a (Fig. 2B, lane 2 vs. lane 1). Thus, myc selectively induced ARF expression within the first 2 days after infection. Ectopic myc expression led to 2-fold increases in p53 mRNA levels (Fig. 2A, lanes 1,2) (Hermeking and Eick 1994; Roy et al. 1994) and to 8- to 10-fold increases in p53 protein (Fig. 2B, lanes 1,2), resulting from p53 stabilization (data not shown). This was accompanied by accumulation of the p53-responsive gene products, Mdm2 (Fig. 2B, lane 2), and p21Cip1 (see Fig. 3, below). In contrast, we did not observe effects of myc overexpression on the levels of the Bcl-2 or Bax proteins (data not shown). Infection of wild-type MEFs with a retrovirus vector encoding oncogenic Ha-ras did not affect expression of p19ARF, p53, or Mdm2, although we did observe a slight increase in p16INK4a levels by 48 hr postinfection (Fig. 2B, lane 3). Virtually all wild-type MEFs infected with E2F-1 virus died by apoptosis within 48 hr after infection (Qin et al. 1994; Shan and Lee 1994; Wu and Levine 1994), preventing us from assaying p19ARF protein levels under these conditions.
Figure 2.
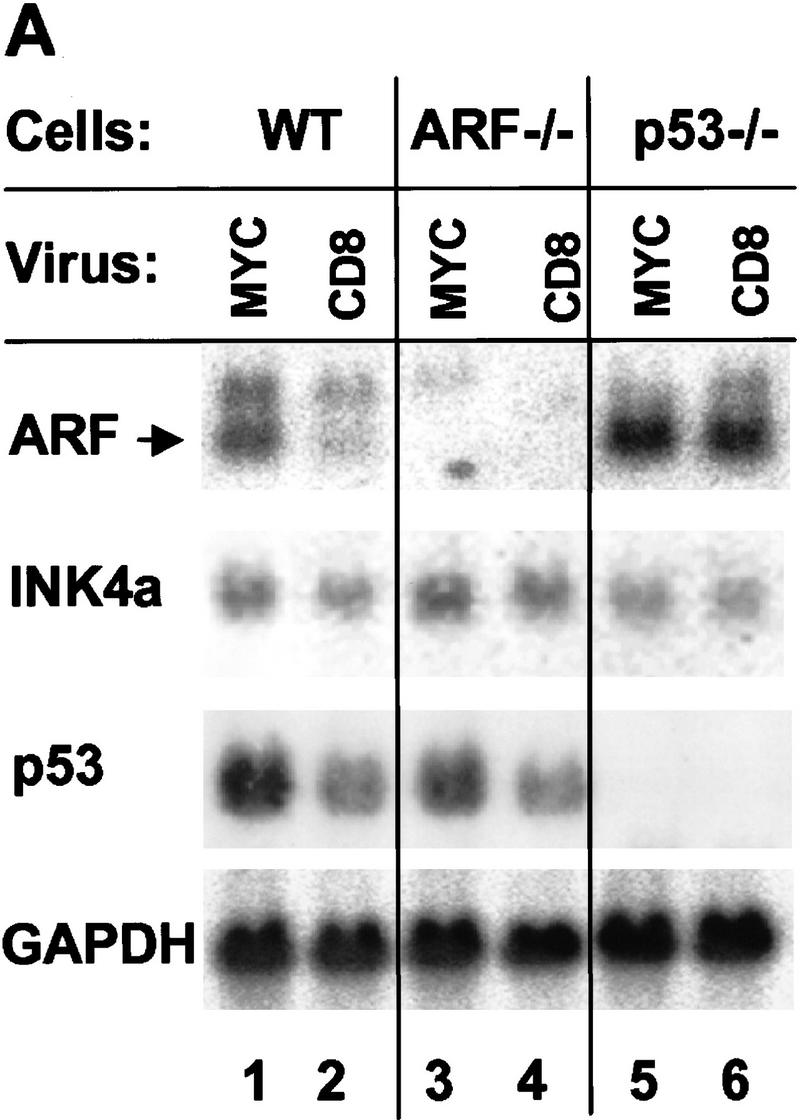
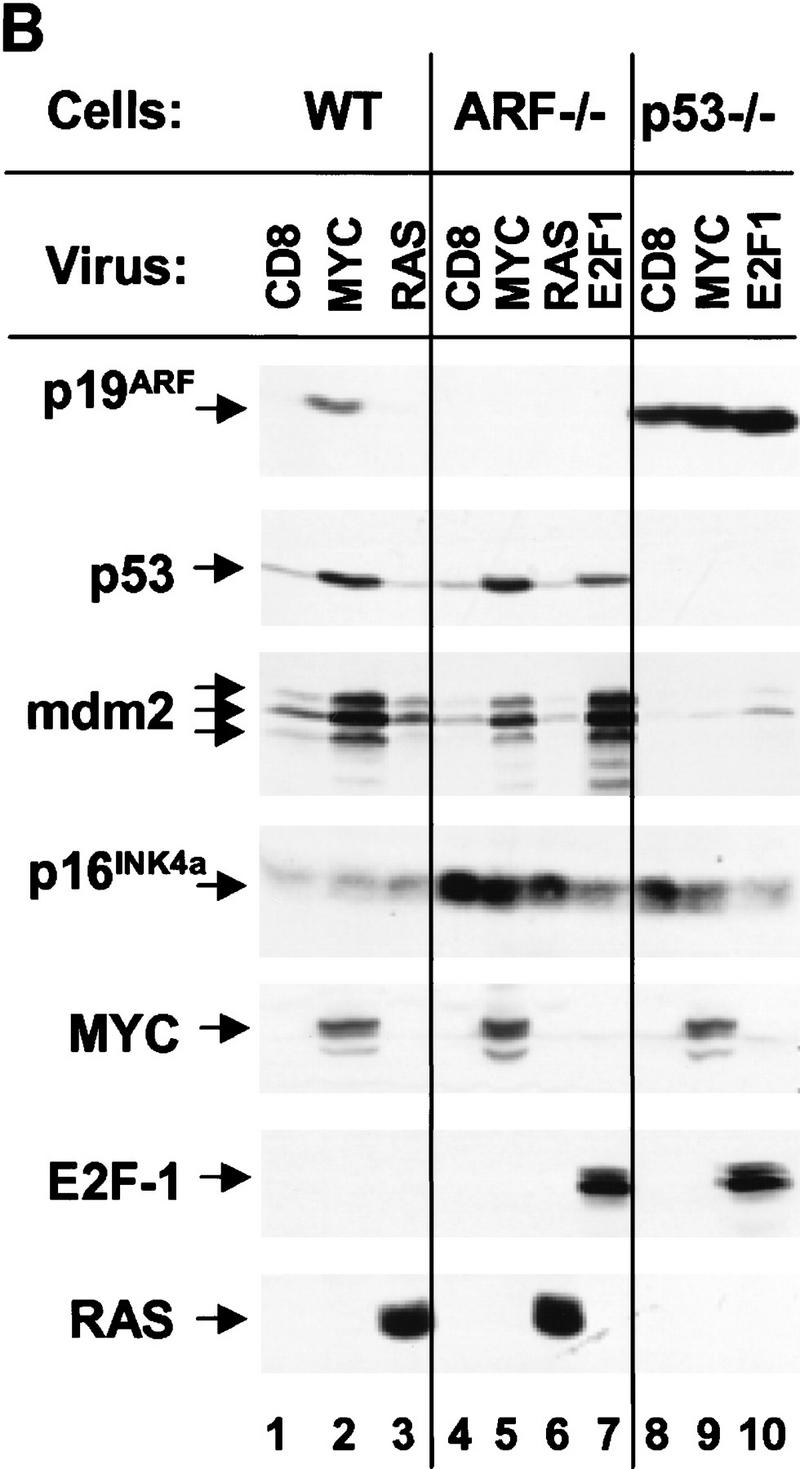
Expression of ARF, p53, and p53 targets in virus-infected MEFs. (A) Wild-type (WT), ARF-null, or p53-null MEFs (top) were infected with either a control (CD8) or myc-expressing retrovirus. At 48 hr postinfection, total RNA was isolated from infected cells, electrophoretically separated, blotted to filters, and hybridized sequentially with 32P-labeled probes specific for ARF (exon 1β), INK4a (exon 1α), p53, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (B) Replicate cultures infected with CD8 or myc viruses, or infected with ras or E2F-1 vectors (top) were lysed 48 hr postinfection and immunoblotted using antibodies directed to the proteins indicated at left. Wild-type cells infected with the E2F-1 virus died and could not be analyzed (see text). Because a smaller fraction of cells from other Myc-infected and E2F-1-infected cultures underwent apoptosis, equal quantities of protein were loaded per lane to provide valid comparisons.
Figure 3.

Induction of ARF, p53, mdm2, and p21Cip1 by Myc–ER. MEFs of the indicated genotypes (top) infected with a myc–ER virus were treated with 4-HT for the indicated intervals (hr), and cell lysates were immunoblotted with antibodies directed to the proteins indicated at left. Levels of Myc–ER expressed in the three cell types were comparable (data not shown).
To assess whether the effects of myc on p53 were ARF-dependent, we infected ARF-null MEFs with myc retrovirus. Here, the introduction of myc also increased expression of p53 mRNA (Fig. 2A, lanes 3,4) and both p53 and Mdm2 proteins (Fig. 2B, lanes 4,5), indicating that the ability of myc to induce p53 was, at least in part, ARF-independent. This activity was observed in response to very high levels of Myc achieved 48 hr post infection, but was less pronounced at later times when Myc levels were reduced (see Fig. 6A, below). Again, oncogenic ras had no such effects (Fig 2B, lane 6). As reported previously, p16INK4a levels are elevated in ARF-null cells (Kamijo et al. 1997), and in this setting, neither Ha-ras nor myc appeared to regulate the protein (Fig. 2B, lanes 5,6).
Figure 6.
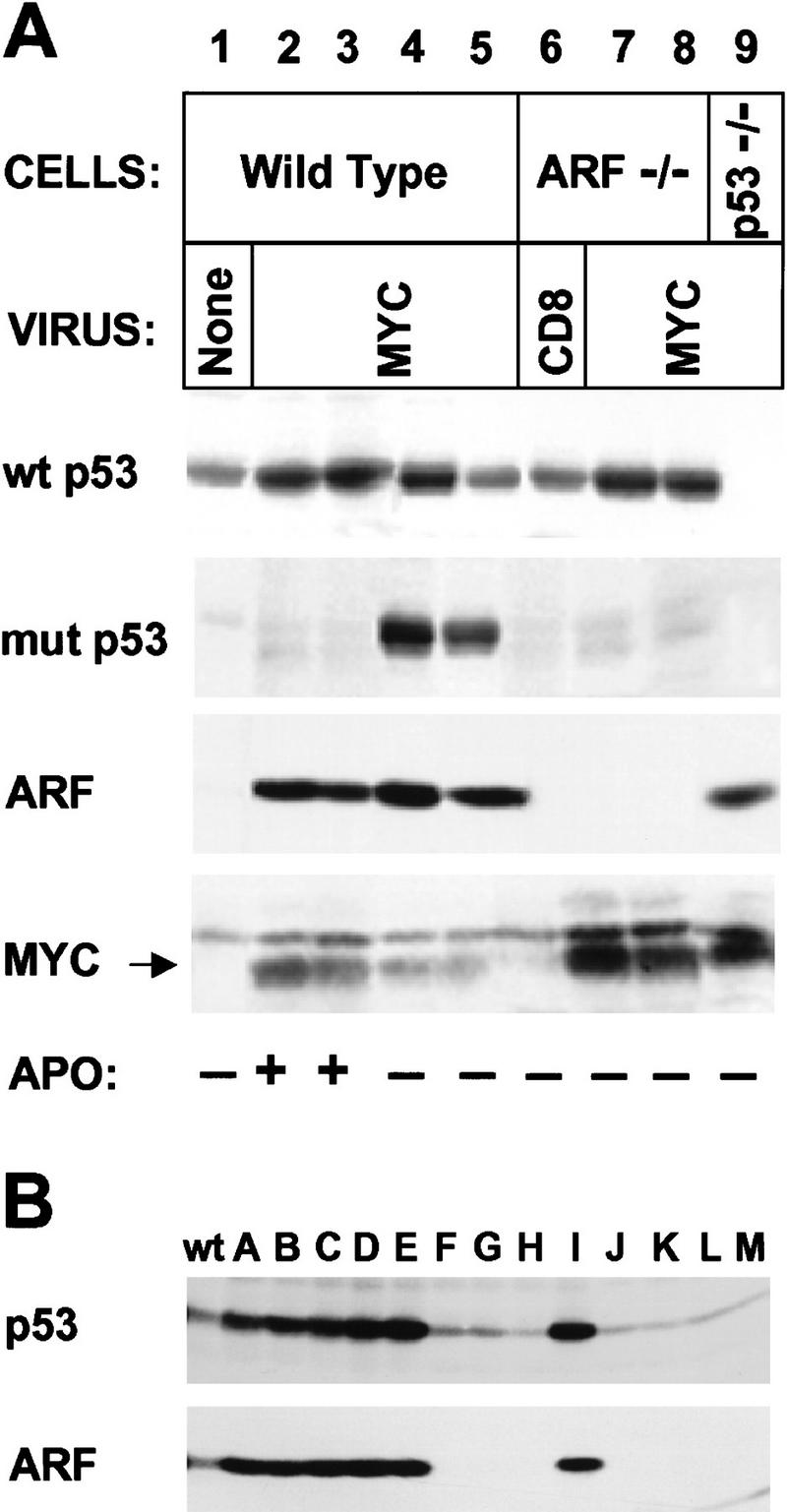
Myc-“immortalized” MEFs lose p53 or ARF function. (A) MEFs of the indicated genotype were infected with CD8 or myc retroviruses at passage 5 after explantation and propagated on a 3T9 protocol. Wild-type cells tested 7–10 days after myc virus infection (lanes 2,3) expressed relatively high levels of p19ARF and wild-type (wt) p53, and were initially sensitive to apoptosis (APO +) when transferred into serum-free medium (see text). However, by 14–21 days postinfection, rapidly growing derivatives were isolated that could grow under serum-free conditions (APO −) and expressed mutant (mut) p53 (lanes 4,5). ARF-null cells infected at passage 5 and transferred 14 days after selection in serum-free medium were resistant to apoptosis but expressed only wild-type p53 (wt) (lanes 7,8). Note that Myc protein levels were significantly higher in ARF-null (lanes 7,8) and p53-null (lane 9) cells than in wild-type MEFs (lanes 2–5). Apoptosis was determined by FACS analysis of propidium iodide- and Hoescht 33342-stained cells. (B) Cells containing a single wild-type ARF allele were infected with myc virus for 4 days and transferred into serum-free medium for 2 days to select for variants resistant to apoptosis. Surviving cells were diluted in microtiter wells and subclones were expanded from single cells in serum-containing medium. Lysates were then blotted for p19ARF and p53. Results with 13 clones (designated A–M) are compared with those obtained with wild-type (wt) uninfected MEFs.
Although p53-null cells express relatively high basal levels of p19ARF, their infection by myc retrovirus further augmented the levels of ARF mRNA (Fig. 2A, lane 5 vs. lane 6) and protein (Fig. 2B, lane 9 vs. lane 8). In four such experiments using p53-null MEFs, enforced myc expression reproducibly elevated p19ARF levels 1.5- to 3-fold, implying that myc can induce ARF via a p53-independent pathway. In contrast, Mdm2 (Fig. 2B, lane 9) and p21Cip1 (shown in Fig. 3, below) were not induced in p53-null cells, indicating that their up-regulation by myc was strictly p53-dependent. Interestingly, ARF-null cells, like p53-null cells (Qin et al. 1994; Shan and Lee 1994; Wu and Levine 1994), were partially resistant to killing by E2F-1, and we were therefore able to document E2F-1 overexpression on a per protein basis (Fig. 2B, lanes 7,10). Like myc, E2F-1 induced both p53 and Mdm2 (lane 7), and Mdm2 induction was p53-dependent (lane 10). Importantly, E2F-1 induced p19ARF in p53-null cells to a level somewhat higher than that seen in myc-infected cells (lanes 10 vs. lane 9), whereas the amounts of p16INK4a were diminished (lanes 7,10). Thus, E2F-1, like myc, induced both p19ARF and p53, and triggered Mdm2 expression in a p53-dependent manner.
To determine the kinetics of the Myc response, we infected MEFs with a retrovirus vector encoding myc fused to the 4-hydroxytamoxifen (4-HT)-responsive domain of the estrogen receptor (ER; Eilers et al. 1991; Littlewood et al. 1995), together with a linked gene encoding resistance to puromycin. Following selection of infected cells for 2 days with puromycin under conditions in which all uninfected MEFs are killed, 4-HT was added to the medium and cells were assayed for p19ARF and p53 protein expression as Myc activity was induced. Figure 3 shows a representative experiment comparing wild-type, ARF-null, and p53-null cells. Ectopic Myc–ER protein levels were equivalent in the three cell lines (data not shown). In wild-type MEFs, 1.8-fold induction of p19ARF was observed within 3 hr of 4-HT treatment, rising to 8.5-fold above the basal level by 24-hr (Fig. 3, lanes 1–5). Induction of p53 was more protracted with a significant elevation (1.8-fold) occurring 6 hr after addition of 4-HT and reaching a maximum (3-fold above basal levels) by 12 hr of treatment. Both Mdm2 and p21Cip1 were induced with kinetics similar to that of p53 (lanes 1–5) but, as expected, were not induced in p53-null cells (lanes 11–15). In these experiments, the constitutively high levels of p19ARF expressed in p53-null cells were not increased further upon 4-HT treatment (lanes 11–15).
In ARF-null cells expressing Myc–ER, p53 levels rose only twofold during the same induction period, in agreement with the concept that p53 induction is partially ARF-dependent (Fig. 3, lanes 6–10). In accord with these findings, induction of the p53-responsive Mdm2 protein was attenuated (lanes 6–10). These results are not consistent with the idea that p19ARF stabilizes p53 by accelerating Mdm2 turnover (Zhang et al. 1998). Basal levels of p21Cip1 are significantly reduced in ARF-null cells (lane 6; see Kamijo et al. 1997), and, surprisingly, no induction of p21Cip1 was seen in response to 4-HT treatment (lanes 6–10). These differences in p53 response between wild-type and ARF-null MEFs were observed in independent experiments using two different Myc–ER-containing vectors (see Materials and methods). Therefore, Myc rapidly induced p19ARF, but in its absence, p53, Mdm2, and p21Cip1 induction were all significantly impaired. Taken together, the above data indicate that (1) myc induces ARF via p53- and Mdm2-independent pathways; (2) myc likely up-regulates p53 through both ARF-dependent and independent pathways; and (3) myc induction of Mdm2 and p21Cip1 is strictly dependent on p53.
ARF loss attenuates myc-induced apoptosis
Apoptosis induced by myc in fibroblasts deprived of serum survival factors (Evan et al. 1992) depends on p53 (Hermeking and Eick 1994; Wagner et al. 1994). Because myc increased the levels of both p19ARF and p53, myc’s ability to trigger apoptosis might also be ARF-dependent. Cells in which the biochemical consequences of myc overexpression had been documented 2 days postinfection (Fig. 2) were expanded in culture for 2 additional days and then shifted into chemically defined medium containing insulin, transferrin, and BSA as the only exogenously added proteins. Under serum-free conditions, myc-infected wild-type MEFs rapidly underwent apoptosis as defined by propidium iodide staining for subdiploid DNA content, visualization of nuclear condensation and blebbing in Hoescht 33342-stained cells, determination of membrane integrity by vital dye exclusion, and TUNEL FACS analysis (Fig. 4A). Similarly, a majority of wild-type MEFs that had been infected with myc–ER virus and induced with 4-HT for 24 hr (Fig. 3) died by apoptosis within a day after transfer to serum-free medium (Fig. 4B, solid symbols). Previously uninduced cells that were shifted into serum-free medium containing 4-HT for 24 hr also died (Fig. 4B, open symbols). In each case, p53-null cells were highly resistant to myc-induced apoptosis, whereas the apoptotic response of ARF-null cells was less compromised (Fig. 4A,B).
Figure 4.
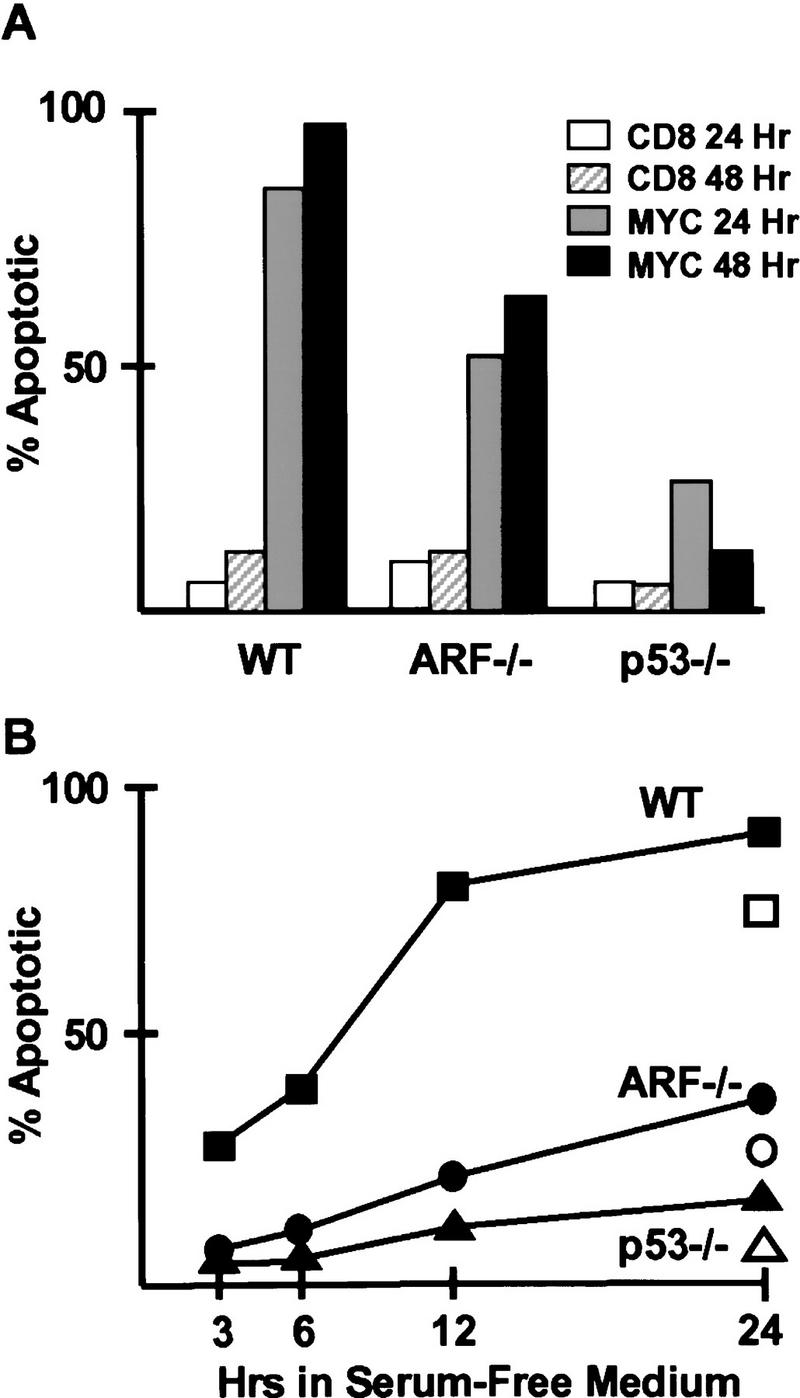
Myc-induced apoptosis. (A) MEFs of the indicated genotypes infected with myc or CD8 virus for 48 hr (the same populations as in Fig. 2) were cultured for 2 more days and then transferred into serum-free medium for an additional 48 hr. Apoptosis was scored using a propidium iodide-based FACS assay to quantitate cells with subdiploid DNA content 24 and 48 hr after serum starvation. Viruses and times of infection are indicated (top right). All standard deviations were within 10% of the means shown. (B) Cells of the indicated genotypes infected with myc–ER virus and pretreated with 4-HT for 24 hr (the same populations as in Fig. 3) were shifted into serum-free medium (solid symbols), and apoptosis was scored by propidium iodide FACs assay at the indicated times (abscissa). Untreated, viable cells were also shifted into serum-free medium containing 4-HT and scored 24 hr later (open symbols).
To explore longer-term effects, cells infected for 4 days with myc- or control CD8 vectors were propagated in either serum-containing or serum-free medium, and their growth rates were determined (Fig. 5). Early passage (p5) wild-type MEFs infected with control CD8 virus proliferated in medium containing serum (Fig. 5A, •) yet underwent only one population doubling in 7 days when serum was removed (Fig. 5A, ○). More than 85% of serum-deprived cells remained viable and arrested in the G1 phase of the cell cycle. However, as first reported by others (Evan et al. 1992), myc-infected MEFs harvested 4 days after infection proliferated less well in the presence of serum (Fig. 5A, ▪) and had a considerably higher apoptotic index (10%–15% TUNEL-positive), so that their rate of growth was in part counterbalanced by cell death. When they were shifted to serum-free medium, cells ectopically expressing myc underwent apoptosis rapidly (Fig. 5A, ). After only 24 hr, the majority were already dead (Figs. 4A and 5), and by 4 days, no viable cells remained.
Figure 5.

Rates of proliferation of virus-infected MEFs. Wild-type (A), ARF-null (B), and p53-null (C) MEFs infected with control CD8 virus were transferred to serum-containing (•) or defined serum-free (○) media 4 days postinfection and counted every day thereafter. Wild-type cells infected with myc virus grew more slowly in serum-containing medium (A, ▪) and died in medium lacking serum (A, ). A significant number of myc-infected ARF-null and p53-null cells survived in serum-free conditions (B, C, ). When reseeded 14 days postinfection, these myc-infected cells grew continuously in serum-free medium (B,C, ▵). All data points represent averages of six to eight determinations using at least three independently derived MEF strains with s.d. less than ±25% of the mean (highly significant on log scale).
Passage 5 ARF-null and p53-null MEFs grew somewhat more rapidly than their wild-type counterparts in the presence of serum (Fig. 5, B and C, •), but still exited the cell cycle when deprived of serum (Fig. 5, B and C, ○). Although myc-infected ARF-null cells transferred to serum-free medium initially underwent apoptosis, a significant fraction survived and continued to proliferate (Fig. 5B, ). By 14 days after infection, these cells were completely resistant to myc-induced apoptosis (data not shown) and grew as rapidly in serum-free medium as did uninfected cells propagated in the presence of serum (Fig. 5B, ▵). Cells lacking p53 were even more resistant to myc-induced apoptosis, undergoing less cell death than ARF-null cells in the first few days after infection (Fig. 5C). All resistant populations continued to express MYC protein ectopically (see Fig. 6, below), confirming that they had been infected. Hence, the effects of myc on apoptosis were significantly attenuated in the absence of ARF or p53 function, and after a few days of selection in serum-free medium, myc ultimately acted as a pure growth promoter.
Although ARF-null and p53-null cells were relatively resistant to myc-induced apoptosis, their response was biphasic. Significant fractions were killed in the first few days after myc virus infection and serum withdrawal, after which resistant cells grew out. Acute phase killing was more severe in ARF-null than in p53-null cells (Fig. 5, B and C, ), consistent with Myc’s ability to target p53 through an ARF-independent pathway (see above). In addition, very high levels of Myc were achieved in the first 1–3 days after infection, but declined as infected MEFs were propagated and were reduced by almost 80% by the time that cells became completely resistant to apoptosis (about day 14) (data not shown, but see Fig. 6, below). Acutely elevated levels of Myc also killed a fraction of p53-null cells (Fig. 4A), accounting for their initial growth lag in serum-free medium (Fig. 5C, ). A critical issue is whether myc overexpression could have selected for additional genetic changes that obviated a requirement for ARF function. To test this possibility, we reintroduced the ARF gene into surviving ARF-null myc overexpressors that had acquired the ability to proliferate in serum-free medium. Reinfection of these cells with an ARF but not control CD8 retrovirus resensitized them to apoptosis in serum-free medium (42% viability in ARF-infected cells vs. >90% in CD8-infected cells at 24 hr postinfection). Therefore, resistance to apoptosis was a direct consequence of ARF loss and was not due to mechanisms that bypass ARF function. Similar effects were observed using E1A in lieu of myc (de Stanchina et al. 1998).
Myc-induced apoptosis selects for cells that lose either p53 or ARF function
Because myc overexpression in wild-type MEFs induces apoptosis and slows their overall proliferative rate in serum-containing medium (Fig. 5A, closed symbols), we reasoned that continued passage of these cells might select for resistant, more rapidly proliferating variants that spontaneously lose ARF or p53 function. myc-virus-infected wild-type strains maintained in serum-containing medium and studied 7–10 days after infection initially remained sensitive to apoptosis when deprived of serum. By this time, the cells synthesized very high levels of p19ARF (Fig. 6A, lanes 2,3) equivalent to those seen in p53-null cells (lane 9). To distinguish wild-type from mutant p53, cells were metabolically labeled with [35S]-methionine for 2 hr, and lysates were precipitated using conformation-specific antibodies (Yewdell et al. 1986; Gannon et al 1990). myc infection increased the rate of wild-type p53 synthesis (Fig. 6A, lanes 2 and 3 vs. lane 1), consistent with induction of p53 mRNA (Fig. 2A). Because of its longer half-life, the steady-state levels of p53 in myc-infected versus CD8 virus-infected cells, as judged by immunoblotting, differed even more significantly (Fig. 2B). At this time after infection, no mutant forms of p53 were detected (lanes 2,3).
By 14–21 days after infection, wild-type MEFs infected with myc virus and maintained in medium containing serum no longer underwent apoptosis when transferred to serum-free medium and continued to proliferate as established cell lines. Emerging variants were readily identified by their much smaller size (1.11 pL vs. 3.3 pL mean corpuscular volume), accelerated growth rate, and ability to proliferate in serum-free medium. Four such independently derived cell lines expressed mutant, dominant-negative forms of p53 in addition to the wild-type form of the protein (Fig. 6A, lanes 4 and 5 show results with two such lines). In contrast, myc-infected ARF-null cell lines growing in serum-free conditions expressed only wild-type p53 (Fig. 6A, lanes 7,8). Therefore, myc-induced immortalization of wild-type cells selected for p53 loss of function, but such selection was obviated in cells lacking ARF.
In general, ARF-null cells tolerated higher levels of ectopic Myc protein than did wild-type MEFs that had acquired p53 mutations in the course of infection (cf. Myc levels in lanes 7 and 8 with those in lanes 4 and 5). Moreover, wild-type MEFs that were initially sensitive to Myc-induced apoptosis expressed higher levels of Myc than did resistant variants (cf. Myc levels in Fig. 6, lanes 2 and 3 vs. lanes 4 and 5). These results are consistent with the idea that high levels of Myc are selected against by apoptosis until resistant variants emerge.
In continuing studies of spontaneously immortalized wild-type MEFs that emerged from crisis on a 3T9 protocol, we determined that 23 of 28 individually derived cell lines had sustained p53 mutations, whereas the remainder exhibited biallelic loss of ARF. In principle, biallelic ARF loss might also occur during myc-induced establishment, but this should again be a less frequent event than p53 mutation, involving two-hit versus one-hit kinetics (Kamijo et al. 1997; Zindy et al. 1997). Moreover, because myc virus-infected populations are polyclonal, attempts to demonstrate biallelic ARF loss in a subset of cells would be occluded by the presence of other cells in the population containing mutant p53 and expressing high levels of p19ARF. To determine whether ARF loss can also occur in response to enforced Myc expression, MEFs hemizygous for a wild-type ARF allele were infected with myc virus, propagated in serum-free medium for 2 days, and then shifted back into medium containing serum. Surviving cells were subcloned by limiting dilution, expanded, and then assayed for p19ARF expression and for the presence of wild-type and mutant p53. Of 26 clones, eleven exhibited p53 mutations, whereas the other 15 lacked detectable p19ARF. Figure 6B shows results with 13 representative clones designated A–M. Mutant p53 was expressed at high levels (clones A–E and I) compared to those in uninfected wild-type MEFs. As expected, clones with mutant p53 also expressed higher levels of p19ARF than wild-type cells. In contrast, ARF-null variants (clones F–H and J–M) expressed low levels of wild-type p53. Southern blotting confirmed the loss of the wild-type ARF allele in the latter cases (data not shown). Therefore, immortalization of wild-type MEFs by myc leads to either ARF or p53 loss and confers resistance to Myc-induced apoptosis.
Discussion
Signaling to ARF and p53
Cells protect themselves from mutant cancer genes (i.e., mutated oncogenes or loss of tumor suppressors) through compensatory mechanisms that arrest cell growth or induce cell suicide (for review, see Sherr 1996; Weinberg 1997). Expression of activated ras in primary MEFs inhibits cell growth (Serrano et al. 1997), whereas overexpression of myc in these same cells triggers apoptosis, a process further aggravated by withdrawal of serum survival factors (Evan et al. 1992). Yet, introduction of myc and ras together into primary rodent embryo fibroblasts elicits cell transformation (Land et al. 1983). myc must somehow block ras-mediated inhibition of cell proliferation, whereas conversely, ras may play a role in attenuating the apoptotic function of Myc (Weinberg 1997).
One hypothesis is that cultured cells achieve replicative immortality by inactivating their INK4a or p53 genes (for review, see Weinberg 1997). To some extent, this idea was based on the ability of MEFs from INK4a/ARF-null mice to grow continuously after explantation into culture and to be transformed by oncogenic ras alone (Serrano et al. 1996; 1997). Yet, MEFs from mice lacking ARF alone exhibit the immortalized features previously attributed to disruption of INK4a, implying that loss of p19ARF in lieu of p16INK4a enables oncogenic ras alleles to transform these cells. Given that Ras positively regulates the synthesis of D-type cyclins and their assembly with CDK4 (Cheng et al. 1998 and references therein), a role for p16INK4a in antagonizing these growth promoting activities of Ras would be expected (see Fig. 7 for schematic). Pomerantz et al. (1998) recently demonstrated that overexpression of ARF in rat embryo fibroblasts transformed by myc plus ras was significantly more potent than INK4a in suppressing transformation. Moreover, they found that p19ARF, but not p16INK4a, suppressed transformation by E1A plus Ras in a p53-dependent manner, consistent with the idea that p19ARF acts downstream of pRb (and E2F-1) in countering oncogenic signaling (Fig. 7). We therefore propose that either p19ARF or p53 inactivation provides an immortalizing function that mimics certain actions of Myc and E1A and renders primary MEFs more susceptible to ras-induced transformation. Clearly, this model does not preclude a requirement for other growth-promoting functions of Myc and E1A in immortalizing wild-type cells.
Figure 7.

Model for ARF signaling. ARF is activated via Myc and E2F-1 and acts in turn to trigger p53-dependent cell cycle arrest or apoptosis, depending on the presence of extracellular survival factors. Ras acts through cyclin D-dependent kinases to stimulate pRB phosphorylation, resulting in release of E2F from pRb constraint and activation of E2F-responsive genes. Activation of ARF by MYC and E2F-1 need not be direct, although both transcription factors have been demonstrated to increase ARF mRNA levels (see text). Like Myc, different E2F isoforms are proposed to regulate both cell growth and cell death. In inhibiting cyclin D-dependent kinases, p16INK4a can modulate certain growth-promoting functions of Ras. Other functions of Myc and Ras are not detailed in the schematic.
Overexpressed myc can signal through p19ARF and p53 to trigger apoptosis, although its effects can be overridden by serum survival factors (Fig. 7). Overexpression of myc induces the accumulation of p19ARF, at least in part by increasing ARF gene expression. Induction of p19ARF synthesis by a conditionally active Myc–ER fusion protein occurred within 3 hr of 4-HT treatment and temporally preceded p53 accumulation and p53-dependent expression of Mdm2 and p21Cip1. Although we have found that myc can induce p53 through an ARF-independent pathway, its induction of p53 and p53-responsive gene products is significantly compromised in ARF-null cells. For unexplained reasons, we observed greater attenuation of the p21Cip1 response than that of Mdm2 in ARF-null cells, implying that not all p53-responsive genes are equally affected by ARF loss. In contrast, other signals that induce p53, such as DNA damage by radiation, are effective in the complete absence of ARF (Kamijo et al. 1997). Therefore, myc signals to p53 at least in part through an ARF-dependent pathway, which is distinct from that triggered by DNA damage.
ARF and Myc-induced apoptosis
Because enforced expression of ARF itself arrests wild-type MEFs but does not kill them (Quelle et al. 1995b), a function of Myc other than ARF induction is required to trigger apoptosis. Nonetheless, the loss of either ARF or p53 confers significant resistance to myc-induced cell death, and these effects of ARF, like its ability to induce cell cycle arrest, are p53-dependent. In cultures of wild-type MEFs acutely infected with myc retrovirus, a significant proportion of the cells underwent apoptosis even when grown in the presence of serum. In the face of Myc overexpression, there was a strong selective advantage for cells that sustained p53 mutations, and once such variants emerged, these soon predominated and were able to continuously proliferate in chemically defined medium lacking serum. Results using myc-infected MEFs containing a single functional ARF allele demonstrated that ARF loss, rather than p53 mutation, could also lead to establishment, in agreement with previous observations made with cells that had undergone spontaneous immortalization (Kamijo et al. 1997). As in the latter cases, loss of ARF or p53 function appeared to be mutually exclusive events, indicating that ARF loss can relieve myc-induced selective pressure for p53 mutation.
A conceptual dilemma is posed by observations that ARF-null cells infected with myc virus were initially sensitive to apoptosis when shifted into serum-free medium, although significantly less so than wild-type MEFs. After several days in serum-free medium, apoptosis was no longer detected, and the myc virus-infected, ARF-null cells again grew rapidly. This raised the possibility that myc overexpression selected for additional cryptic genetic changes that rendered the cells resistant to apoptosis. However, when cells that had resumed proliferation in serum-free medium were infected with an ARF virus, they promptly died, implying that attenuation of apoptosis was a direct consequence of ARF loss. Therefore, we favor the interpretation that the high levels of myc expression achieved acutely after virus infection were able to kill cells through an ARF-independent pathway, likely involving p53 directly. Myc levels fell as infected MEFs were propagated, and because both ARF-null and p53-null cells tolerate higher levels of Myc than wild-type cells, they appear to become resistant to apoptosis without further selection.
ARF function in tumor surveillance
Other immortalizing oncogenes, such as adenovirus E1A, can act like myc in triggering apoptosis in an ARF-dependent manner (see de Stanchina et al. 1998). Among its many effects, E1A releases E2F1, E2F2, and E2F3 from pRb constraint; E2F-1 can selectively induce the ARF gene (DeGregori et al. 1997) and protein expression, and trigger apoptosis in a p53-dependent manner (Wu and Levine 1994; Qin et al. 1994; Shan and Lee 1994; Kowalik et al. 1995). In agreement with these findings, MEFs lacking Rb exhibited relatively high levels of p19ARF expression, and E1A mutants that are unable to interact with pRb were handicapped in their ability to induce p19ARF (de Stanchina et al. 1998). Because Rb-null MEFs undergo replicative senescence in culture (F. Zindy et al., unpubl.), high p19ARF levels should sensitize them to apoptosis as long as p53 function is intact. Similarly, in an in vivo mouse model using the developing murine lens, Rb deficiency triggers apoptosis in a largely p53-dependent manner (Morgenbesser et al. 1994). Lenses from animals lacking exon 2 of the INK4a gene, and hence likely disrupted for both INK4a and ARF function, exhibited less apoptosis than wild-type lenses but more than that observed in a p53-null background (Pomerantz et al. 1998).
Unlike ARF, p53 also integrates signals emanating from DNA-damage response pathways. Cancer cells are generally considered to have conserved normal p53 function if they retain wild-type p53 and exhibit an intact p53-dependent DNA damage checkpoint response. However, if such cells lack ARF, they are still compromised in their p53 response, because they would fail to respond to hyperproliferative signals induced by oncogenes such as myc. The fact that hyperproliferative signals and DNA damage pathways can collaborate to induce p53 suggests that cells sustaining oncogenic stimulation would initially be more susceptible than their normal counterparts to chemotherapeutic drugs and to radiotherapeutic regimens that induce DNA damage (de Stanchina et al. 1998). Loss of ARF would disable this synergy, making tumor cells more resistant to treatment and ultimately selecting for p53 loss in the face of higher dose therapy. ARF function may have evolved to harness the apoptotic machinery precisely for the purpose of preventing abnormal cell growth in response to oncogenic signals. This would explain why its loss is such a common event in many different forms of cancer.
Materials and methods
Cell culture
MEFs from day 14.5 embryos (wild-type, ARF-null, p53-null, p21-null) or day 13.5 embryos (Rb-null) were explanted and maintained on a 3T9 protocol (9 × 105 cells transferred at 3-day intervals) (Kamijo et al. 1997) and propagated in Dulbecco’s modified Eagle’s medium (DMEM) plus 10% fetal bovine serum, 2 mm glutamine, 0.1 mm nonessential amino acids, 55 μm 2-mercaptoethanol, and 10 μg/ml gentamycin (GIBCO). ARF-null cells were established in our laboratory. MEFs from p53-null cells were derived from mice purchased from the Jackson Laboratories. Timed p21-null and Rb-null pregnant females were generously provided by Stephen Elledge (Baylor College of Medicine, Houston, TX) and Tyler Jacks (MIT, Cambridge, MA), respectively. Where indicated, cultured MEFs at passage 5 were switched to defined, serum-free medium containing insulin, transferrin, and BSA as the only added proteins (Roussel and Sherr 1989). At the same time, cells infected for 48 hr with the indicated retroviruses were diluted and plated at 2 × 104 cells/60-mm-diam. dish in 4 ml of complete medium. The following day, fresh medium containing or lacking serum was added, and cells from replicate cultures were counted every day thereafter. Viability was determined by trypan blue exclusion, and DNA fragmentation was monitored using a terminal deoxynucleotidyl transferase (FACS TUNEL) assay (Gorczya et al. 1993) and by measurement of subdiploid (<2N) DNA content of propidium iodide-stained nuclei (Askew et al. 1991). Where indicated, cells grown for 10–14 days post-infection in complete serum-containing medium were diluted as above and their kinetics of proliferation and survival in serum-free medium were reassessed.
Cells hemizygous for a functional ARF allele were infected with myc retrovirus, and 4 days postinfection were transferred into serum-free medium for 2 days. Survivors were plated in complete medium at limiting dilution in 96-well microtiter plates, and 26 clones derived from single cells were expanded and assayed for p19ARF and p53 proteins as shown in Figure 6B. Cells lacking ARF were confirmed by Southern blotting to have segregated the residual wild-type allele. The presence of mutant and wild-type p53 was confirmed by immunoprecipitation of metabolically labeled cell lysates with conformation-specific antibodies (see below).
Virus infection
Human kidney 293T cells were from Dr. David Baltimore (California Institute of Technology, Pasadena). A helper ecotropic retrovirus plasmid defective in Θ2 packaging sequences, and pSRα vectors containing human c-myc (Roussel et al. 1995) and CD8 (Quelle et al. 1995b) were provided by Dr. Charles Sawyers (UCLA). The human E2F1 cDNA, provided by Dr. Scott Hiebert (Vanderbilt University, Nashville, TN), or oncogenic Ha-ras (Val12) cDNA, from Dr. Michael M. White (Southwestern Medical Center, Dallas, TX), was cloned into the same vector in place of c-myc. A myc–ER retroviral vector containing a linked gene for puromycin-resistance was provided by Drs. Dean Felsher and J. Michael Bishop (UCSF). The ER moiety is unable to bind estrogen yet retains its affinity for the synthetic ligand, 4-HT (Littlewood et al. 1995). The cDNA cassette encoding myc–ER was also expressed in the pSRα vector and MEFs infected with this retrovirus yielded similar results to those shown in Figures 3 and 4B. Viruses produced by cotransfection of 293T cells with vector and helper virus plasmids (Roussel et al. 1995) were harvested every 6 hours, 24–72 hours after transfection. Pooled, filtered supernatants (three successive additions of 2 ml at 4-hr intervals) were used to infect naive primary MEF strains (2 × 105 cells plated/100-mm-diam. dishes) in the presence of 10 μg/ml Polybrene (Sigma, St. Louis, MO). At 12 hr postinfection, 10 ml of fresh medium was added, and medium was changed 24 hr later. Cells infected with Myc–ER virus were selected in 2 μg/ml puromycin for 48 hr prior to treatment of surviving cells with 1 μm 4-HT for times indicated in Figure 3.
RNA and protein expression
Total RNA was separated electrophoretically in gels containing formaldehyde (20 μg/lane), blotted to nitrocellulose, and detected using 32P-labeled probes specific for exons 1α (INK4a) and 1β (ARF) of the mouse ARF–INK4a locus (Quelle et al. 1995b; Zindy et al. 1997). Proteins were detected by direct immunoblotting. Frozen cell pellets (∼ 2 mg protein) were lysed on ice in Tween 20 lysis buffer (50 mm HEPES at pH 7.5, 150 mm NaCl, 1 mm EDTA, 2.5 mm EGTA, 0.1% Tween 20, 1 mm PMSF, 0.4 U/ml aprotinin, 1 mm NaF, 10 mm β-glycerophosphate, and 0.1 mm Na orthovanadate), sonicated 2 × 7 sec (Virtis VirSonic 475, 12%–14% power), and left on ice for 30 min. Debris was removed by sedimentation at 4°C in a microcentrifuge (5 min at 15,000 rpm), and protein was quantitated using a BCA kit (Pierce, Rockford IL). Samples (200 μg of protein per lane) were separated by SDS–PAGE and transferred to nitrocellulose membranes (MSI, Westboro MA). Filters were washed in TBS-Tween (10 mm Tris HCl at pH 7.4, 150 mm NaCl, 0.1% Tween 20) and blocked in the same solution with 10% (wt/vol) nonfat dry milk. Filters were then exposed for 1–2 hr at room temperature to either 0.2 μg/ml affinity-purified rabbit antibody to the mouse p19ARF carboxyl terminus (Quelle et al. 1995b) or the p16INK4a carboxyl terminus (Quelle et al. 1995a); rabbit antiserum to E2F-1 (from Scott Hiebert); or monoclonal antibodies directed to p53 (Ab-7, Calbiochem La Jolla, CA), Mdm2 (2A10 provided by Gerard Zambetti, St. Jude Children’s Reseach Hospital), p21Cip1 (F-5, Santa Cruz Biochemicals, CA), human Myc (06340, Upstate Bio., Inc) or p21ras (rat mAb 259, Santa Cruz Biochemicals). Those filters exposed to affinity-purified rabbit antibodies to p19ARF were washed for 45 min in TBS–Tween and incubated 45 min with a 1/2000 dilution of donkey antibodies to rabbit IgG (Amerham) in TBS–Tween containing 5% milk. All filters were then rewashed as described above and antibody binding sites were visualized by enhanced chemiluminescence using appropriate second antibody conjugates or horseradish peroxidase-conjugated protein A (for p16INK4a) as per the manufacturer’s instructions (ECL kit, Amersham). For discrimination of mutant and wild-type forms of p53, cells were metabolically labeled with [35S]methionine and lysed, and cleared lysates were precipitated with antibodies that detect either mutant or wild-type forms of the protein (Yewdell et al. 1986; Gannon et al. 1990) as described previously (Kamijo et al. 1997).
Acknowledgments
We thank Tyler Jacks and Stephen Elledge for pregnant female Rb-null and p21-null mice, respectively; Scott Hiebert for E2F-1 cDNA and antiserum to the protein; Michael White for ras cDNA; Arnold Levine and Gerard Zambetti for antibodies to Mdm2; Charles Sawyers and J. Michael Bishop for retroviral vectors; David Baltimore for 293T cells; and Richard A. Ashmun and Richard Cross for assistance with FACS analysis. We also appreciate the excellent technical support of Joseph Watson, Carol Bockhold, Esther Van de Kamp, Rose Mathew, and Zhen Lu, and helpful suggestions from other members of our laboratory. This work was supported in part by National Institutes of Health grants CA-71907 and CA-56819 (M.F.R.), DK-44158 (J.L.C.), Cancer Center Core grant (CA-21765), and by the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL sherr@stjude.org; FAX (901) 495-2381.
References
- Askew DS, Ashmun RA, Simmons BC, Cleveland JL. Constitutive c-mycexpression in an IL3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene. 1991;6:1915–1922. [PubMed] [Google Scholar]
- Barak Y, Juven T, Haffner R, Oren M. Mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–468. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1regulated by mitogen-activated protein kinase kinase (MEK1) Proc Natl Acad Sci. 1998;95:1091–1096. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes & Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins J. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Stanchina, E., M.E. McCurrach, F. Zindy, S-Y. Shieh, G. Ferbeyre, A.V. Samuelson, C. Prives, M.F. Roussel, C.J. Sherr, and S.W. Lowe. 1998. E1A signaling to p53 involves the p19ARF tumor suppressor. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Eilers M, Schirm S, Bishop JM. The MYC protein activates transcription of the α-prothymosin gene. EMBO J. 1991;10:133–141. doi: 10.1002/j.1460-2075.1991.tb07929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer E, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Water CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-mycprotein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Franza BR, Maruyama K, Garrels JI, Ruley HE. In vitro establishment is not sufficient prerequisite for transformation by activated ras oncogene. Cell. 1986;44:409–418. doi: 10.1016/0092-8674(86)90462-9. [DOI] [PubMed] [Google Scholar]
- Gannon JV, Greaves R, Iggo R, Lane DP. Activating mutations in p53 produce a common conformational effect: A monoclonal antibody specific for the mutant form. EMBO J. 1990;9:1595–1602. doi: 10.1002/j.1460-2075.1990.tb08279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorczya W, Gong J, Darzynkiewicz Z. Detection of DNA strand breaks in individual apoptotic cells by the in situ terminal deoxynucleotidyl transferase and nick translation assays. Cancer Res. 1993;53:1945–1951. [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- Hicks GC, Egan SE, Greenberg AH, Mowat M. Mutant p53 tumor suppressor alleles release ras-induced cell cycle growth arrest. Mol Cell Biol. 1991;11:1344–1352. doi: 10.1128/mcb.11.3.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa T, Ruley HE. Rescue of cells from ras oncogene-induced growth arrest by a second, complementing oncogene. Proc Natl Acad Sci. 1991;85:1519–1523. doi: 10.1073/pnas.85.5.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Letts. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Kern SE, Pietenpol JA, Thiagalingam S, Seymour A, Kinzler K, Vogelstein B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science. 1992;256:827–830. doi: 10.1126/science.1589764. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:641–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Interactions of the ARF tumor suppressor with p53 and Mdm2: Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci. 1998;95:8292–8297. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalik TF, DeGregori J, Schwarz JK, Nevins JR. E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J Virol. 1995;69:2491–2500. doi: 10.1128/jvi.69.4.2491-2500.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- Lin H-J, Eviner V, Prendergast GC, White E. Activated H-ras rescues E1A-induced apoptosis and cooperates with E1A to overcome p53-dependent growth arrest. Mol Cell Biol. 1995;15:4536–4544. doi: 10.1128/mcb.15.8.4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE. Stabilization of the p53 tumor suppressor is induced by adenovirus-5 E1A and accompanies apoptosis. Genes & Dev. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- Morgenbesser SD, Williams BO, Jacks T, DePinho RA. p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature. 1994;371:72–74. doi: 10.1038/371072a0. [DOI] [PubMed] [Google Scholar]
- Newbold RF, Overell RO. Fibroblast immortality is a prerequisite for transformation by EJ c-Ha-ras oncogene. Nature. 1983;304:648–651. doi: 10.1038/304648a0. [DOI] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Lígeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee H-W, Cordon-Cardo C, DePinho R. The Ink4a tumor suppressor gene product, p19ARF, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Qin XQ, Livingston DM, Kaelin WG, Jr, Adams PD. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci. 1994;91:10918–19022. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quelle DE, Ashmun RA, Hannon GJ, Rehberger PA, Trono D, Richter H, Walker C, Beach D, Sherr CJ, Serrano M. Cloning and characterization of murine p16INK4a and p15INK4bgenes. Oncogene. 1995a;11:635–645. [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995b;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Rao L, Debbas M, Sabbatini P, Hockenbery D, Korsmeyer S, White E. The adenovirus E1A proteins induce apoptosis which is inhibited by the E1B 19K and Bc1-2 proteins. Proc Natl Acad Sci. 1992;89:7742–7746. doi: 10.1073/pnas.89.16.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel MF, Sherr CJ. Mouse NIH/3T3 cells expressing human CSF-1 receptors overgrow in serum-free medium containing human CSF-1 as their only growth factor. Proc Natl Acad Sci. 1989;86:7924–7927. doi: 10.1073/pnas.86.20.7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel MF, Theodoras AM, Pagano M, Sherr CJ. Rescue of defective mitogenic signaling by D-type cyclins. Proc Natl Acad Sci. 1995;92:6837–6841. doi: 10.1073/pnas.92.15.6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B, Beamon J, Balint E, Reisman D. Transactivation of the human p53 tumor suppressor gene by c-Myc/Max contributes to elevated mutant p53 expression in some tumors. Mol Cell Biol. 1994;14:7805–7815. doi: 10.1128/mcb.14.12.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruley HE. Adenovirus early region 1A enables viral and cellular transforming genes to transform primary cells in culture. Nature. 1983;304:602–606. doi: 10.1038/304602a0. [DOI] [PubMed] [Google Scholar]
- ————— . Transforming collaborations between ras and nuclear oncogenes. In: Vande Woude GF, Levine AJ, Topp WC, Watson JD, editors. Cancer cells 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. pp. 258–268. [PubMed] [Google Scholar]
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H-W, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p153 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shan B, Lee WH. Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol Cell Biol. 1994;14:8166–8173. doi: 10.1128/mcb.14.12.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Wagner AJ, Kokontis JM, Hay N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21 waf1/cip1. Genes & Dev. 1994;8:2817–2830. doi: 10.1101/gad.8.23.2817. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The cat and mouse games that genes, viruses, and cells play. Cell. 1997;88:573–575. doi: 10.1016/s0092-8674(00)81897-8. [DOI] [PubMed] [Google Scholar]
- White E, Cipriani R, Sabbatini P, Denton A. The adenovirus E1B 19-kilodalton protein overcomes the cytotoxicity of E1A proteins. J Virol. 1991;65:2968–2978. doi: 10.1128/jvi.65.6.2968-2978.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Levine AJ. p53 and E2F-1 cooperate to mediate apoptosis. Proc Natl Acad Sci. 1994;91:3602–3606. doi: 10.1073/pnas.91.9.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes & Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- Yewdell JW, Gannon JV, Lane DP. Monoclonal antibody analysis of p53 expression in normal and transformed cells. J Virol. 1986;59:444–452. doi: 10.1128/jvi.59.2.444-452.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4alocus deletion impairs both the Rb and p53 tumor suppressor pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4atumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]