

Abstract
The adenovirus E1A oncogene activates p53 through a signaling pathway involving the retinoblastoma protein and the tumor suppressor p19ARF. The ability of E1A to induce p53 and its transcriptional targets is severely compromised in ARF-null cells, which remain resistant to apoptosis following serum depletion or adriamycin treatment. Reintroduction of p19ARF restores p53 accumulation and resensitizes ARF-null cells to apoptotic signals. Therefore, p19ARF functions as part of a p53-dependent failsafe mechanism to counter uncontrolled proliferation. Synergistic effects between the p19ARF and DNA damage pathways in inducing p53 may contribute to E1A’s ability to enhance radio- and chemosensitivity.
Keywords: E1A signaling, p53, p19ARF tumor suppressor
Tumor-specific mutations identify genes essential for normal growth control and reveal fundamental processes involved in tumorigenesis. Similarly, viral oncoproteins target cellular proteins critical for malignant transformation—often the same activities altered by spontaneous mutation in cancer cells. For example, many DNA tumor viruses encode proteins that bind and inactivate both p53 and the retinoblastoma (Rb) protein, and inactivation of both is essential for viral transformation (Lane and Crawford 1979; Linzer and Levine 1979; DeCaprio et al. 1988; Whyte et al. 1988a; Dyson et al. 1989; Werness et al. 1990). Consistent with the relevance of these interactions, p53 and Rb are frequently mutated in human tumors (for review, see Greenblatt et al. 1994; Weinberg 1995).
Although the high frequency of p53 mutations in human cancer implies a central role for p53 in tumorigenesis, the signals that trigger p53 in suppressing tumor growth remain poorly defined. p53 is a sequence-specific DNA-binding protein that promotes cell-cycle arrest or apoptosis in response to a variety of cellular stresses (for examples, see Kastan et al. 1991; Graeber et al. 1994; Linke et al. 1996; for review, see Ko and Prives 1996; Levine 1997). For example, p53 levels and activity increase following DNA damage owing, in part, to de novo phosphorylation and the accompanying conformational changes (Shieh et al. 1997; Siliciano et al. 1997). Phosphorylation at serine-15 prevents p53’s interaction with Mdm2 (Shieh et al. 1997), a protein that can down-regulate p53 via ubiquitin-mediated proteolysis (Haupt et al. 1997; Kubbutat et al. 1997). In principle, failure of p53 to suppress proliferation following DNA damage might indirectly promote tumor development by allowing the growth and survival of cells with mutations (Livingstone et al. 1992; Yin et al. 1992; Griffiths et al. 1997), but whether this provides the primary driving force for p53 mutation in tumors is unclear.
Oncogenes can also induce p53, leading to increased apoptosis or premature senescence (Lowe and Ruley 1993; Hermeking and Eick 1994; Wagner et al. 1994; Serrano et al. 1997). For example, the adenovirus E1A oncogene induces p53 and promotes apoptosis in primary cells (Debbas and White 1993; Lowe and Ruley 1993; Querido et al. 1997; Samuelson and Lowe 1997), which is reflected by E1A’s remarkable ability to enhance radio- and chemosensitivity (Lowe et al. 1993). Although E1A is a mitogenic oncogene, p53 acts to limit its oncogenic potential. Thus, p53-deficient primary fibroblasts expressing E1A are resistant to apoptosis and become oncogenically transformed (Lowe et al. 1994b). Two E1A domains act in concert to promote p53 accumulation and apoptosis in primary cells; the first inactivates Rb, whereas the second binds the p300/CBP transcriptional coactivators (Samuelson and Lowe 1997). Interestingly, the integrity of both domains is required for E1A’s oncogenic potential (Whyte et al. 1988b, 1989). The ability of E1A to activate p53 is not unique, as c-Myc activates p53 to promote apoptosis (Hermeking and Eick 1994; Wagner et al. 1994) and oncogenic ras induces p53 leading to premature senescence (Serrano et al. 1997). How oncogenic signals activate p53 is not known, although it is conceivable that they induce p53 by inadvertently damaging DNA. Nevertheless, the general involvement of p53 in the cellular response to oncogenes raises the possibility that these stimuli are fundamental to p53’s tumor suppressor activity.
The INK4a/ARF locus is second only to p53 in the frequency of its disruption in human cancer (for review, see Haber 1997). This locus encodes p16INK4a, a cyclin-dependent kinase inhibitor (CDKI) that acts upstream of Rb to promote cell-cycle arrest (Serrano et al. 1993). Although compelling evidence indicates that p16INK4a is an important tumor suppressor, the INK4a/ARF locus encodes a second protein translated in an alternate reading frame, designated p19ARF (Quelle et al. 1995). p19ARF and p16INK4a are often codeleted in tumor cells, but mice lacking p19ARF alone are highly cancer prone (Kamijo et al. 1997; for review, see Haber 1997). p19ARF promotes cell-cycle arrest (Quelle et al. 1995), whereas ARF-null primary mouse embryo fibroblasts (MEFs) do not undergo replicative senescence and are transformed by oncogenic ras alone (Kamijo et al. 1997). Thus, ARF is a bona fide tumor suppressor.
p19ARF may function in a genetic and biochemical pathway that involves p53. At the organismal level, the consequences of deleting p53 and ARF are remarkably similar (Donehower et al. 1992; Kamijo et al. 1997). In either case, the mutant mouse develops normally but is highly predisposed to malignant tumors of a similar overall pattern and latency. At the cellular level, enforced expression of p19ARF can induce cell-cycle arrest in cells harboring wild-type but not mutant p53 (Kamijo et al. 1997). In turn, p19ARF can physically associate with p53 itself and/or Mdm2 to alter p53 levels and activity (Kamijo et al. 1998; Pomerantz et al. 1998; Zhang et al. 1998). Nevertheless, ARF is not required for the p53 response following DNA damage, as radiation induces G1 arrest in ARF-deficient fibroblasts and apoptosis in ARF-deficient thymocytes (Kamijo et al. 1997, 1998). Thus, an understanding of the signals that activate p19ARF may help to explain its role as a tumor suppressor as well as that of p53.
In this study we compared the mechanism whereby DNA damaging agents and the E1A oncogene activate p53. We demonstrate that E1A activates p53 through a fundamentally different mechanism than DNA damage, which is dependent on the presence of p19ARF. Furthermore, simultaneous activation of p53 through oncogenes and DNA damage synergize to promote apoptosis and thereby enhance radio- and chemosensitivity. These data imply that p19ARF acts to suppress tumor growth in response to hyperproliferative signals. Conversely, as p19ARF mediates activation of p53 by an oncogene and is frequently lost in human tumors, these data strongly support the view that p53’s tumor suppressor activity can arise from its ability to eliminate oncogene-expressing cells.
Results
E1A and DNA damage induce p53 through distinct mechanisms
The E1A oncogene induces p53 through a mechanism involving inactivation of Rb gene product, and up-regulation of p53 correlates with the ability of E1A to promote apoptosis (Lowe and Ruley 1993; Lowe et al. 1994b; Samuelson and Lowe 1997). DNA damage produced by radiation and certain cytotoxic drugs also activates p53, at least in part, through a kinase that phosphorylates p53 on serine-15 (Shieh et al. 1997; Siliciano et al. 1997). To determine whether DNA damage and E1A induce p53 through similar mechanisms, we examined the phosphorylation status of p53 on serine-15 in cells expressing or lacking E1A. E1A was introduced into normal diploid human fibroblasts (IMR90 cells) by retroviral-mediated gene transfer. After a 3-day drug selection to eliminate uninfected cells, p53 levels and phosphorylation status were assessed by Western blot analysis using antibodies that recognize total p53 or only that fraction phosphorylated on serine-15 (Shieh et al. 1997; Siliciano et al. 1997). For comparison, IMR90 cells were treated with ionizing radiation or with the calpain/proteosome inhibitor LLnL, both of which are also known to stabilize p53 (Maki et al. 1996). Total p53 was examined by Western blotting; alternatively, p53 was immunoprecipitated and scored for the presence of serine-15 phosphate using antibodies that detect this epitope.
As expected, ionizing radiation produced a large increase in p53 protein (Fig. 1A, lane 2) accompanied by p53 phosphorylation on serine 15 (Fig. 1B, lane 2). LLnL also induced p53 but without serine-15 phosphorylation (Fig. 1, A, lane 3, and B, lane 1). E1A produced even greater increases in p53 levels (Fig. 1A, lane 4) without detectable phosphorylation of p53 on serine 15 (Fig. 1B, lane 3). However, E1A did not inhibit p53 phosphorylation on serine-15, as γ-irradiation of cells expressing E1A produced little, if any, additional increase in p53 protein (Fig. 1A, lane 5) but led to a marked increase in anti-phosphoserine-15 reactivity (Fig. 1B, lane 5). Induction of p53 in the absence of serine-15 phosphorylation argues that E1A does not produce DNA damage indirectly but, rather, suggests that E1A and ionizing radiation activate p53 through distinct mechanisms.
Figure 1.
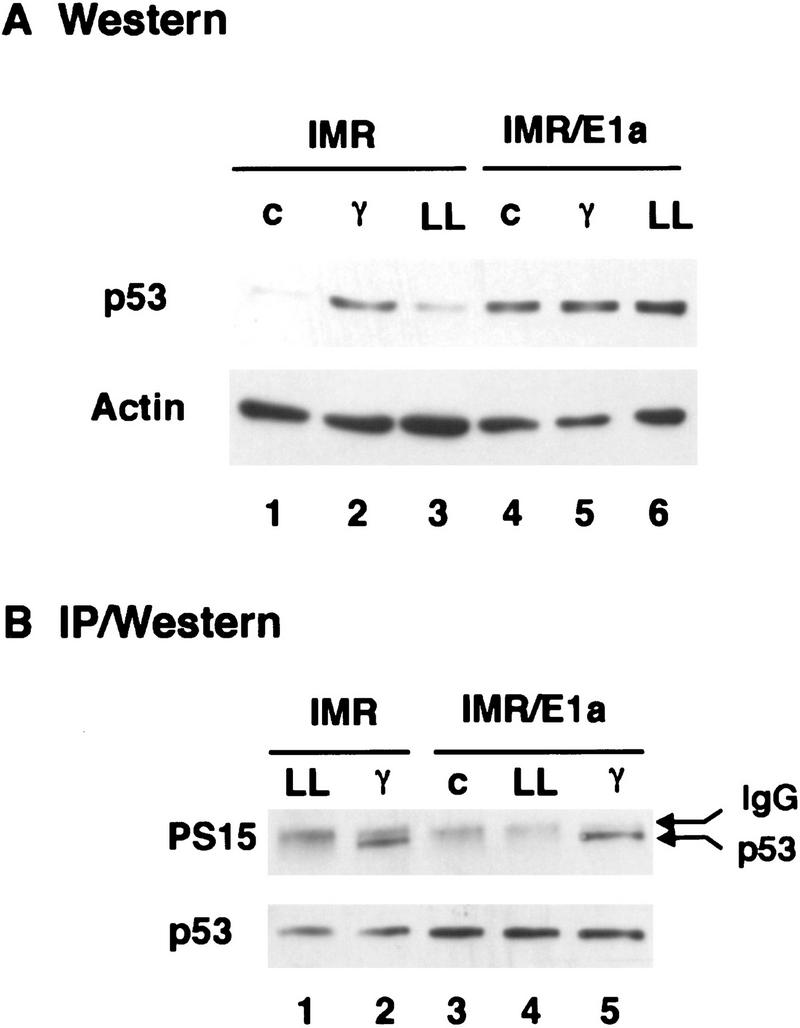
E1A induces p53 in the absence of phosphorylation on serine-15. IMR90 fibroblasts were infected with control (IMR) or E1A-expressing (IMR/E1A) retroviruses. Extracts were prepared from untreated cells (c), or from cells treated 3 hr earlier with 7 Gy γ radiation (γ) or 2 hr earlier with 50 μm LLnL (LL). (A) p53 levels were determined by Western blot analysis using pAb 1801 and DO1. Equal loading of the gel was confirmed by stripping the blot and reprobing with anti-β-actin antiserum. (B) p53 was immunoprecipitated from extracts corresponding to 100 μg (IMR) or 35 μg (IMR/E1A) total protein using pAb 1801, and Western blots were probed with antibodies specific for p53 phosphoserine-15 (αp53-P–Ser-15).
E1A induces p19ARF through domains required for p53 accumulation and apoptosis
Enforced expression of p19ARF stabilizes p53 and arrests proliferation in a p53-dependent manner, yet ARF is not required for radiation-induced cell-cycle arrest or apoptosis (Kamijo et al. 1997; Pomerantz et al. 1998; Zhang et al. 1998). The fact that E1A also stabilizes p53 through a DNA damage-independent mechanism is consistent with the possibility that E1A acts through p19ARF to induce p53. E1A or various E1A mutants were introduced into primary MEFs, and p19ARF expression was monitored 3 days later. E1A caused a dramatic induction of p19ARF, correlating with p53 accumulation (Fig. 2, A and B, cf. lanes 2 and 1). A similar increase was also observed in ARF mRNA expression, indicating that E1A was affecting ARF transcription or message stability (Fig. 2B, cf. lanes 2 and 1). As demonstrated previously (Kamijo et al. 1997), ARF is constitutively upregulated in p53−/− MEFs (Fig. 2B, lane 5), suggesting the presence of a negative feedback loop. However, E1A still induced p19ARF expression in p53-deficient cells (two- to threefold), implying that p53 is not required for p19ARF up-regulation by E1A (Fig. 2B, lane 6).
Figure 2.
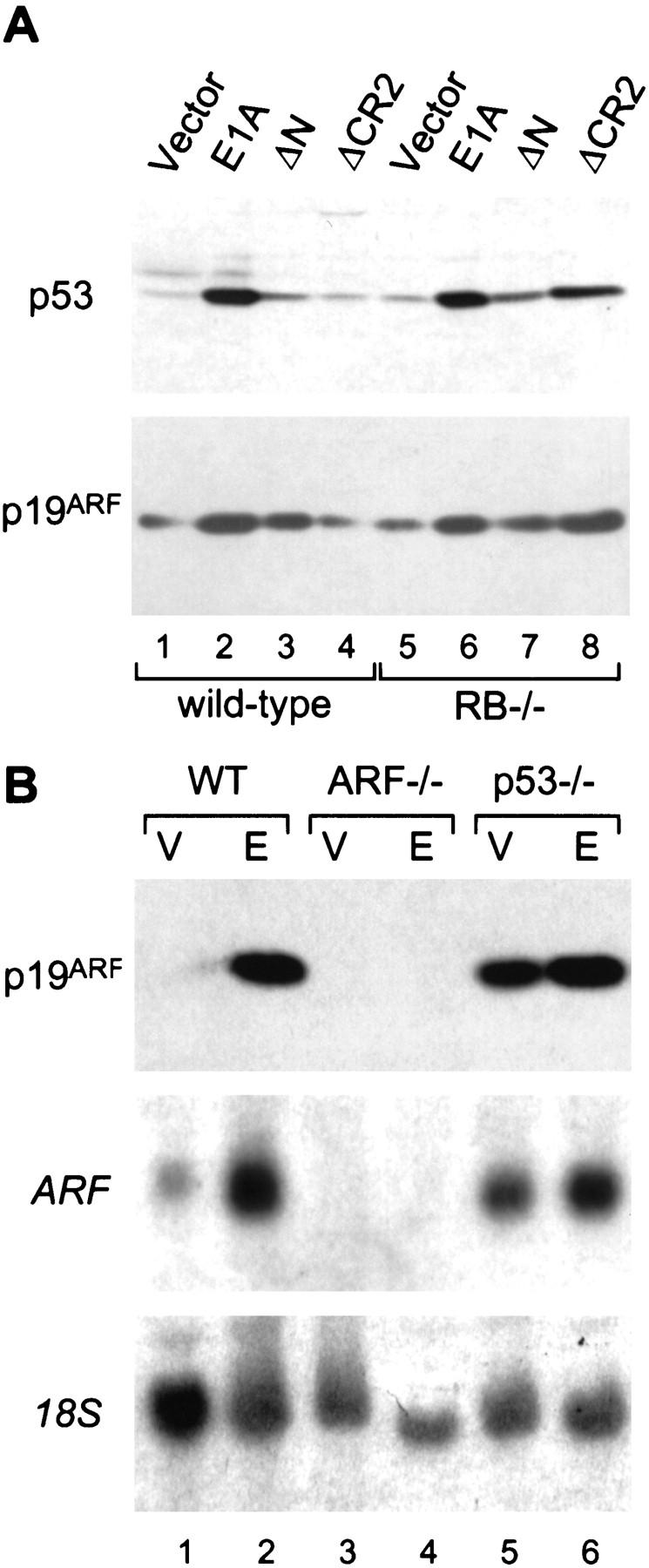
E1A induces p19ARF and p53 through a similar mechanism. (A) Early passage (about three to four) wild-type and Rb−/− MEFs from littermates embryos were infected with retroviruses expressing full-length E1A or E1A mutants unable to bind p300/CBP (ΔN) or the Rb-related proteins (ΔCR2). An empty retroviral vector was used as a control (vector). Immunoblotting was performed using polyclonal antibodies against p19ARF or p53. Using this procedure, each E1A mutant is efficiently expressed at comparable levels (Samuelson et al. 1997). (B) Wild-type (WT), ARF-null (ARF−/−), and p53-null (p53−/−) MEFs were infected with a control vector (V) or a retrovirus expressing full-length E1A (E). Lysates were derived from whole populations passaged minimally in culture (<1 week) and analyzed for ARF protein (top) or mRNA (middle) expression by Western or Northern blotting, respectively. Northern blots were rehybridized using a probe to the 18S rRNA to confirm equal loading (bottom).
E1A associates with a series of cellular proteins, including Rb, the Rb-related proteins p107 and p130, and the transcriptional coactivators p300 and CBP (for review, see Flint and Shenk 1997). E1A mutants unable to bind either p300/CBP (E1A ΔN) or the Rb-family proteins (E1A ΔCR2) were impaired in their ability to induce p19ARF and p53 (Fig. 2A, lanes 3,4), implying that E1A’s ability to bind both sets of cellular proteins is required for maximal p19ARF accumulation. In agreement, p19ARF protein induction was restored in cells coinfected with both E1A mutants (data not shown). p19ARF levels were slightly elevated in Rb-deficient MEFs (Fig. 2A, lane 5) although this difference was more pronounced in later passage MEFs (data not shown; see also Zindy et al. 1998). Importantly, p19ARF levels were further increased by expression of E1A (Fig. 2A, lane 6) or, in contrast to normal cells, the E1A ΔCR2 mutant (Fig. 2A, cf. lanes 4 and 8). However, p19ARF was not elevated in p107- and p130-deficient MEFs, nor was it induced by E1A ΔCR2 (data not shown). Thus, among the Rb-family proteins that bind E1A, the recognized ability of E1A to inactivate Rb solely contributes to p19ARF accumulation. These data demonstrate that at least two E1A functions contribute to p19ARF induction: inactivation of Rb and, possibly, binding to p300/CBP. Notably, these are the same domains of E1A that are necessary for its ability to induce p53 and promote apoptosis (Samuelson and Lowe 1997).
ARF promotes p53 accumulation in response to E1A
p53 activation is typically accompanied by increased expression of its transcriptional targets, including p21 and Mdm2. p21 is a CDKI involved in p53-dependent cell-cycle arrest (El Deiry et al. 1993; Harper et al. 1993; Xiong et al. 1993). Mdm2 acts in a negative feedback loop to down-regulate p53 and is expressed from two promoters, one of which is regulated by p53 (Barak et al. 1993, 1994; Wu et al. 1993). To determine whether ARF is required for p53 induction by E1A, the expression of p53, p21, and Mdm2 were examined in wild-type, ARF−/−, and p53−/− MEFs. In wild-type MEFs, E1A increased p53 protein expression, which was accompanied by accumulation of p21 and several forms of Mdm2 (Fig. 3A, lane 2). Induction of p21 and Mdm2 was p53-dependent, as neither protein was induced by E1A in p53-deficient cells (Fig. 3A, lane 6). Remarkably, expression of equivalent levels of E1A did not induce p53 in ARF-deficient cells, nor affect its targets p21 and Mdm2 (Fig. 3A, lane 4). Of note, wild-type and ARF−/− MEFs infected with a control vector displayed similar p53 levels, indicating that p19ARF loss does not markedly affect basal p53 expression (compare lanes 1 and 3). Therefore, ARF facilitates the up-regulation of p53 protein and its associated transcriptional activity following expression of E1A.
Figure 3.
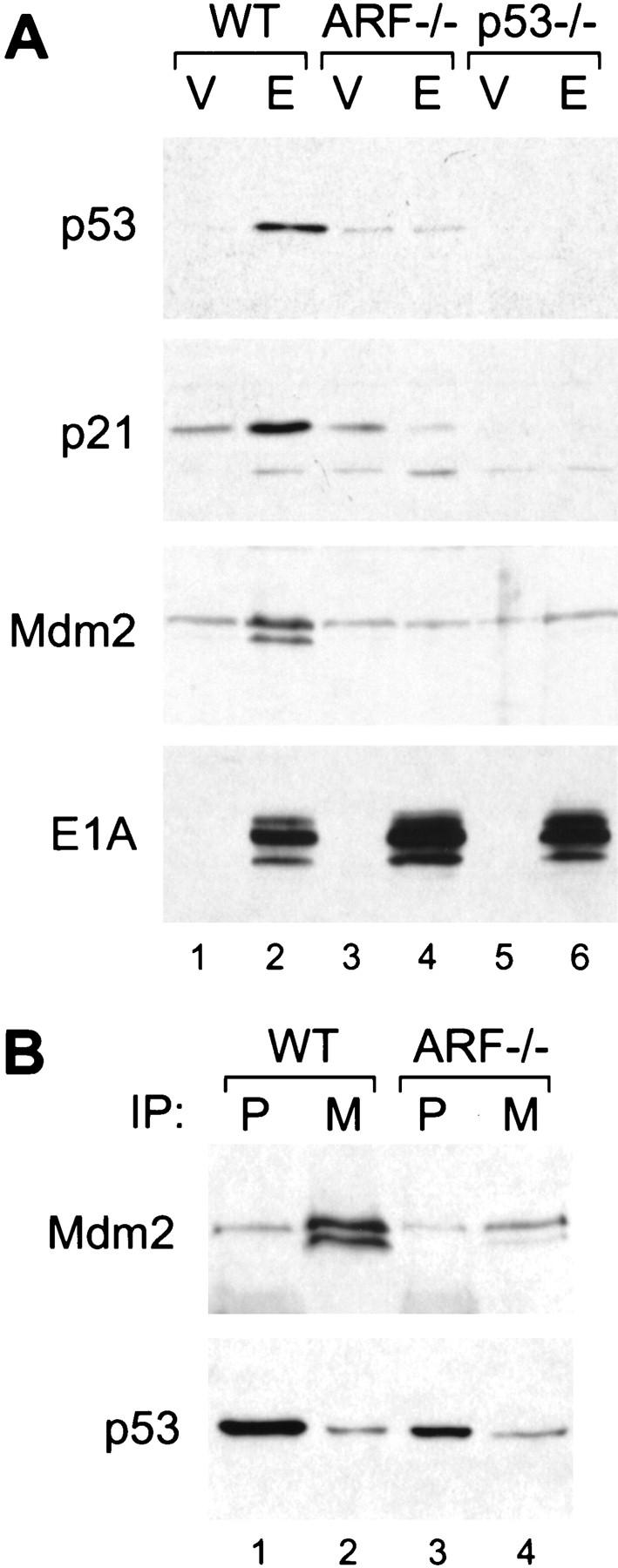
p19ARF mediates p53 induction by E1A and interferes with the p53/Mdm2 interaction. Wild-type (WT), ARF-null (ARF−/−) and p53-null (p53−/−) cell populations harboring a control vector (V) or expressing E1A (E) were prepared by retroviral gene transfer. Protein expression was analyzed in whole cell populations passaged minimally in culture (<1 week). (A) p53 protein levels along with the levels of its transcriptional targets p21 and Mdm2 were determined by immunoblotting. (B) Mdm2/p53 complexes were examined in wild-type and ARF-null populations expressing E1A by immunoprecipitation with monoclonal antibodies directed against p53 (P) or Mdm2 (M), followed by immunoblotting with a polyclonal rabbit antibody against p53. The blots were then reprobed using the same monoclonal antibody against Mdm2. Note that the p53 blot was overexposed to allow visualization of the amount associated with Mdm2.
When activated by DNA damage, Mdm2 is induced as part of a negative feedback loop that facilitates p53 degradation. However, wild-type MEFs expressing E1A accumulate p53 despite a large increase in Mdm2 levels (see Fig. 3A, lane 2). We examined the ability of Mdm2 to associate with p53 in MEFs expressing E1A by use of sequential immunoprecipitation and Western blotting. Despite the fact that wild-type MEFs expressing E1A displayed an ∼10-fold increase in p53 and Mdm2 levels as compared to their ARF-deficient counterparts, the absolute amount of Mdm2 bound to p53 was comparable in both cell types (Fig. 3B, cf. p53, lanes 2 and 4). Thus, p53 associates poorly with Mdm2 in wild-type cells expressing E1A. This implies that p19ARF, either directly or indirectly, contributes to p53 accumulation by preventing Mdm2-mediated degradation of p53 (Pomerantz et al. 1998; Zhang et al. 1998).
Inactivation of ARF attenuates apoptosis
E1A sensitizes primary fibroblasts to apoptosis induced by diverse stimuli, including serum depletion and treatment with chemotherapeutic drugs. The fact that ARF-deficient cells are unable to induce p53 in response to E1A suggests that ARF−/− MEFs expressing E1A might be resistant to apoptosis. Consistent with this possibility, the ability of Rb deficiency to trigger apoptosis was attenuated in developing mouse lenses disrupted for both ARF and INK4a (Pomerantz et al. 1998). Therefore, we compared the sensitivity of various virus-infected populations to cell death following serum withdrawal and treatment with adriamycin, a chemotherapeutic drug that produces double-stranded DNA breaks (Ross and Bradley 1981) and induces p53-dependent apoptosis in this setting. Two criteria were used to monitor apoptosis: annexin V staining followed by flow cytometry to assay membrane changes, and DAPI staining followed by fluorescence microscopy to visualize the characteristic chromatin condensation in apoptotic cells.
Concordant with previous results, wild-type MEFs expressing E1A lost viability following serum depletion or adriamycin treatment, whereas p53−/− MEFs expressing E1A did not (Fig. 4A,B). ARF−/− MEFs were significantly more resistant to E1A-induced apoptotic signals as compared to their wild-type counterparts but were somewhat more sensitive than cells lacking p53. In all cases, cell death was due to apoptosis, as measured by annexin V binding as well as chromatin condensation (Fig. 4C). Uninfected MEFs of all genotypes remained viable following serum depletion or adriamycin treatment at these doses, indicating that E1A was required for apoptosis under these conditions (data not shown). Therefore, p19ARF contributes to p53’s apoptotic potential in cells expressing E1A. However, the fact that p53 loss is more protective than ARF loss implies that some apoptotic signals address p53 through a p19ARF-independent pathway. For example, adriamycin might also exert some of its effects through the DNA damage pathway (see below).
Figure 4.

E1A-expressing cells lacking ARF are defective in apoptosis. Wild-type (•), ARF-null (▴), and p53-null (▪) early passage MEFs were infected with control retroviruses (not shown) or retroviruses expressing E1A. Within a week of gene transfer, the resulting cell populations were examined for cell death at various times following serum depletion (A) or 24 hr after treatment with the indicated doses of adriamycin (B). Cell viability was assessed by trypan blue exclusion. Each point represents the mean±s.d. from at least three separate experiments. Fibroblasts of all genotypes infected with a control vector retained viability (>90%) following serum depletion or adriamycin treatment (data not shown). (C) Wild-type (WT), ARF-null (ARF−/−) and p53-null (p53−/−) MEFs expressing E1A were examined for apoptosis 18 hr after transfer to 0.1% serum conditions. Annexin V binds phosphotidylserine. Apoptotic changes in membrane biochemistry lead to increased concentration of phosphotidylserine on the outer plasma membrane, where it becomes accessible to annexin V (Andree et al. 1990). Propidium iodide fluorescently stains late apoptotic cells that have lost membrane integrity. Shown are representative dot plots from two-color flow cytometry: (Bottom left quadrant) Viable; (bottom right quadrant) early apoptotic; (top right quadrant) late apoptotic. DAPI staining allows visualization of the chromatin condensation characteristic of apoptotic cells. Note that there was little apoptosis in E1A-expressing populations in 10% serum nor in vector-only control populations in 0.1% serum (data not shown).
If ARF loss protects cells from apoptosis in a p53-dependent manner, a clear prediction is that reintroduction of ARF into E1A-expressing cells containing wild-type p53 should resensitize them to the effects of serum deprivation and adriamycin. Conversely, cells lacking p53 should be unaffected by ARF. Hemagluttinin (HA)-tagged ARF was introduced by retroviral gene transfer into wild-type, ARF−/−, and p53−/− MEFs expressing E1A. Cells were infected at high multiplicity to bypass a need for drug selection. Exogenous p19ARF expression caused a 5- to 10-fold increase in p53 expression in both wild-type and ARF−/− MEFs expressing E1A (Fig. 5A), consistent with previous results (Kamijo et al. 1997, 1998). E1A-expressing wild-type MEFs infected with a control vector did not undergo apoptosis in high serum conditions but upon transfer to low serum conditions, underwent similar levels of apoptosis as uninfected E1A-expressing MEFs (Fig. 5B). As shown above (see Fig. 4), vector-infected cells lacking ARF or p53 were resistant to apoptosis when transferred to serum-depleted medium (Fig. 5B). Following infection with ARF retrovirus, both wild-type and ARF−/− MEFs expressing E1A displayed a modest increase in apoptosis when maintained in serum and underwent massive apoptosis upon serum depletion. Importantly, the same levels of exogenous p19ARF had little effect on p53−/− MEFs (Fig. 5B). Hence, depending upon the growth conditions, p19ARF can act upstream of p53 to induce either cell cycle arrest (Kamijo et al. 1997) or apoptosis. The fact that restoration of ARF function can resensitize ARF−/− MEFs to the combined effects of E1A and low serum provides compelling evidence that attenuation of apoptosis in ARF−/− cells is a direct consequence of ARF loss and not due to additional genetic changes.
Figure 5.
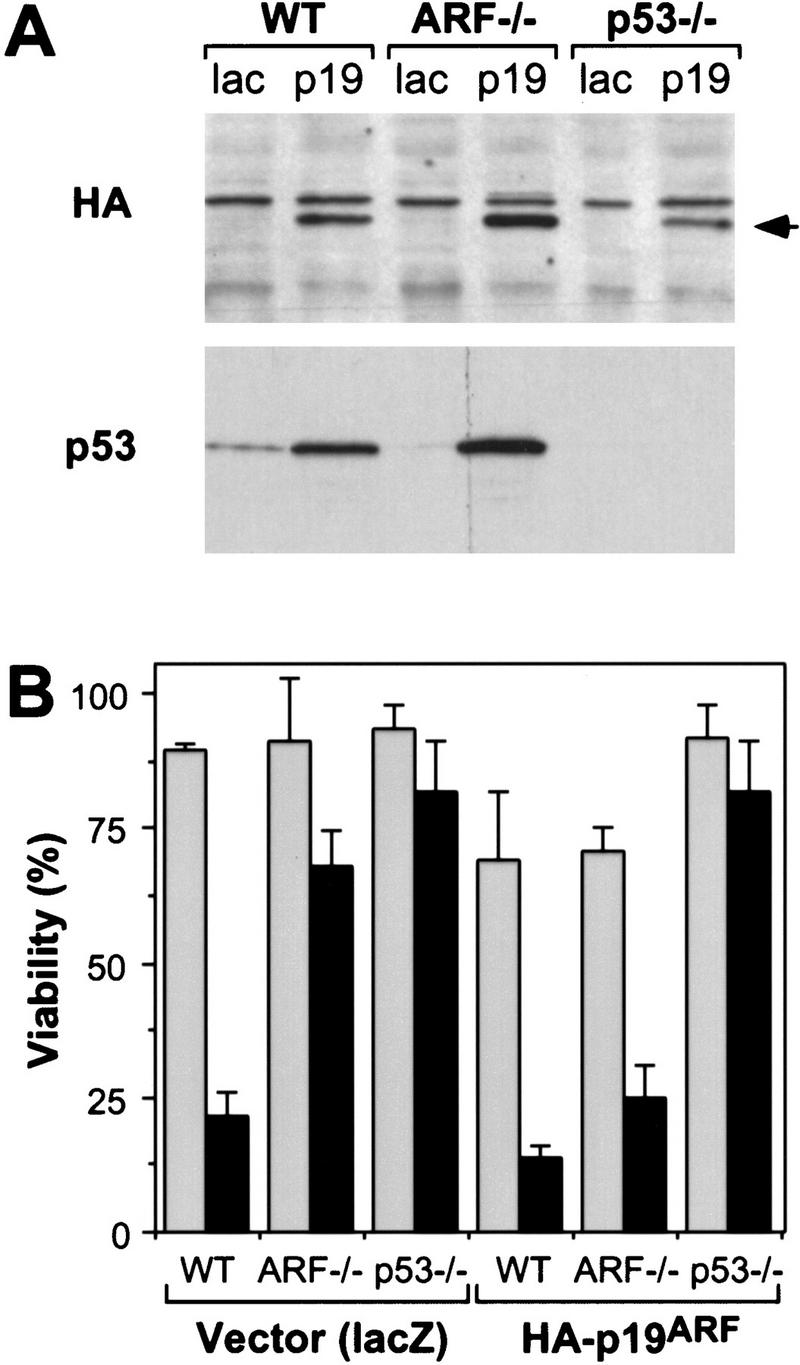
Reintroduction of p19ARF restores apoptosis. Control and E1A-expressing populations derived from wild-type (WT), ARF-null (ARF−/−) and p53-null (p53−/−) populations were infected with retroviruses expressing lacZ or an HA-tagged ARF cDNA (Quelle et al. 1995). Thirty-six hours later, the resulting cell populations were analyzed for p53 and exogenous p19ARF protein expression or treated with apoptotic stimuli. (A) Immunoblotting of infected populations using a monoclonal antibody recognizing the HA epitope fused to p19ARF or a polyclonal antibody directed against p53. The arrow denotes the migration of HA-tagged p19ARF. (B) The indicated cell populations were placed in 10% (shaded bars) or 0.1% (solid bars) serum for 24 hr and cell viability was measured by trypan blue exclusion. The values represent the mean and s.d. of at least three separate infections.
Synergy between p19ARF-dependent and -independent pathways targeting p53
Because DNA damage and E1A can activate p53 through distinct mechanisms, they might act synergistically to enhance cellular chemo- or radiosensitivity. Consistent with this possibility, enforced expression of p19ARF caused a marked increase in apoptosis induced by adriamycin when expressed in either wild-type or ARF−/− MEFs expressing E1A (Fig. 6A). Similar results were obtained following treatment of the cells with ionizing radiation (Fig. 6B). Importantly, the enhanced chemosensitivity produced by enforced p19ARF expression required both E1A and a cytotoxic insult. Hence, wild-type MEFs lacking E1A did not undergo apoptosis following adriamycin treatment and remained insensitive to low doses of the drug upon enforced expression of p19ARF (Fig. 6A, squares). ARF−/− cells expressing E1A were relatively resistant to drug-induced apoptosis (see also Fig. 4) but were resensitized when ARF was reintroduced (Fig. 6A, triangles). Importantly, introduction of ARF into wild-type cells expressing E1A also enhanced apoptosis in response to low doses of adriamycin (Fig. 6A, circles) or ionizing radiation (Fig. 6B), demonstrating that activation of the ARF–p53 pathway promotes both chemo- and radiosensitivity in the face of an oncogenic signal.
Figure 6.
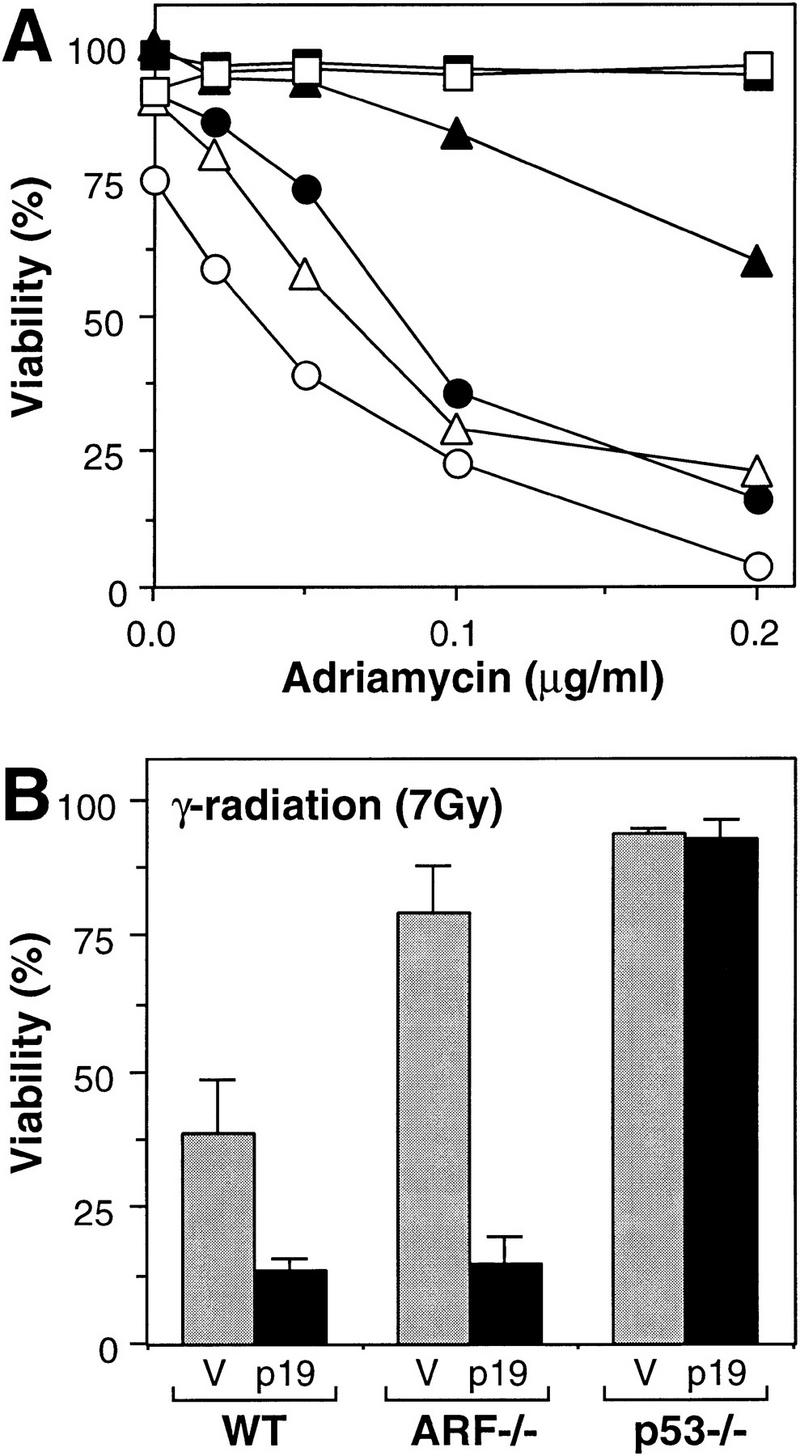
Synergy between p19ARF-dependent and -independent pathways targeting p53. (A) lacZ (solid symbols)- and HA–ARF (open symbols)-expressing cell populations were treated with the indicated doses of adriamycin, and cell viability was determined 24 hr later by trypan blue exclusion. The cell populations were as follows: Wild-type MEFs lacking E1A (squares); wild-type MEFs expressing E1A (circles); ARF−/− MEFs expressing E1A (triangles). Note that ARF−/− and p53−/− MEFs lacking E1A, as well as p53-deficient MEFs expressing E1A, remained viable in adriamycin whether or not they expressed HA–p19ARF (data not shown). (B) lacZ (V, shaded bars) and HA–p19ARF (p19, solid bars) expressing cell populations were treated with 7 Gy ionizing radiation and cell viability was determined 24 hr later by trypan blue exclusion. The values represent the mean and s.d. of at least three separate populations. MEFs not expressing E1A were resistant to apoptosis under these conditions (data not shown; see also Lowe et al. 1993).
Discussion
Oncogenic signaling through the ARF–p53 pathway
A variety of cellular stresses activate p53, including DNA damage, hypoxia, and expression of mitogenic oncogenes (for review, see Ko and Prives 1996; Levine 1997). Following DNA damage, p53 becomes phosphorylated by kinases such as DNA–PK or ATM, leading to changes in p53 conformation and activity. In contrast, the E1A oncogene activates p53 through a fundamentally different mechanism, mediated largely by the tumor suppressor p19ARF. Importantly, the DNA damage and E1A signaling pathways act in parallel: E1A does not produce p53 phosphorylation at serine-15 and DNA damage activates p53 independently of p19ARF (Kamijo et al. 1997). Moreover, p53 is phosphorylated on serine-15 following irradiation of ARF-deficient cells (data not shown). Therefore, these data provide a clear example of how p53 integrates upstream signaling pathways emanating from diverse stimuli (Fig. 7).
Figure 7.
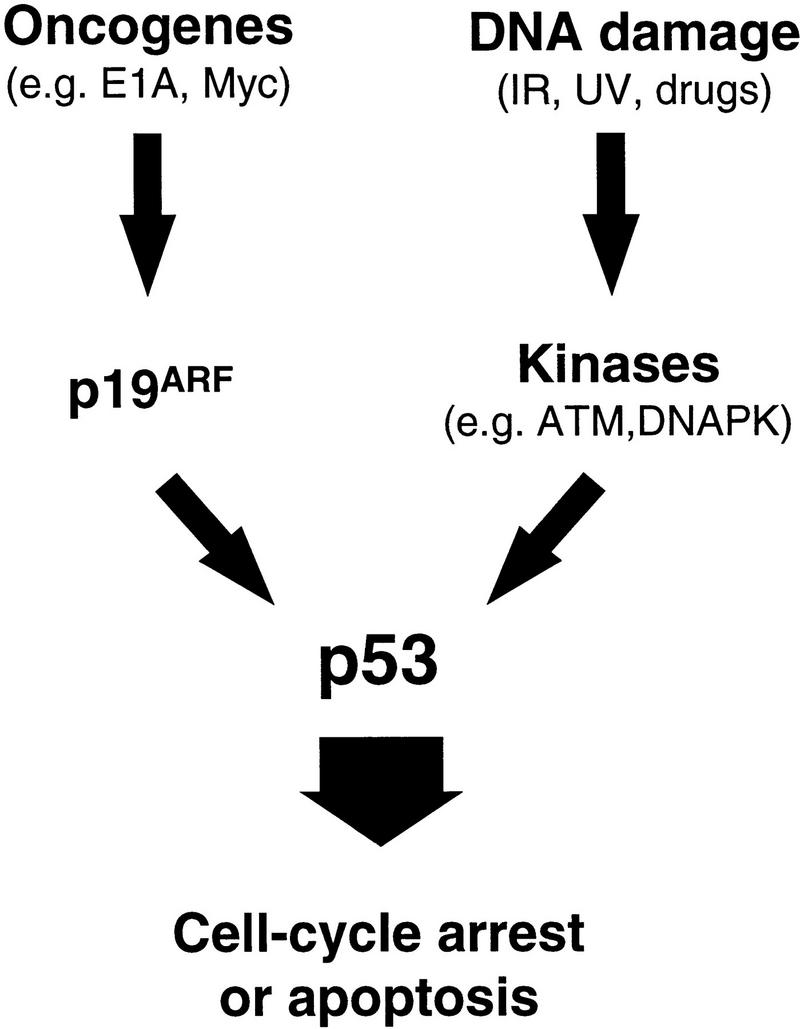
Oncogenes and DNA damage activate p53 through distinct mechanisms. p19ARF acts as an intermediary in p53 activation by mitogenic oncogenes such as E1A and myc. In contrast, activation of p53 following DNA damage involves de novo phosphorylation of p53 on serine-15 (and other residues) by kinases such as the DNA-dependent protein kinase (DNA–PK) or the product of the ataxia-telangiectasia gene (ATM) (Shieh et al. 1997; Siliciano et al. 1997). Activation of p53 by oncogenes does not involve phosphorylation on serine-15, and both serine-15 phosphorylation (not shown) and p53 activation (Kamijo et al. 1997) following DNA damage are unimpaired in the absence of ARF. Therefore, the two upstream signaling pathways to p53 are fundamentally distinct.
Activation of p53, in turn, can produce several cellular responses, including transient cell-cycle arrest, senescence or apoptosis. Each signaling pathway to p53 may produce subtle differences in p53 activity or function, and perhaps the diversity achieved by a combination of these signals accounts for the complex biology of p53. For example, simultaneous activation of p53 by p19ARF and DNA damage synergize to promote apoptosis in the presence of the E1A oncogene (Fig. 6; see also Lowe et al. 1993; Samuelson and Lowe 1997). If similar processes occur in human cancer, therapeutic strategies to exploit p19ARF activation may enhance the radiosensitivity or chemosensitivity of p53-expressing tumors.
Like p53, the outcome of p19ARF activation is dependent on cellular context. For example, enforced ARF expression in MEFs induces cell cycle arrest, but cells overexpressing p19ARF, together with E1A or Myc (Zindy et al. 1998), undergo apoptosis, which is potentiated by withdrawal of serum survival factors (Evan et al. 1992; Lowe and Ruley 1993; Lowe et al. 1994b). ARF-null MEFs are resistant to both E1A- and Myc-induced apoptosis, bypassing the p53-dependent fail-safe mechanism that normally protects them from these oncogenic signals, and thereby enabling E1A and Myc to function as pure growth promoters. Myc’s action as an “immortalizing gene” depends in part on its ability to dismantle the ARF–p53 pathway by selecting for surviving cells that have lost either gene (Zindy et al. 1998). In turn, ARF-null MEFs do not undergo replicative senescence and can be transformed by oncogenic ras alone (Kamijo et al. 1997). We suspect that E1A’s immortalizing activity involves similar mechanisms.
Also like p53, ARF has no overt role in normal cell cycle control or development; hence, the physiologic circumstances in which it would become activated to inhibit proliferation or suppress tumor growth were not obvious. Studies here with E1A mutants suggest that p19ARF can be activated to suppress proliferation by the E1A oncogene through mechanisms that correlate with its binding to both p300/CBP and Rb. These same functions are required for E1A to induce p53 and to promote apoptosis in primary fibroblasts (Samuelson and Lowe 1997) and, remarkably, are also required for E1A’s transforming potential (Whyte et al. 1988b, 1989). Loss of Rb contributes to ARF induction consistent with the possibility that ARF is an E2F-responsive gene (DeGregori et al. 1997). Enforced expression of E2F-1 induces p19ARF, and conversely, ARF-null cells are resistant to E2F-1-induced apoptosis (Zindy et al. 1998). Consequently, p19ARF function, like p53, depends upon the mutational status of Rb, and upon both c-myc and ras proto-oncogene activities. Irrespective of the precise outcome, ARF mutations compromise p53 activation and reduce its ability to counter uncontrolled proliferation.
The data presented here provide additional insights into p53’s role in tumor suppression. The predominant view of p53 action centers around its ability to function in the cellular response to DNA damage. Although this stimulus is undoubtedly important for p53’s tumor suppressor activity and may contribute to the outcome of cancer therapy (Lowe et al. 1993, 1994a), p53 activation in response to oncogenes provides an alternative pressure to mutate p53 during tumorigenesis (Lowe and Ruley 1993; Lowe et al. 1994b; Symonds et al. 1994). In this view, p53 normally acts to limit the consequences of uncontrolled mitogenesis by promoting cell-cycle arrest or apoptosis, while its loss allows proliferation to continue unabated. The fact that disruption of the ARF-p53 pathway occurs in the majority of human cancers underscores its global importance in suppressing proliferation of oncogene-expressing cells.
Materials and methods
Cells and cell culture
IMR90 fibroblasts (early–mid passages) expressed the ecotropic retrovirus receptor to allow infection with murine retroviruses (Serrano et al. 1997). Primary MEFs derived from wild-type, p53−/− (Jacks et al. 1994), and ARF−/− (Kamijo et al. 1997) day 13.5 embryos were prepared as described previously (Serrano et al. 1997). All cultures were maintained in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO) supplemented with 10% fetal bovine serum (FBS; Sigma) and 1% penicillin G/streptomycin sulfate (Sigma). To induce DNA damage, cells were either irradiated with 7 Gy ionizing radiation using a J.L. Shepherd Mark I irradiator with a 137Cs source or treated with 0.1–0.5 μg/ml adriamycin. To induce p53 independently of DNA damage, cells were treated for 2 hr with 50 μm LLnL (Sigma).
Retroviral vectors and infection
For most experiments, high-titer ecotropic retroviruses were generated by transient transfection using the Phoenix retrovirus packaging system (G. Nolan, Stanford University, CA) as described previously (Serrano et al. 1997). Virus supernatants were used to infect either IMR90 fibroblasts or early-passage MEFs (≤passage 5), and pure populations of E1A-expressing cells were isolated by selection for 2 days in the presence of 2 μg/ml puromycin. Infection was typically between 70% and 90% of cells as judged using a control virus expressing β-galactosidase (not shown). For ectopic expression of p19ARF, a protocol designed to achieve nearly complete infection of cells (Zindy et al. 1998) was used. Retroviral vectors were as follows: LPC, control vector expressing puromycin phosphotransferase (puro); LPC-12S, a 12S E1A cDNA in LPC (McCurrach et al. 1997); LPC-12S.ΔN and LPC-12S.ΔCR2, E1A mutants that fail to associate with p300/CBP or the Rb-related proteins, respectively (Samuelson and Lowe 1997). The retroviral vector encoding HA–p19ARF coexpressed a CD8 cell surface marker (Quelle et al. 1995). pBabePuro–lacZ (a gift of J. Morgenstern, Millenium Pharmaceutical, Cambridge, MA) was used to monitor infection efficiencies and, in some experiments, as a control vector.
Gene expression
Analysis of p53 phosphorylation on serine-15 was performed exactly as described (Shieh et al. 1997). p53 levels were determined by Western blots using PAb1801 and DO1. p53 immunoprecipitations were performed using pAb 1801 followed by immunoblotting with αp53-P–Ser-15 to identify p53 proteins phosphorylated on serine-15. Western blots to detect p19ARF were performed using antibodies to the carboxyl terminus as described (Kamijo et al. 1998); HA-tagged p19ARF was detected using mAb 12CA5 (1:5000 dilution). All other Western blots were carried out as described previously with minor modifications (Serrano et al. 1997). Whole-cell lysates were derived by lysing cell pellets in SDS sample buffer (60 mm Tris-HCl at pH 6.8, 10% glycerol, 2% SDS, 5% 2-mercaptoethanol). Samples corresponding to 30 μg of protein (Bio-Rad protein assay) were separated on SDS-PAGE gels and transferred to Immobilon-P membranes (Millipore). p53 was detected using polyclonal antibody CM5 (1:8000 dilution) (a gift of Peter Hall, Dundee University, UK); Mdm2 using mAb 2A10 (provided by G. Zambetti, St. Jude Children’s Research Hospital); p21 using polyclonal antibody C-19 (1: 500 dilution) (Santa Cruz), and E1A using mAb M58 (Harlow et al. 1985). Proteins were visualized by ECL (Amersham) and equal sample loading was confirmed by India Ink or Ponseau S staining of the membrane.
For p53/Mdm2 immunoprecipitations, cell pellets were disrupted in ice-cold NP-40 lysis buffer (50 mm Tris-HCl at pH 8, 5 mm EDTA, 150 mm NaCl, 0.5% NP-40, 1 mm PMSF, 0.4 U/ml aprotinin, 10 mm β-glycerophosphate, 1 mm NaF, 0.1 mm Na3VO4) on ice for 1 hr. Cleared lysates were incubated for 2 hr at 4°C with two monoclonal antibodies directed against p53 (pAb 421 and pAb 248) or Mdm2 (2A10), plus 10 mg/ml BSA. Complexes precipitated with protein A–Sepharose (Amersham) were washed three times with ice-cold NP-40 lysis buffer. Immunoprecipitates were separated on 7.5% SDS–polyacrylamide gels and transferred to nitrocellulose. Mdm2 was detected by immunoblotting using the same antibody, whereas p53 was detected with CM5 polyclonal antibody as described above.
For Northern blots, total RNA was extracted from cells using RNAzolB (Cinna/Biotecx) ∼1 week postinfection and 30 μg was loaded per lane. Following agarose gel electrophoresis and transfer to Hybond membranes (Amersham), blots were hybridized with a 32P-labeled probe specific for INK4a exon 1β [the portion of the INK4a/ARF locus unique to ARF (Quelle et al. 1995)]. A probe specific for 18S rRNA was used to confirm equal loading.
Cell viability and apoptosis
Cells were distributed into 12-well plates (105 cells/22-mm well) 12–24 hr prior to serum withdrawal, radiation, or drug treatment. Adherent and nonadherent cells were pooled 24 hr after treatment with γ-radiation, adriamycin, or 0.1% FBS and analyzed for viability by trypan blue exclusion; ⩾200 cells were scored for each point. Apoptotic cell death was confirmed by staining with DAPI or FITC–annexin V. Cells (∼1 × 105) were fixed in 5% paraformaldehyde (Mallinckrodt) and DNA was stained with DAPI (1 μg/ml). Images were digitized using a fluorescence microscope coupled to a Photometrics PXL CCD camera (Photometrics Ltd.). For annexin staining, cells were incubated in DMEM with 0.1% FBS for 18 hr, after which adherent and nonadherent cells were pooled. Staining with FITC–annexin V and PI were performed according to the manufacturer’s instructions (BioWhitaker) and the cells were analyzed by two-color flow cytometry.
Acknowledgments
We thank Y. Taya for the generous gift of immunopurified antibodies directed against phosphoserine-15 p53 and N. Dyson for p107- and p130-deficient MEFs. We also thank Maria Coronesi and Esther Van de Kamp for technical assistance. E.S. is supported by the Italian Ph.D. program and thanks Dr. G. Biamonti and Professor A. Galizzi for support; G.F. is a Tularik postdoctoral fellow; A.V.S. is supported by an Army Breast Cancer Research fellowship; S.W.L. is supported by a Kimmel Scholarship Award. This work was funded in part by American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital, and by grants CA58316 (C.P.); CA56819, CA71907 (M.F.R.), and CA13106 (S.W.L) from the National Institutes of Health.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lowe@cshl.org; FAX (516) 367-8454.
References
- Andree HA, Reutelingsperger CP, Hauptmann R, Hemker HC, Hermens WT, Willems GM. Binding of vascular anticoagulant alpha (VAC alpha) to planar phospholipid bilayers. J Biol Chem. 1990;265:4923–4928. [PubMed] [Google Scholar]
- Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild-type p53 activity. EMBO J. 1993;12:461–468. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak Y, Gottlieb E, Juvengershon T, Oren M. Regulation of mdm2 expression by p53: Alternative promoters produce transcripts with nonidentical translation potential. Genes & Dev. 1994;8:1739–1749. doi: 10.1101/gad.8.15.1739. [DOI] [PubMed] [Google Scholar]
- Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes & Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- DeCaprio JA, Ludlow JW, Lynch D, Furukawa Y, Griffin J, Piwnica-Worms H, Huang CM, Livingstone DM. SV40 large T antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–283. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JA, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–220. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- El Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters C, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Flint J, Shenk T. Viral transactivating proteins. Annu Rev Genet. 1997;31:177–212. doi: 10.1146/annurev.genet.31.1.177. [DOI] [PubMed] [Google Scholar]
- Graeber TG, Peterson JF, Tsai M, Monica K, Fornace AJ, Giaccia AJ. Hypoxia induces accumulation of p53 protein, but activation of a G(1)-phase checkpoint by low-oxygen conditions is independent of p53 status. Mol Cell Biol. 1994;14:6264–6277. doi: 10.1128/mcb.14.9.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54:4855–4878. [PubMed] [Google Scholar]
- Griffiths SD, Clarke AR, Healy LE, Ross G, Ford AM, Hooper ML, Wyllie AH, Greaves M. Absence of p53 permits propagation of mutant cells following genotoxic damage. Oncogene. 1997;14:523–531. doi: 10.1038/sj.onc.1200871. [DOI] [PubMed] [Google Scholar]
- Haber DA. Splicing into senescence: The curious case of p16 and p19ARF. Cell. 1997;91:555–558. doi: 10.1016/s0092-8674(00)80441-9. [DOI] [PubMed] [Google Scholar]
- Harlow E, Franza BR, Jr, Schley C. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J Virol. 1985;55:533–546. doi: 10.1128/jvi.55.3.533-546.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Khandan K, Elledge SJ. The p21 cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Eick D. Mediation of c-myc induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Weber JS, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci. 1998;95:8292–8297. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- Ko LJ, Prives C. p53: Puzzle and paradigm. Genes & Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Linke SP, Clarkin KC, Di Leonardo A, Tsou A, Wahl GM. A reversible, p53-dependent G0/G1 cell cycle arrest induced by ribonucleotide depletion in the absence of detectable DNA damage. Genes & Dev. 1996;10:934–947. doi: 10.1101/gad.10.8.934. [DOI] [PubMed] [Google Scholar]
- Linzer DIH, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40 transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- Livingstone LR, White A, Sprouse J, Livanos E, Jacks T, Tlsty TD. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 1992;70:923–935. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE. Stabilization of the p53 tumor suppressor is induced by adenovirus E1A and accompanies apoptosis. Genes & Dev. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:954–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher D, Housman DE, Jacks T. p53 status and the efficacy of cancer therapy in vivo. Science. 1994a;266:807–810. doi: 10.1126/science.7973635. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Jacks T, Housman DE, Ruley HE. Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc Natl Acad Sci. 1994b;91:2026–2030. doi: 10.1073/pnas.91.6.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki CG, Huibregtse JM, Howley PM. In vivo ubiquitination and proteasome-mediated degradation of p53(1) Cancer Res. 1996;56:2649–2654. [PubMed] [Google Scholar]
- McCurrach ME, Connor TM, Knudson CM, Korsmeyer SJ, Lowe SW. bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc Natl Acad Sci. 1997;94:2345–2349. doi: 10.1073/pnas.94.6.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW, Cordon-Cardo C, DePinho RA. The INK4a tumor suppressor gene product, p19ARF, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Querido E, Teodoro JG, Branton PE. Accumulation of p53 induced by the adenovirus E1A protein requires regions involved in the stimulation of DNA synthesis. J Virol. 1997;71:3526–3533. doi: 10.1128/jvi.71.5.3526-3533.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross WE, Bradley MO. DNA double-stranded breaks in mammalian cells after exposure to intercalating agents. Biochim Biophys Acta. 1981;654:129–134. doi: 10.1016/0005-2787(81)90145-3. [DOI] [PubMed] [Google Scholar]
- Samuelson AV, Lowe SW. Selective induction of p53 and chemosensitivity in RB-deficient cells by E1A mutants unable to bind the RB-related proteins. Proc Natl Acad Sci. 1997;94:12094–12099. doi: 10.1073/pnas.94.22.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA damage induces phosphorylation of the amino terminus of p53. Genes & Dev. 1997;11:3471–3481. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symonds H, Krall L, Remington L, Saenzrobles M, Lowe S, Jacks T, Vandyke T. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78:703–711. doi: 10.1016/0092-8674(94)90534-7. [DOI] [PubMed] [Google Scholar]
- Wagner AJ, Kokontis JM, Hay N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1/cip1. Genes & Dev. 1994;8:2817–2830. doi: 10.1101/gad.8.23.2817. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA, Harlow E. Association between an oncogene and an anti-oncogene: The adenovirus E1A proteins bind to the retinoblastoma gene product. Nature. 1988a;334:124–129. doi: 10.1038/334124a0. [DOI] [PubMed] [Google Scholar]
- Whyte P, Ruley HE, Harlow E. Two regions of the adenovirus early region 1A proteins are required for transformation. J Virol. 1988b;62:257–265. doi: 10.1128/jvi.62.1.257-265.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte P, Williamson NM, Harlow E. Cellular targets for transformation by the adenovirus E1A proteins. Cell. 1989;56:67–75. doi: 10.1016/0092-8674(89)90984-7. [DOI] [PubMed] [Google Scholar]
- Wu X, Bayle JH, Olson D, Levine AJ. The p53–mdm-2 autoregulatory feedback loop. Genes & Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–705. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- Yin Y, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell. 1992;70:937–948. doi: 10.1016/0092-8674(92)90244-7. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zindy, F., C.M. Eischen, D.H. Randle, T. Kamijo, J.L. Cleveland, C.J. Sherr, and M.F. Roussel. 1998. MYC-induced immortalization and apoptosis targets the ARF-p53 pathway. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]