

Abstract
The retinoblastoma tumor suppressor protein, RB, is a negative regulator of cell proliferation. Growth inhibitory activity of RB is attenuated by phosphorylation. Mutation of a combination of phosphorylation sites leads to a constitutively active RB. In Rat-1 cells, the phosphorylation-site-mutated (PSM)-RB, but not wild-type RB, can inhibit S-phase entry. In PSM-RB-arrested G1 cells, normal levels of cyclin E and cyclin E-associated kinase activity were detected, but the expression of cyclin A was inhibited. The ectopic expression of cyclin E restored cyclin A expression and drove the PSM-RB expressing cells into S phase. Interestingly, Rat-1 cells coexpressing cyclin E and PSM-RB could not complete DNA replication. Microinjection of cells that have passed through the G1 restriction point with plasmids expressing PSM-RB also led to the inhibition of DNA synthesis. The S-phase inhibitory activity of PSM-RB could be attenuated by the coinjection of SV40 T-antigen, adenovirus E1A, or a high level of E2F-1 expression plasmids. However, the S-phase inhibitory activity of PSM-RB could not be overcome by the coinjection of cyclin E or cyclin A expression plasmids. These results reveal a novel role for RB in the inhibition of S-phase progression that is distinct from the inhibition of the G1/S transition, and suggest that continued phosphorylation of RB beyond G1/S is required for the completion of DNA replication.
Keywords: Cyclin A, cyclin E, p16ink4a, p21cip1, T-antigen
RB, the retinoblastoma tumor suppressor protein, is a negative regulator of cell proliferation (for review, see Wang et al. 1994; Weinberg 1995; Beijersbergen and Bernards 1996; Sidle et al. 1996; Kaelin et al. 1997). Observations of cell cycle-dependent oscillation in RB phosphorylation led to the hypothesis that phosphorylation inactivates RB. RB is a major substrate for the cyclin-dependent kinases (cdks), cdk4, cdk6, cdk2, and cdc2 (Lin et al. 1991; Akiyama et al. 1992; Meyerson and Harlow 1993; Matsushime et al. 1994). A number of phosphorylation sites have been mapped in RB (Lees et al. 1991; Lin et al. 1991; Hamel et al. 1992; Zarkowska et al. 1997; Connell-Crowley et al. 1997). Recent evidence has demonstrated that specific phosphorylation sites regulate distinct protein-binding functions of RB (Knudsen and Wang 1996, 1997; Zarkowska et al. 1997). The phosphorylation-inactivation model predicts that an unphosphorylatable RB would act as a constitutive inhibitor of cell cycle progression. Indeed, several combinations of phosphorylation-site-mutations (PSMs) have resulted in RB proteins that inhibit the proliferation of cells which are insensitive to wild-type RB (Knudsen and Wang 1997; Lukas et al. 1997).
Phosphorylation of RB during transition from G1 to S is initiated by the D-type cyclin/cdk complexes (D/cdk4 or D/cdk6) (Sherr 1996; Sidle et al. 1996). The activity of cdk4/6 is stimulated by D-type cyclins and inhibited by a family of low molecular weight inhibitors, exemplified by the tumor suppressor protein p16ink4a (Bartek et al. 1997; Palmero and Peters 1996; Sherr 1996). Components of the RB phosphorylation pathway, that is, p16ink4a, D-type cyclins, cdk4, and cdk6 are altered in tumor cells at a high frequency (Palmero and Peters 1996; Sherr 1996; Lukas et al. 1997). In particular, loss of p16ink4a is observed in a variety of tumor types. Ectopic expression of p16ink4a blocks cells in G1, and this is critically dependent on RB (Koh et al. 1995; Lukas et al. 1995; Medema et al. 1995). Ectopic expression of p16ink4a does not arrest RB-deficient cells in G1, suggesting that RB function is necessary for p16ink4a to induce G1 arrest.
The ability of RB to inhibit the G1/S transition is well established by multiple lines of evidence. Physiological growth inhibitors such as TGF-β inhibit RB phosphorylation to block cells in G1 (Wang et al. 1994; Herrera et al. 1996a). Embryo fibroblasts derived from RB-deficient mice have a shortened G1 phase and a weakened response to anti-mitogenic signals (Herrera et al. 1996a,b). Ectopic expression of wild-type RB arrests some RBdeficient tumor cells in G1 (Hamel et al. 1993; Wang et al. 1994). A major mechanism by which RB inhibits the G1/S transition is through the repression of E2F-regulated genes, among which are cyclin E and cyclin A (Ohtani et al. 1995; Weinberg 1995; Geng et al. 1996; Kaelin 1997; Sidle et al. 1996). Both cyclin E and cyclin A can bind to and activate cdk2, and cdk2 activity is rate-limiting for S-phase entry (Del Sal et al. 1996; Sherr 1996). Using a tetracycline-regulated promoter, Resnitzky et al. have shown that overproduction of cyclin E or cyclin A in Rat-1 cells shortens the G1 interval (Resnitzky and Reed 1995; Resnitzky et al. 1995). In RB-deficient mouse embryo fibroblasts, the timing of cyclin E expression is advanced, in keeping with the faster progression of these cells from quiescence into S phase (Herrera et al. 1996a; Hurford et al. 1997). Thus, RB-mediated repression of cyclin E expression has been hypothesized to underlie its G1/S inhibitory activity.
Previously, we have reported the construction of two PSM-RB proteins that can inhibit the proliferation of Rat-1 cells (Knudsen and Wang 1997). In the context of full-length RB, mutation of nine phosphorylation sites in PSM.9I-RB is required to generate a constitutively active growth suppressor (Kundsen and Wang 1997). In the context of an amino-terminal deleted RB, which is commonly known as the large pocket (LP; Qin et al. 1992), mutation of seven phosphorylation sites (PSM.7-LP) is sufficient to block the inactivation of RB (Knudsen and Wang 1997). In this study we compared the effect of PSM-RB and p16ink4a on cell cycle progression in Rat-1 cells. Although both p16ink4a and PSM-RB induce G1 arrest, they do so at different execution points along the G1/S transition. Our study also uncovered an unexpected inhibitory effect of PSM-RB on S-phase progression, which was not observed with p16ink4a. Our findings show that RB can inhibit both G1/S transition and S-phase progression, and provide an explanation for the continued hyperphosphorylation of RB throughout the S phase of the cell cycle.
Results
PSM-RB arrests Rat-1 cells in G1
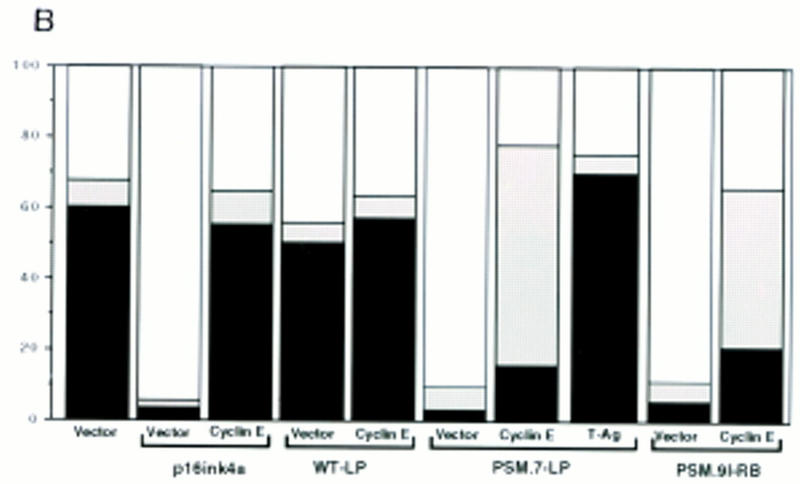
Ectopic expression of WT-RB, or the previously described pΔ34-phosphorylation site mutant RB, does not inhibit the growth of Rat-1 cells (Knudsen and Wang 1997; Lukas et al. 1997; Alevizopoulos et al. 1997). However, PSM.9I-RB, PSM.7LP (Knudsen and Wang 1997) and the RBΔcdk (Lukas et al. 1997) can block Rat-1 cell cycle progression. Because p16ink4a and RB are in the same G1-inhibitory pathway, we compared the G1inhibitory activity of PSM.7-LP to that of p16ink4a. Rat-1 cells were transiently transfected with vector, WT-LP, PSM.7-LP or p16ink4a expression plasmids and a plasmid encoding the green fluorescent protein (GFP). Cell cycle progression of the transfected, GFP-positive, cells was measured by BrdU-incorporation. With vector and WT-LP transfected cells, between 60% and 70% incorporated BrdU (Fig. 1A). In contrast, only 5% of the cells transfected with p16ink4a or PSM.7-LP incorporated BrdU (Fig. 1A).
Figure 1.
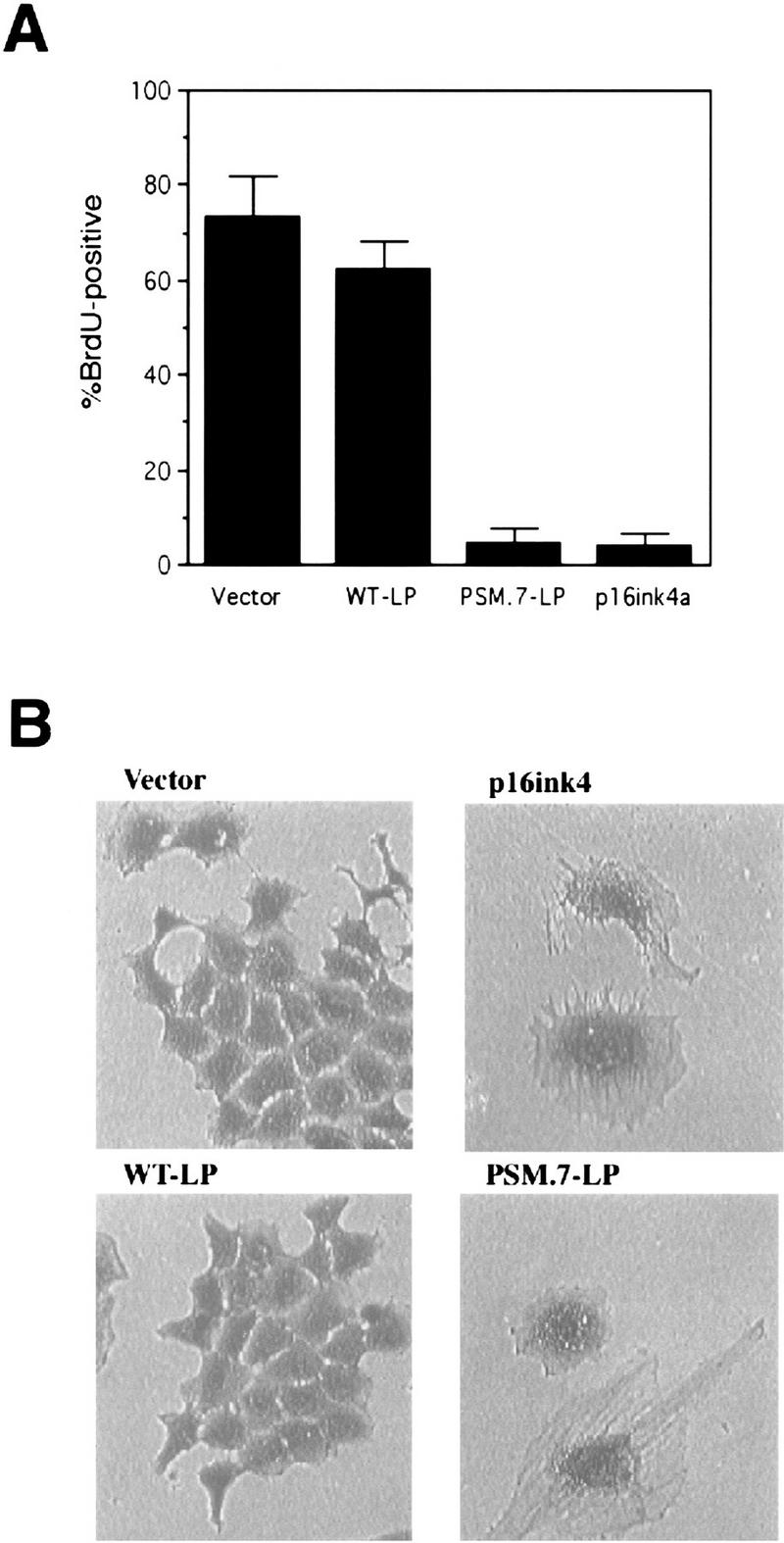



PSM-RB arrests cell growth without inhibiting the phosphorylation of p107/p130. (A) Rat-1 cells were cotransfected with a GFP expression plasmid (1.0 μg) and the indicated plasmids (5.0 μg). BrdU was added 24 hr post-transfection for a total labeling period of 20 hr. Cells were stained with anti-BrdU antibody, and the percentage of GFP-positive cells exhibiting BrdU incorporation was determined by indirect immunofluorescence. The average and deviation values shown are from two independent experiments with at least 200 GFP-positive cells counted per experiment. (B) Rat-1 cells were cotransfected with a puromycin-resistance plasmid (0.5 μg) and the indicated expression plasmids (5.0 μg). Transfected cells were selected with puromycin for 7 days and then stained with crystal violet. Pictures were taken at 20× magnification with a phase-contrast microscope. (C) Rat-1 cells were cotransfected with a puromycin-resistance plasmid (0.5 μg) and vector (lane 1), WT-LP (lane 2), or PSM.7-LP (lane 3) expression plasmids (5.0 μg). Alternatively, PSM.7-LP (2.5 μg) and either vector (lane 4) or p16ink4a (lane 5) expression plasmids (2.5 μg) were cotransfected with a puromycin-resistance plasmid (0.5 μg). Transfected cells were selected with puromycin for 72 hr and then lysed. Total protein (15 μg) was resolved by SDS-PAGE and the transfected RB protein was detected by immunoblotting with anti-RB antibody. (ppLP) Hyperphosphorylated large-pocket fragment of RB; (pLP) hypophosphorylated large-pocket fragment. (D) Rat-1 cells were cotransfected with a puromycin-resistance plasmid (0.5 μg) and vector (lane 3), WT-LP (lane 4), or PSM.7-LP (lane 5) plasmids (5.0 μg). Transfected cells were selected with puromycin for 72 hr and then harvested. As controls, quiescent (lane 1) or asynchronously growing (lane 2) Rat-1 cells were also harvested. Total protein (15 μg) was resolved by SDS-PAGE and the endogenous p107 or p130 proteins were detected by immunoblotting with the respective antibodies as indicated. (E) Rat-1 cells were cotransfected with a puromycin-resistance plasmid (0.5 μg) and vector (lanes 1,4), PSM.7-LP (lanes 2,5), or p16ink4a (lanes 3,6) plasmids (5.0 μg). Transfected cells were selected with puromycin for 72 hr and then harvested. Total protein (15 μg) was resolved by SDS-PAGE, and the endogenous p107 proteins were detected by immunoblotting.
To examine the long-term effect, cells were cotransfected with a plasmid expressing puromycin resistance, and either vector, WT-LP, PSM.7-LP, or p16ink4a expression plasmids. Transfected cells were subjected to selection with high concentrations of puromycin, through which all untransfected cells were killed within 72 hr (not shown). Labeling of puromycin-selected cells with [3H]thymidine showed robust incorporation with vector or WT-LP-transfected cells, but virtually no incorporation with PSM.7-LP-transfected cells (not shown). Following 7 days of culture, both vector and WT-LP-transfected cells grew into colonies, staining darkly with crystal violet (Fig. 1B). In contrast, PSM.7-LP and p16ink4a suppressed colony formation. Microscopic viewing showed the presence of single, flat cells in either PSM.7-LP or p16ink4a-transfected cultures (Fig. 1B). The morphologies of the PSM.7-LP and p16ink4a-arrested cells were indistinguishable from one another. These results are consistent with the role of RB as a negative regulator of cell proliferation.
RB, p107, and p130 are hyperphosphorylated in PSM-RB-arrested Rat-1 cells
The expression and phosphorylation status of the LP proteins was examined after 72 hr of puromycin selection. The WT-LP protein was predominantly hyperphosphorylated, which was consistent with the continued growth of these transfected cells (Fig. 1C, lane 2). The PSM.7-LP protein migrated during SDS-PAGE as a doublet (Fig. 1C, lanes 3,4), indicating that the remaining three cdk sites are phosphorylated (Knudsen and Wang 1997). Therefore, RB kinase activity does not appear to be inhibited in the PSM.7-LP-arrested cells. Although PSM.7-LP from cells arrested by its own expression migrated as a doublet (Fig. 1C, lane 3,4), it migrated as a single band with the coexpression of p16ink4a (Fig. 1C, lane 5). These observations suggest that cdk4/6 is active in PSM.7-LP-arrested cells but not in p16ink4a-arrested cells.
We also examined the phosphorylation status of p107 and p130 in the PSM-RB-arrested cells (Fig. 1D,E). In asynchronously growing Rat-1 cells, p107 and p130 were hyperphosphorylated and migrated slower than the p107 and p130 of quiescent cells (Fig. 1D, cf. lanes 1 and 2). The electrophoretic mobilities of p107 and p130 in the vector, WT-LP- or PSM.7-LP-transfected cells were similar to that of proliferating Rat-1 cells (Fig. 1D, lanes 3–5). Overproduction of p16ink4a has been shown to inhibit the phosphorylation of p107 and p130 (Del Sal et al. 1996; Sherr 1996; Alevizopoulos et al. 1997). The p107 protein from p16ink4a-arrested Rat-1 cells migrated faster than that from PSM.7-LP-arrested cells (Fig. 1E, cf. lanes 3 and 6 to lanes 2 and 5). Therefore, p16ink4a inhibits the phosphorylation of p107 and p130, whereas PSM-RB does not interfere with these phosphorylation events.
Cyclin A expression is suppressed in PSM-RB-arrested Rat-1 cells
We also examined the effect of PSM-RB on the expression and activity of several cdks, cyclins, and their inhibitors. The levels of cyclin D1, cyclin E, cdk2, p21cip1, and p27kip1 in PSM.7-LP-expressing cells were similar to those found in proliferating Rat-1 cells and vector- or WT-LP-transfected cells (Fig. 2A,B). In contrast, the levels of cyclin A, cyclin B, and cdc2 were reduced in PSM.7-LP-transfected cells (Fig. 2A,B). Consistent with the normal levels of cyclin E, cdk2, p21cip1, and p27kip1, we found that cyclin E-associated kinase activity was not inhibited by PSM.7-LP (Fig. 2C, αCyclin E). The total cdk2 activity was reduced in PSM.7-LParrested cells (Fig. 2C, αCdk2); this is likely a result of the inhibition of cyclin A expression.
Figure 2.


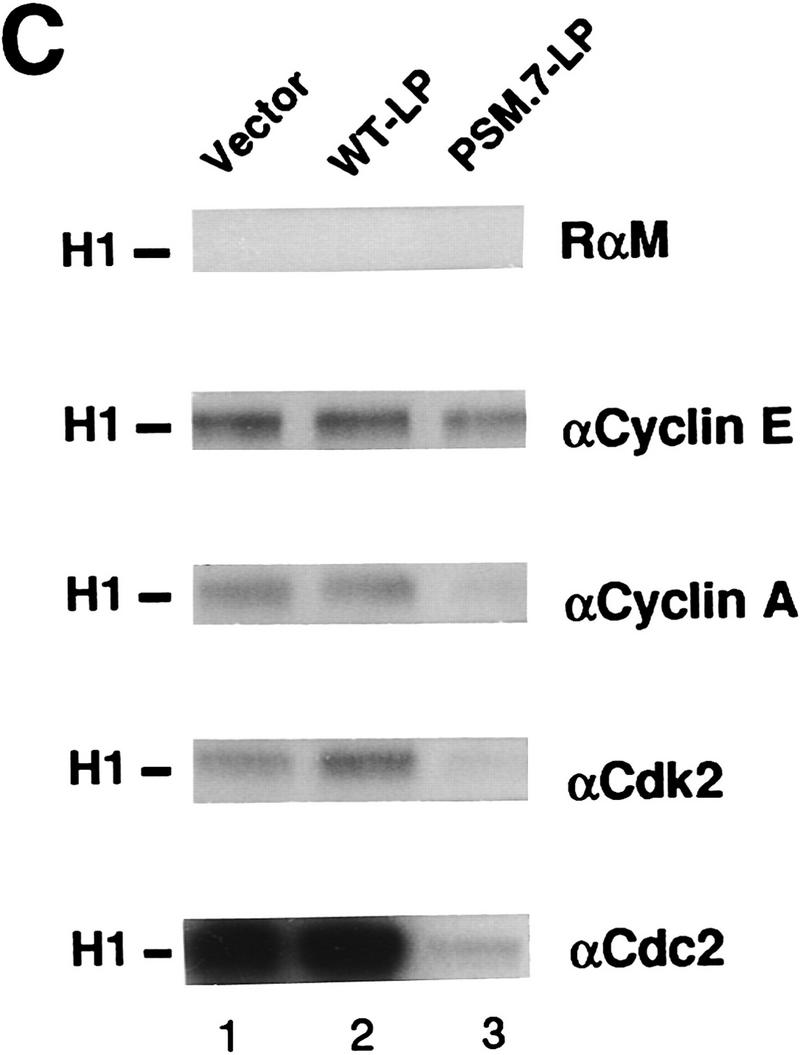

PSM-RB inhibits cyclin A expression in Rat-1 cells. (A) Rat-1 cells were cotransfected with a puromycin-resistance plasmid (0.5 μg) and vector (lane 3), WT-LP (lane 4), or PSM.7-LP (lane 5) expression plasmids (5.0 μg). Transfected cells were selected with puromycin for 72 hr and then harvested. As controls, quiescent (lane 1) or asynchronously growing (lane 2) Rat-1 cells were also harvested. Total protein (15 μg) was resolved by SDS-PAGE, and the endogenous cyclin D1, cyclin E, cyclin A, and cyclin B proteins were detected by immunoblotting with the respective antibodies. (B) Rat-1 cells were cotransfected with a puromycinresistance plasmid and the indicated expression plasmids, selected, and harvested as in A. Total protein (15 μg) was resolved by SDS-PAGE, and the endogenous cdk2, cdc2, p21cip1, and p27kip1 proteins were detected by immunoblotting with the respective antibodies. (C) Rat-1 cells were cotransfected with a puromycin-resistance plasmid and the indicated expression plasmids, selected, and harvested as in A. Twenty micrograms of total protein was utilized in in vitro kinase reactions with histone H1 as a substrate. Cdk/cyclin complexes were recovered by immunoprecipitation with the indicated antibodies against cyclin E, cyclin A, cdk2, or cdc2 and protein A–Sepharose. A nonspecific rabbit anti-mouse antibody was utilized as a negative control. Incorporation of 32P into histone H1 was visualized by autoradiography. (D) Rat-1 cells were cotransfected with a puromycin-resistance plasmid and the indicated expression plasmids, selected, and harvested as in A. Quiescent and asynchronous growing Rat-1 cells were used as controls. Total RNA was prepared. Quantitative RT-PCR was performed with 1–16 ng of total RNA as template. The level of cyclin A PCR product was normalized to an internal control PCR with primers that amplify the sequence of GAPDH (see Materials and Methods). Relative cyclin A mRNA level from five independent PCR reactions is shown, with the level found in asynchronous growing cells set to 100%.
To determine whether cyclin A was down-regulated at the RNA level, quantitative RT–PCR was used to measure the level of cyclin A mRNA. In Rat-1 cells transfected with vector and selected with puromycin, the level of cyclin A mRNA was similar to that of asynchronously growing cells (Fig. 2D). In contrast, in cells transfected with PSM.7-LP, the level of cyclin A mRNA was reduced to that of quiescent cells (Fig. 2D). Therefore, the expression of cyclin A mRNA is inhibited in PSM-RB arrested cells. Together, these results suggest that cells transfected with PSM-RB are predominantly blocked in late G1 prior to the expression of cyclin A.
We have shown previously that the ectopic expression of cyclin A can override the wild-type RB-mediated arrest of SAOS-2 cells (Knudsen and Wang 1996), or the v-Abl-mediated arrested of the NIH-3T3 subclone N-3T3 cells (Chen et al. 1996). Therefore, we attempted to override the G1-inhibitory effect of PSM-RB with the ectopic expression of cyclin A. When transfected into Rat-1 cells, the cyclin A expression plasmid did not lead to an increase in the overall levels of cyclin A protein (not shown). The inability of the cells to overproduce cyclin A in these cotransfection experiments could be attributable to an induction of cell death, as the cyclin A-transfected cells were found to exhibit condensed nuclei (not shown). Because the ectopic expression of cyclin A could not be achieved, we were unable to conclude whether cyclin A expression could rescue the PSM-RB-mediated G1 arrest in Rat-1 cells.
Cyclin E expression partially rescues the negative effect of PSM-RB on DNA synthesis
The ectopic expression of cyclin E abrogated the PSM-RB-induced G1 arrest. This was detected as a decrease in the G1 content and a concomitant increase in S phase by FACS analysis (Fig. 3A). However, cells transfected with cyclin E plus PSM.7-LP exhibited a peculiar profile of S-phase DNA content with no increase in G2 cells (Fig. 3A). This is in direct contrast to cells transfected with cyclin E plus p16ink4a, wherein cyclin E expression caused a return to a cell cycle profile similar to that of nontransfected (CD20-negative) cells (Fig. 3A).
Figure 3.
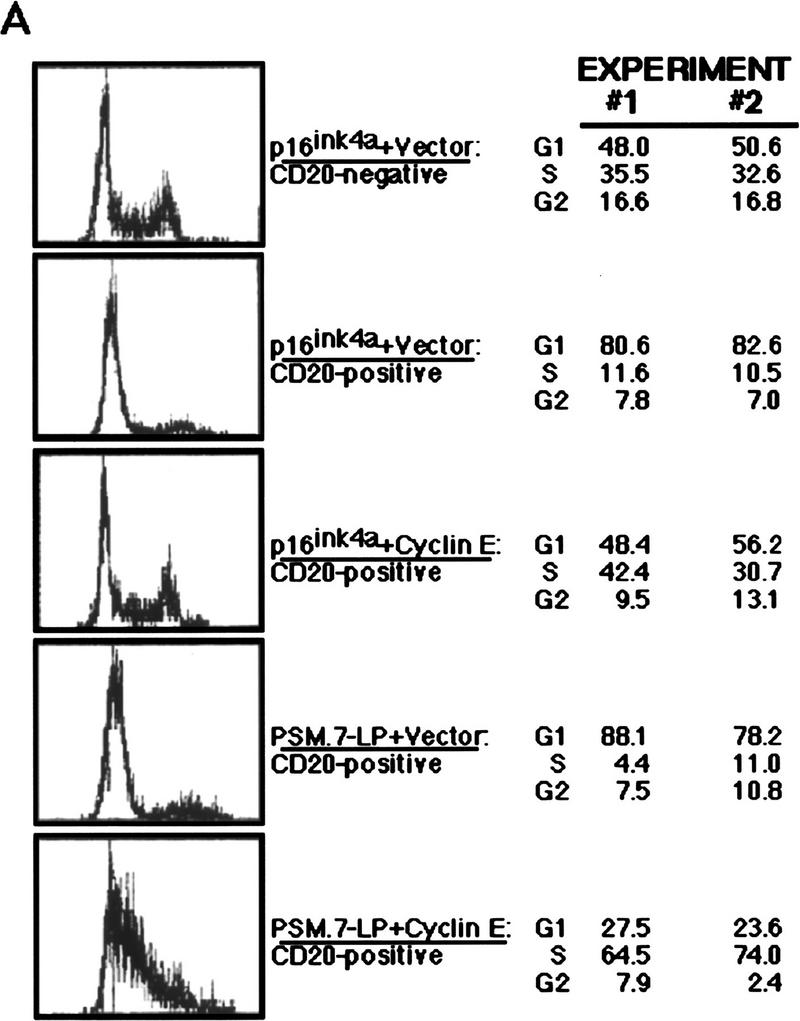

Ectopic expression of cyclin E overcomes the PSM-RB G1 arrest. (A) Rat-1 cells were cotransfected with a CD20 expression plasmid (1 μg) and expression plasmids encoding the indicated proteins (PSM.7-LP or p16ink4a, 5 μg; cyclin E, 10 μg). Cells were harvested 48 hr post-transfection by trypsinization, stained with FITC anti-CD20, fixed in ethanol, and then stained with propidium iodide. The DNA content of at least 2500 CD20-positive, or the bulk CD20-negative, cells was determined by use of a FACScan cytometer with Cellfit software. Representative histograms and the cell cycle distributions from two independent experiments are shown. (B) Cells cotransfected with a puromycin-resistance plasmid (0.4 μg) and the indicated expression plasmids: 2.0 μg of PSM.7-LP plus 4.0 μg each of vector (lanes 1,4), cyclin E (lane 2), or SV40 T-Ag (lane 5), and vector alone (lane 3), were selected with puromycin for 72 hr and then harvested. From these selected cells, 15 μg of total protein was resolved by SDS-PAGE and cyclin A protein was detected by immunoblotting.
Because cyclin A is required for DNA synthesis (Hunter and Pines 1994), we determined whether the expression of cyclin E could rescue the negative effect of PSM.7-LP on the expression of cyclin A. Following 72 hr of selection with puromycin, cells coexpressing cyclin E and PSM.7-LP had cyclin A protein at a level comparable to that of proliferating cells (Fig. 3B). Nevertheless, these transfected cells remained as single flat cells through the selection period, similar to those arising from the transfection of PSM.7-LP alone (not shown). The expression of SV40 large T-antigen (T-Ag) with PSM-RB also rescued the expression of cyclin A (Fig. 3B), and these cotransfected cells resumed proliferation, leading to the formation of colonies (not shown). Therefore, the ectopic expression of cyclin E overcame the inhibitory effect of RB on cyclin A expression and drove cells into S phase. However, cyclin E did not overcome the growth arrest activity of PSM-RB, whereas T-Ag completely abrogated the PSM-RB-induced growth arrest.
We also examined the incorporation of BrdU by cells transfected with cyclin E plus PSM-RB. Cells transfected with PSM.7-LP did not incorporate BrdU (Fig. 4A). When cotransfected with cyclin E, a punctate BrdU staining pattern was observed (Fig. 4A). The same result was obtained when the full-length PSM.9I-RB was used in the experiment. Again, PSM.9I-RB blocked BrdU incorporation, which was partially rescued by the coexpression of cyclin E. To determine quantitatively the amount of BrdU incorporation, a digital CCD camera was used to compare the intensity of BrdU signal, and these analyses showed that the intensity of punctate staining was ∼25% of the BrdU incorporation observed in homogeneously labeled nuclei (data not shown). This result was consistent with the FACS profile (Fig. 3A) and suggested that cells cotransfected with cyclin E and PSM-RB initiated DNA synthesis but incorporated BrdU at a significantly reduced rate.
Figure 4.

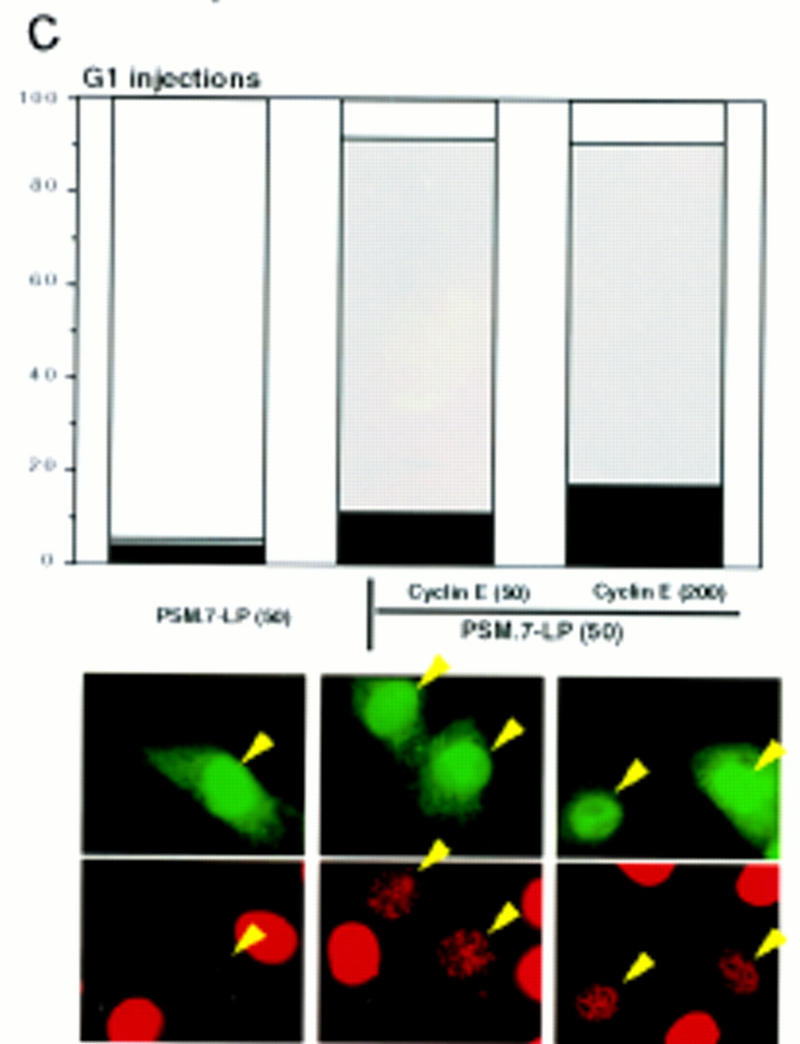
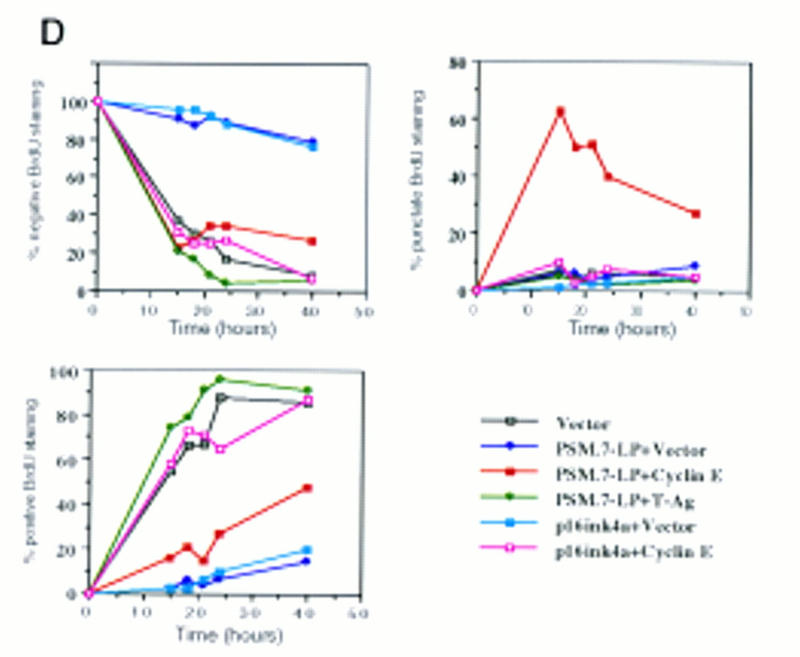
Progression of cyclin E-induced DNA synthesis is inhibited by PSM-RB. (A) Rat-1 cells were cotransfected with a GFP expression plasmid (0.4 μg) and the indicated expression plasmids (2.0 μg of PSM.7-LP, PSM.9I-RB or p16ink4a; 4.0 μg of cyclin E or vector). BrdU was added at 24 hr post-transfection and cells were labeled for 18 hr. Cells were fixed and stained with anti-BrdU antibody (red). Photographs were taken at 63× magnification. The lower four panels were printed in blow-up to better visualize the punctate BrdU labeling. (Arrows) GFP-positive (green) cells. (B) Rat-1 cells were cotransfected with GFP and the indicated expression plasmids as described in A. BrdU was added 24 hr post-transfection, and cells were labeled for 15 hr. The percentage of GFP-positive cells exhibiting homogenous (positive, solid bars), punctate (bottom open bars), or no (negative, top open bars) BrdU labeling was determined. Values are the average from two independent experiments with at least 200 GFP-positive cells counted per experiment. (C) Rat-1 cells were made quiescent by culturing in serum-free media for 72 hr. These cells were stimulated with 10% serum for 2 hr and then microinjected with the indicated plasmids at the following concentrations: GFP, 100 ng/μl; PSM.7-LP, 50 ng/μl; cyclin E, 50 or 200 ng/μl. Immediately following injection, BrdU was added, cells were labeled for 18 hr, and then fixed and stained for BrdU incorporation. The percentage of productively injected (GFP-positive) cells exhibiting homogenous (positive), punctate, or no (negative) BrdU-labeling was determined (see B for explanation). Values shown are the average from two independent experiments with at least 50 injected cells counted per experiment. Representative photographs taken at 63× magnification are shown. (Arrowheads) GFP-positive cells. (D) Rat-1 cells were cotransfected GFP and the indicated expression plasmids as described in A. BrdU was added 24 hr post-transfection. Cells were collected at 15, 18, 21, 24, and 40 hr after BrdU addition and processed for indirect immunofluorescence detection of BrdU incorporation. The percentage of GFP-positive cells exhibiting homogenous (positive), punctate, or no (negative) BrdU labeling was determined for each time point. Values shown are the average from two independent experiments with at least 200 GFP-positive cells counted per experiment. Note that punctate staining was only observed with the combined transfection of PSM.7-LP and cyclin E.
Quantitation of cells with homogenous (positive), punctate, or negative BrdU staining was determined for several different transfections (Fig. 4B). Coexpression of cyclin E with p16ink4a resulted in strong homogenous BrdU staining at a level comparable to that of vector-transfected cells (Fig. 4B). The punctate BrdU staining was not observed when cyclin E was coexpressed with WT-LP (Fig. 4B). In contrast to cyclin E, coexpression of T-Ag with PSM-RB resulted in strong, homogenous BrdU staining (Fig. 4B). Thus, T-Ag but not cyclin E can completely overcome the negative effect of PSM-RB on DNA synthesis.
To determine whether the reduced BrdU incorporation was the result of a limitation on the amount of cyclin E, we carried out microinjection experiments with cells synchronized in early G1 (Fig. 4C). Identical to the result observed with transfection, microinjection of PSM.7-LP blocked the incorporation of BrdU, while coinjection with PSM.7-LP and cyclin E gave rise to punctate BrdU staining (Fig. 4C). The punctate staining was observed at both 1:1 and 1:4 ratios of PSM-RB to cyclin E (Fig. 4C), indicating that a limitation on cyclin E is not likely to be responsible for the retardation in S-phase progression.
To determine whether the reduced BrdU incorporation represented a block in S phase or a slower rate of DNA synthesis, cells were labeled for up to 40 hr and the percentage of cells exhibiting positive, punctate, or negative BrdU staining was determined (Fig. 4D). We found that after 40 hr of continuous labeling, punctate staining persisted in cells expressing PSM.7-LP and cyclin E (closed red squares). There was a gradual decline in punctate staining and a concomitant increase in the percentage of cells exhibiting homogenous BrdU labeling, suggesting a slow progression through S phase. In contrast, cells coexpressing p16ink4a plus cyclin E (open red squares) or PSM-RB plus T-Ag (closed green diamonds) showed BrdU labeling similar to vector transfected cells over the experimental time course (Fig. 4D). Following extended periods of culture, those cells that coexpressed p16ink4a and cyclin E gave rise to microcolonies of two to eight adjoining GFP-positive cells, indicating completion of the cell cycle (not shown). However, cells coexpressing PSM.7-LP and cyclin E remained as single GFP-positive cells, suggesting that they did not undergo cell division during the course of the experiment (not shown). Taken together, the results presented in Figures 3 and 4 show that increased expression of cyclin E can drive PSM-RB-arrested cells into S phase but DNA synthesis is retarded in the presence of PSM-RB.
PSM-RB inhibits DNA synthesis when expressed in S-phase-committed cells
To demonstrate directly that PSM-RB can inhibit DNA synthesis, we performed microinjection experiments with cells that have passed through the G1 restriction point (Fig. 5). The DNA polymerase inhibitor aphidicolin was used to reversibly arrest asynchronous Rat-1 cells in S phase. After the removal of aphidicolin, these cells completed DNA replication and entered G2 phase within 6 hr, as determined by flow cytometry (not shown). Aphidicolin-treated Rat-1 cells were arrested beyond the G1 restriction point, because they completed DNA replication after release in the presence or absence of serum (Fig. 5A, flow cytometry not shown). Furthermore, aphidicolin-arrested cells contained a high level of cyclin A protein and hyperphosphorylated RB (Fig. 5B). These aphidicolin-arrested cells were coinjected with the expression plasmids of interest and GFP. The accumulation of plasmid-encoded proteins was allowed to occur over 16 hr in the presence of aphidicolin. Cells were then released from the aphidicolin block, and the effect of the plasmid-encoded proteins was measured by monitoring of BrdU incorporation in the GFP-positive cells (Fig. 5C).
Figure 5.
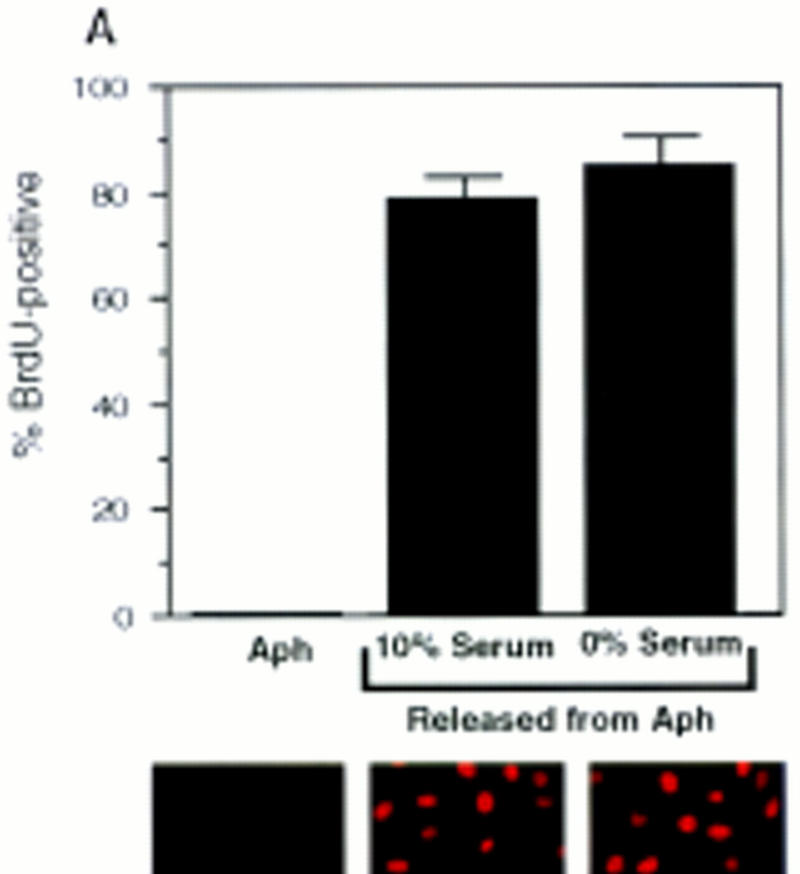

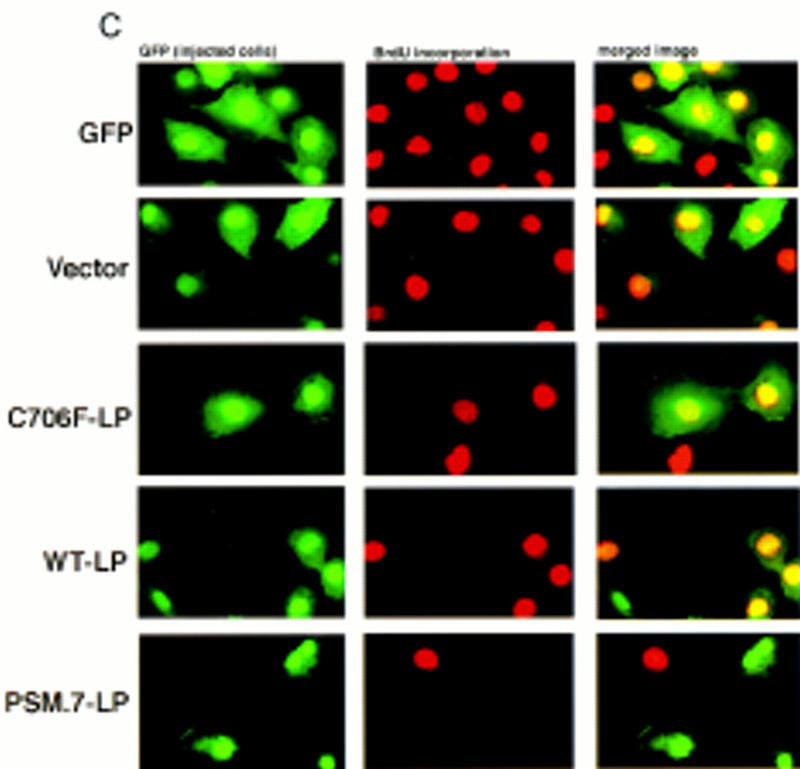



PSM-RB inhibits both the G1/S transition and S-phase progression. (A) Rat-1 cells were synchronized in early S phase by culturing for 24 hr in growth medium supplemented with aphidicolin. Cells were then labeled with BrdU for 6 hr, either in the presence of aphidicolin or released from the aphidicolin block in the presence of 10% or 0% serum. The percentage of BrdU-positive cells was determined. Values shown are the average and deviation from two independent experiments with over 200 cells counted per experiment. Representative photographs taken at 63× magnification are shown below the graph. (B) Rat-1 cells which were quiescent (lane 1), asynchronous growing (lane 2), or arrested in S phase by aphidicolin (lane 3) were harvested. Total protein (15 μg) was resolved by SDS-PAGE, and cyclin A was detected by immunoblotting. Alternatively, 0.5 mg of total protein was subjected to immunoprecipitation with polyclonal anti-RB antibodies (851), immunocomplexes were resolved on SDS-PAGE, and RB protein was then detected by immunoblotting. (C) Rat-1 cells synchronized in S phase by culturing for 24 hr with aphidicolin were comicroinjected with a GFP expression plasmid (100 ng/μl) and the indicated expression plasmids (50 ng/μl). Accumulation of the plasmid-encoded proteins was allowed to occur over an additional 16 hr of culture in aphidicolin. Cells were then washed extensively and cultured in the presence of 10% serum and BrdU. After 6 hr of labeling, cells were fixed and stained for BrdU-incorporation. Representative photographs of cells were taken at 63× magnification. GFP (green) and BrdU (red) images are merged in the right-hand column. (D) Rat-1 cells synchronized in S phase were coinjected as described in C. The wild-type and mutant RB plasmids were injected at the indicated DNA concentration (ng/μl). The percentage of injected (GFP-positive) cells exhibiting BrdU incorporation was determined. Values shown are the average and deviation from at least two independent microinjection experiments with ∼70 GFP-positive cells counted per experiment. With the coinjection of PSM.7-LP, between 20%–25% of the GFP-positive cells appeared to be resistant to the inhibitory effect. Note that WT-LP at a high concentration also inhibited DNA synthesis in S-phase cells. (E) Rat-1 cells synchronized in early G1 at 2 hr after serum stimulation from quiescence were comicroinjected with expression plasmids for GFP (100 ng/μl) and PSM.7-LP at the indicated DNA concentrations (0.4 ng/μl = 1–4 plasmid molecules per injected cell, 2.0 ng/μl = 5–20 plasmid molecules per injected cell, 50 ng/μl = 125–500 plasmid molecules per injected cell). BrdU was immediately added to the medium and cells were labeled for an additional 18 hr. The percentage of GFP-positive cells with BrdU staining was determined. Values shown are the average and deviation from two independent experiments with ∼70 GFP-positive cells counted per experiment. (F) Rat-1 cells synchronized in S phase by culturing for 24 hr with aphidicolin were comicroinjected with expression plasmids for GFP (100 ng/μl) and PSM.7-LP at the indicated DNA concentrations (0.4 ng/μl = 1–4 plasmid molecules per injected cell, 2.0 ng/μl = 5–20 plasmid molecules per injected cell, 50 ng/μl = 125–500 plasmid molecules per injected cell). Accumulation of the plasmid encoded proteins was allowed to occur over an additional 16 hr in aphidicolin. Cells were then washed extensively, labeled for 6 hr with BrdU in 10% serum and processed for indirect immunofluorescence analysis. Values shown are the average and deviation from two independent experiments with ∼70 GFP-positive injected cells counted per experiment.
Coinjection of vector or the defective RB mutant C706F-LP did not inhibit DNA synthesis, as 70%–90% of GFP-positive cells stained positive for BrdU (Fig. 5C,D). This failure to inhibit S-phase progression occurred even at plasmid concentrations up to 200 ng/μl (500–2000 plasmid molecules per injected cell; Fig. 5D). The WT-LP did not inhibit DNA synthesis at 10 or 50 ng/μl (25–100 or 125–500 plasmid molecules per injected cell, respectively); however, WT-LP did interfere with BrdU incorporation when injected at 200 ng/μl (500–2000 plasmid molecules per injected cell; Fig. 5D). The expression of PSM.7-LP, on the other hand, reduced the incorporation of BrdU at 10 ng/μl. Only 20%–25% of the GFP-positive cells were found to incorporate BrdU with the coinjection of PSM-RB (Fig. 5D).
To compare the sensitivity of S-phase cells and G1 cells to the inhibitory effect of PSM.7-LP, titration experiments were performed (Fig. 5E,F). At a DNA concentration of 0.4 ng/μl, which corresponded to the injection of one to four molecules of plasmid per cell, PSM.7-LP reduced BrdU incorporation in both G1 and S-phase cells, albeit not significantly (Fig. 5E,F). At a concentration of 2 ng/μl (5–20 molecules of plasmid per injected cells), PSM.7-LP maximally inhibited BrdU incorporation in G1 and S-phase cells. Injection of DNA at 50 ng/μl concentration did not further increase the extent of inhibition (Fig. 5E,F). These results indicate that PSM-RB can inhibit both the entry and the progression of S phase at similar levels of expression. While a majority of the G1 cells are sensitive to the inhibitory effect of PSM.7-LP (Fig. 5E), a subset of the aphidicolin-arrested cells appeared to be resistant to PSM.7-LP because we consistently observed BrdU incorporation in 20%–25% of GFP-positive cells coinjected with PSM-RB (Fig. 5D,F).
To rule out the possibility that the inhibitory effect of PSM.7-LP on S-phase progression was attributable to the prolonged incubation (40 hr) with aphidicolin, we microinjected S-phase cells that were generated by two other protocols with shorter periods of treatment (20 hr) or no aphidicolin treatment.
To reduce the time in aphidicolin, cells were made quiescent by serum withdrawal, restimulated with serum for 14 hr, and then treated with aphidicolin for 6 hr to block cells in early S phase. These cells were injected, cultured for an additional 14 hr in aphidicolin to allow for the accumulation of proteins encoded by the plasmids, and then released from the aphidicolin block with BrdU. Under these conditions, PSM.7-LP inhibited S-phase progression with efficacy similar to injections performed with cells that had been treated with aphidicolin for 40 hr (Fig. 6A).
Figure 6.

PSM-RB inhibits DNA synthesis in S-phase cells synchronized by different protocols. (A) Rat-1 cells were made quiescent by culturing in serum-free media for 72 hr and then stimulated with 10% serum for 14 hr, at which time aphidicolin was added for an additional 6 hr. These S-phase cells were microinjected with the indicated expression plasmids (100 ng/μl GFP; 50 ng/μl PSM.7-LP, or 50 ng/μl of vector) and cultured for an additional 14 hr in aphidicolin to allow protein accumulation. These cells were released from the aphidicolin arrest, labeled with BrdU for 6 hr, and then processed for the determination of BrdU incorporation. Values shown are the average and deviation from two independent experiments with over 100 productively injected cells (GFP-positive) counted per experiment. (B) Rat-1 cells made quiescent as described in A were stimulated with 10% serum. BrdU was added at 2 or 12 hr postserum stimulation and labeling was conducted in 0% or 10% serum. Cells were harvested at 24 hr postserum stimulation and BrdU incorporation was determined by immunofluorescence microscopy. Values shown are the average and deviation from two independent experiments with >200 cells counted per experiment. These data showed that Rat-1 cells became serum independent for DNA synthesis at 12 hr post-stimulation. (C) Rat-1 cells were microinjected with the indicated expression plasmids at 12 hr postserum addition. BrdU was added at 6 hr postinjection and cells were labeled for an additional 6 hr (harvested at 24 hr postserum stimulation). Cells were processed for the determination of BrdU incorporation by immunofluorescence microscopy. Values shown are the average and deviation from two independent experiments with ∼50 productively injected (GFP-positive) cells counted per experiment.
To eliminate aphidicolin treatment, we injected synchronized cells that have passed through the G1 restriction point with the PSM-RB expression plasmid. To identify the time point suitable for injection, we measured BrdU incorporation in the presence or absence of serum at different times after stimulation of quiescent cells. As shown in Figure 6B, removal of serum at 2 hr inhibited BrdU incorporation, however, removal of serum at 12 hr did not inhibit BrdU incorporation. These results demonstrated that virtually all of the Rat-1 cells had passed through the G1 restriction point at 12 hr after serum stimulation. We therefore microinjected the PSM-RB expression plasmid at 12 hr after serum stimulation and labeled cells with BrdU at 6 hr postinjection (Fig. 6C). When cells were coinjected with vector at 12 hr, >80% of the injected (GFP-positive) cells incorporated BrdU, in the presence or absence of serum (Fig. 6C, and data not shown). When cells were coinjected with the PSM.7-LP expression plasmid, however, only 40%–50% of the GFP-positive cells incorporated BrdU (Fig. 6C). The same extent of inhibition of BrdU incorporation was achieved by the injection of the PSM.7-LP RB expression plasmid whether the BrdU labeling was performed in the presence or the absence of serum. The reduced effect of PSM.7-LP in this experimental protocol could be attributable to the reduced time allowed for protein accumulation and possibly the development of resistance to PSM-RB as cells progress into late S phase. Nevertheless, PSM-RB clearly interfered with DNA synthesis in S-phase-committed cells, even in the complete absence of aphidicolin treatment.
The S-phase inhibitory effect of PSM-RB was not recapitulated with the microinjection of three inhibitors of cdk: p16ink4a, p21cip1, and p27kip1 (Fig. 7A). Each of these inhibitors, however, was capable of inhibiting G1/S transition when they were injected into G1 cells (Fig. 7B). In these experiments, microinjection with PSM.7-LP inhibited BrdU incorporation in either S-phase or G1 cells. These results further support that PSM-RB can inhibit DNA synthesis beyond G1/S transition.
Figure 7.


S-phase inhibitory effect of PSM-RB is not observed with cdk inhibitors. (A) Rat-1 cells arrested in S-phase by culturing for 24 hr with aphidicolin were microinjected with the indicated expression plasmids (GFP, 100 ng/μl; p16ink4a, p21cip1, p27kip1, or PSM,7-LP, 50 ng/μl). Following 16 hr more in aphidicolin to allow the accumulation of the plasmid-encoded protein, cells were released from the aphidicolin block and labeled with BrdU for 6 hr. Cells were then fixed and stained for BrdU incorporation. The percentage of GFP-positive cells with BrdU staining was determined. Values shown are the average and deviation from two independent experiments with ∼70 GFP-positive cells counted per experiment. (B) Rat-1 cells were synchronized in early G1 by a 2-hr stimulation of quiescent cells with 10% serum. These early G1 cells were microinjected with the indicated expression plasmids (GFP, 100 ng/μl; p16ink4a, p21cip1, p27kip1, or PSM,7-LP, 50 ng/μl). BrdU was immediately added to the media, cells were labeled continuously for 18 hr and then processed for immunofluorescence analysis. Values shown are the average and deviation from two independent experiments with ∼70 productively injected cells (GFP-positive) counted per experiment.
T-Ag, E1A, and E2F-1 but not cyclin E or cyclin A can reverse the PSM-RB inhibition of S-phase progression
To determine whether the negative effect of PSM-RB on DNA synthesis was dependent on its protein-binding function, we coinjected T-Ag with PSM.7-LP into aphidicolin-synchronized S-phase cells (Fig. 8A). Wild-type T-Ag effectively reversed the RB-mediated inhibition of BrdU incorporation (Fig. 8A,B). Two T-Ag mutants, K1 (Fig. 8A,B) and H42Q (Fig. 8B), did not overcome the inhibitory effect of PSM-RB. The K1 mutant is defective in binding RB (DeCaprio et al. 1988), while the H42Q mutant binds to RB but cannot disrupt the RB–E2F complex (Zalvide et al. 1998). The E1A protein of adenovirus also reversed the PSM-RB-mediated inhibition of BrdU incorporation. These results suggest that the formation of a PSM-RB–E2F complex may be required for the observed inhibition of S-phase progression.
Figure 8.
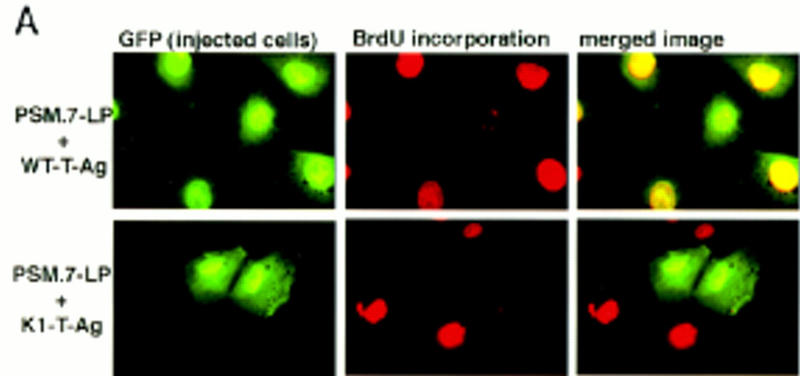


T-Ag, E1A, and E2F-1, but not cyclin E or cyclin A, reverse the S-phase inhibition by PSM-RB. (A,B) Rat-1 cells synchronized in S phase by 24 hr of culturing with aphidicolin were microinjected with a GFP expression plasmid (100 ng/μl) plus the indicated combinations of expression plasmids (50 ng/μl each). Accumulation of the plasmid-encoded proteins was allowed to occur over an additional 16 hr in aphidicolin. Cells were then extensively washed and cultured in medium containing 10% serum and BrdU. Labeling was carried out for 6 hr at which point cells were fixed and stained for BrdU-incorporation. (A) Representative photographs of cells taken at 63× magnification. (B) Quantitation of BrdU-positive injected cells. Values shown are the average and deviation from two independent microinjection experiments with ∼70 productively injected (GFP-positive) cells counted per experiment. The K1 mutant of T-Ag does not bind to RB (DeCaprio et al. 1988). The H42Q mutant of T-Ag binds RB but cannot disrupt RB-E2F complex (Zalvide et al. 1998). (C) Rat-1 cells synchronized in S phase by aphidicolin were microinjected with a GFP expression plasmid (100 ng/μl) and the indicated combinations of expression plasmids at the indicated DNA concentrations (ng/μl). Accumulation of the plasmid-encoded proteins was allowed to occur over an additional 16 hr culture in aphidicolin. Cells were then extensively washed and cultured in medium containing 10% serum and BrdU. Labeling was carried out for 6 hr at which point cells were fixed and stained for BrdU-incorporation. Values shown are the average and deviation from two independent microinjection experiments with ∼70 (GFP-positive) injected cells counted per experiment, with the exception of PSM.7-LP alone, which is from B and shown only for comparative purposes.
We also coinjected PSM.7-LP with an E2F-1 expression plasmid (Fig. 8C). When this plasmid was injected at a DNA concentration of 50 ng/μl, we did not observe a significant rescue of DNA synthesis (Fig. 9C). The GFP-positive cells in this coinjection experiment showed an increased incidence of condensed chromatin, indicative of apoptosis (not shown). This is consistent with previous reports that overproduction of E2F-1 can induce apoptosis in S-phase cells (Qin et al. 1994; Shan and Lee 1994; Kowalik et al. 1995). At a much higher DNA concentration (200 ng/μl), coinjection with the E2F-1 expression plasmid did overcome the PSM.7-LP-mediated inhibition of BrdU incorporation (Fig. 8C), with some BrdU-positive cells also showing condensed chromatin (not shown). Although overproduction of E2F-1 could reverse the negative effect of PSM.7-LP on S-phase progression, coinjection with plasmids expressing either cyclin E or cyclin A at similar DNA concentrations was unable to rescue DNA synthesis (Fig. 8C). Moreover, the coexpression of cyclin E and PSM-RB in aphidicolin-synchronized S-phase cells did not lead to punctate staining (not shown). Taken together, these results suggest that the S-phase inhibitory activity of RB is mediated by its protein-binding function, and this activity cannot be suppressed by the increased expression of cyclin E or cyclin A.
Figure 9.

A schematic summary of the inhibitory activity of PSM-RB on G1/S transition and S-phase progression. Cell cycle inhibitory effects observed in this study are depicted by the vertical T-bars. The lack of any effect is depicted by open circles. The solid arrows indicate an observed rescue of the negative effect. (R) Restriction point. PSM-RB inhibits S-phase entry and this is correlated with the inhibition of cyclin A expression. Three cdk inhibitors: p16ink4a, p21cip1, and p27kip1, each can also inhibit G1/S transition. Unlike p16ink4a, PSM-RB does not interfere with the phosphorylation of p107 and p130. PSM-RB also inhibits DNA synthesis when it is expressed in cells that have traversed the G1 restriction point. These post-R cells are not inhibited by the three cdk inhibitors tested. Overproduction of the SV40 T-Ag can overcome both the G1/S and the S-phase inhibitory function of PSM-RB. Overproduction of cyclin E can overcome the G1 block, induced by either p16ink4a or PSM-RB. However, overproduction of cyclin E cannot overcome the negative effect of PSM-RB on S-phase progression. These results suggest that unphosphorylated RB can interfere with DNA synthesis beyond the G1-restriction point, and this negative function of RB does not appear to be mediated by the inhibition of the known G1/S cyclin/cdk activities.
Discussion
Cyclin E and the RB-related proteins are not the targets of PSM-RB in Rat-1 cells
Two tumor suppressors, p16ink4a and RB, are components of a single growth inhibitory pathway that is mutated in a majority of human tumor cells (Sherr 1996; Palmero and Peters 1996). Overproduction of p16ink4a blocks cells in G1 by preventing the phosphorylation of RB and related proteins p107 and p130 (Sherr 1996; Alevizopoulos et al. 1997). We found that the endogenous p107 and p130 proteins are hyperphosphorylated in PSM-RB-arrested cells but they are hypophosphorylated in p16ink4a arrested cells. This observation suggests that PSM-RB, unlike p16ink4a, does not inhibit the activity of cdk4/cyclin D. Therefore, p16ink4a and PSM-RB have distinct targets during G1 progression (Fig. 9), and there is not a feedback regulatory mechanism associated with the p16ink4a–RB pathway.
Cyclin E has been proposed to be a target of regulation by RB. The repression of cyclin E expression in quiescent cells is thought to be mediated by RB (Herrera et al. 1996a; Hurford et al. 1997). The cyclin E promoter contains E2F-binding sites, and this promoter activity can be inhibited by RB in transient transfection assays (Ohtani et al. 1995; Geng et al. 1996). Furthermore, in RB-deficient murine embryo fibroblasts, cyclin E expression is advanced and correlates with the shortening of G1 (Herrera et al. 1996b; Hurford et al. 1997). In Rat-1 cells, cyclin E protein levels are not reduced in quiescent cells. In these cells, PSM-RB does not inhibit the expression of cyclin E. Similar observations have been reported for p16ink4a-arrested cells (Alevizopoulos et al. 1997; Lundberg and Weinberg 1998). The kinase activity of cyclin E–cdk2 is inhibited as Rat-1 cells exit the cell cycle (Resnitzky and Reed 1995). However, PSM-RB does not inhibit cyclin E/cdk2 kinase activity, most likely because PSM-RB does not influence the levels of cdk-inhibitors such as p21cip1 and p27kip1. Thus, cyclin E expression and cyclin E-associated kinase activity are not the targets of PSM-RB in Rat-1 cells.
PSM-RB causes repression of cyclin A expression that can be relieved by cyclin E
The phosphorylation of RB during G1 progression coincides with the traversal of the G1 restriction point, beyond which cells are committed to DNA synthesis and become insensitive to positive and negative growth signals (Weinberg 1995; Del Sal et al. 1996). The observation that PSM-RB can induce G1 arrest supports the hypothesis that unphosphorylated RB prevents entry into S phase. Our results, together with those reported by Lukas et al. (1997), suggest that the overall activity of cdk2 is rate-limiting for S-phase entry and unphosphorylated RB interferes with the accumulation of active cdk2 complexes. In Rat-1 cells, PSM-RB achieves this goal by suppressing the expression of cyclin A (Fig. 9).
Cyclin A mRNA is down-regulated in quiescent Rat-1 cells and stimulated in late G1 (Resnitzky and Reed 1995; Resnitzky et al. 1995). The cyclin A promoter also contains E2F-binding sites that have been implicated in promoter repression (DeGregori et al. 1996; Slansky et al. 1996). A PSM-RB–E2F complex may directly bind to and repress the cyclin A promoter in PSM-RB-arrested cells. We have shown that the coexpression of SV40 T-Ag can overcome the PSM-RB-mediated inhibition of cyclin A expression (Fig. 9). This is consistent with the idea that a PSM-RB–E2F complex causes the repression of cyclin A promoter.
It is interesting to find that overproduction of cyclin E can also activate the expression of cyclin A in cells expressing PSM-RB (Fig. 9). Lukas et al. (1997) have shown that cyclin E cannot induce the E2F transactivating function in cells expressing an unphosphorylatable RB protein (RBΔcdk, which is similar but not identical to PSM-RB). Thus, the activation of E2F activity is not responsible for the observed cyclin A expression. There are two possible mechanisms by which the overproduction of cyclin E may relieve the repressive function of PSM-RB. First, increased cyclin E kinase activity may inactivate the DNA-binding function of E2F and thus remove the PSM-RB–E2F complex from the cyclin A promoter. Alternatively, cyclin E kinase may disrupt the transcription repression function of PSM-RB. Three recent reports have shown that RB can recruit a histone deacetylase to repress transcription (Brehm et al. 1998; Luo et al. 1998; Magnaghi-Jaulin et al. 1998). The cyclin A promoter could conceivably be derepressed by a direct inhibition of the histone deacetylase activity through the increased expression of cyclin E.
PSM-RB can inhibit DNA synthesis beyond the G1 restriction point
The previous model predicts that once a threshold level of cdk2 activity is achieved and DNA synthesis has been initiated, then RB would have no further effect on cell cycle progression (Weinberg 1995). This notion is based on two lines of evidence: First, microinjection of RB protein into G1 SAOS-2 cells can inhibit DNA synthesis, but no inhibition was observed when it was injected into S-phase SAOS-2 cells (Goodrich et al. 1991). Second, overproduction of cyclin E can overcome the effect of p16ink4a and RBΔcdk and drive the completion of the cell cycle (Alevizopoulos et al. 1997; Lukas et al. 1997). Because S-phase cells contain a high level of cdk/cyclin kinase activity, RB may become phosphorylated and inactivated under those previous experimental conditions.
As demonstrated here, PSM-RB is capable of inhibiting S-phase progression beyond the G1 restriction point (Fig. 9). Several lines of evidence support this conclusion. First, overproduction of cyclin E cannot overcome the negative effect of PSM-RB on DNA synthesis. The punctate BrdU labeling indicates that cyclin E can drive the initiation of DNA synthesis, but DNA synthesis cannot proceed at a normal rate in the presence of PSM-RB. Second, expression of PSM-RB in aphidicolin-synchronized S-phase cells inhibits BrdU incorporation. Aphidicolin-synchronized cells have passed through the G1 restriction point as indicated by (1) their ability to complete the cell cycle in serum-free media; (2) the accumulation of cyclin A and the hyperphosphorylation of the endogenous RB; and (3) their resistance to several cdk inhibitors: p16ink4a, p21cip1, and p27kip1. Others have reported that microinjection of anti-cyclin A antibodies into aphidicolin-synchronized S-phase cells does not interfere with DNA synthesis (Pagano et al. 1992), and that the addition of p21cip1 into aphidicolin-released Xenopus egg extracts does not inhibit DNA synthesis (Strausfeld et al. 1994). These observations are consistent with our results that microinjection of p21cip1 does not interfere with S-phase progression from aphidicolin release, and the ectopic expression of cyclin A cannot rescue the negative effect of PSM-RB. The amount of PSM-RB required to achieve a maximal level of inhibition of DNA synthesis in S-phase cells is similar to that required to induce G1 arrest. While the G1/S inhibition by PSM-RB is overcome by cyclin E, the S-phase inhibition cannot be overcome by cyclin E. Finally, at a high level of expression (500–2000 molecules of plasmids per injected cell), the wild-type RB was found to inhibit DNA synthesis in S-phase cells (Fig. 5D). Taken together, these results indicate that unphosphorylated RB can inhibit DNA synthesis through a mechanism that is independent of the inhibition of the known G1/S cdk–cyclin activities.
Possible mechanisms for the PSM-RB-mediated inhibition of S-phase progression
The negative effect of PSM-RB on S-phase progression requires the protein-binding function of RB. Because the LP fragment of PSM-RB can inhibit S-phase progression the amino-terminal region of RB is dispensable for the function. The SV40 large T-Ag can overcome both the G1/S and the S-phase inhibitory functions of PSM-RB. The K1 mutant of T-Ag, unable to bind RB (DeCaprio et al. 1988), does not override the inhibitory effect of RB on DNA synthesis. Interestingly, the H42Q mutant, which binds RB but cannot disrupt the RB–E2F complex (Zalvide et al. 1998), is also unable to overcome the negative effect of PSM-RB. Moreover, a high level of E2F-1 expression can override the effect of PSM-RB on S-phase progression. These results implicate the inhibition of E2F activity by PSM-RB as a potential mechanism. E2F-binding sites are found in increasing number of promoters that are activated for DNA synthesis (DeGregori et al. 1995; Slansky et al. 1995; Ohtani et al. 1996; Kaelin 1997). The negative effect of PSM-RB on S-phase progression may be attributable to the suppression of E2F-activated genes that have to be expressed continuously throughout S phase to achieve the efficient replication of genomic DNA. Alternatively, the S-phase inhibitory activity of RB might be mediated by its interaction with cellular targets other than E2F. T-Ag may disrupt the formation of these non-E2F RB complexes. The overproduction of E2F-1 may also displace RB from its physiological targets in S-phase cells. Whether the S-phase inhibitory function of RB is mediated by transcription regulation through E2F or by other mechanisms awaits further investigation.
Regulation of RB phosphorylation during S phase
The continued phosphorylation of RB in S-phase cells has previously been thought to be a bystander effect resulting from the high levels of cdk/cyclin activity. However, RB protein levels have been observed to increase as cells enter S phase (Chen 1988; Gill et al. 1994; Chen et al. 1996), which is a wasteful regulation if RB is without any function beyond the restriction point. Our finding that PSM-RB impedes DNA synthesis suggests that the continued phosphorylation of RB is a requirement for the completion of DNA replication. It is paradoxical to observe that PSM-RB, but not p21cip1 or p27kip1, can inhibit DNA synthesis when expressed in aphidicolin-synchronized S-phase cells. There are two possible interpretations for these results. First, the amount of p21cip1 or p27kip1 expressed might not be sufficient to inhibit the cdk/cyclin kinase activities in S-phase cells. Alternatively, other RB kinases that are insensitive to p21cip1 and p27kip1 may exist in S-phase cells to phosphorylate RB.
Our results predict that dephosphorylation of RB during S phase will retard the rate of DNA synthesis. We have observed a rapid dephosphorylation of RB in S-phase cells that are exposed to the DNA cross-linking agent cisplatin (E.S. Knudsen, D. Booth, and J.Y.J. Wang, unpubl.). The dephosphorylation of RB occurs in cisplatin-treated S-phase cells despite high levels of cdk activity and correlates with a retardation of DNA synthesis. Regulated dephosphorylation of RB may therefore be utilized by DNA damage and/or other yet unknown signals to control the progression of cells through S phase.
Materials and methods
Cell culture and plasmids
Rat-1 cells were kindly provided by Dr. D. Green (La Jolla Institute for Allergy and Immunology, CA). Asynchronously growing Rat-1 cells were cultured in DMEM supplemented with 10% FBS and antibiotics. Rat-1 cells were made quiescent by culture in defined minimal medium (DMM) for 72 hr. For synchronization in S phase, cells were cultured in the presence of 2 μg/ml aphidicolin for 24 hr. Alternatively, quiescent Rat-1 cells were stimulated with 10% FBS for 14 hr and then exposed to aphidicolin for 6 hr.
Transfections of Rat-1 cells were carried out by use of either calcium phosphate precipitation, Lipofectamine (Gibco-BRL), or Superfect (Qiagen). Transfected Rat-1 cells were selected 24 hr post-transfection with 2.5 μg/ml puromycin (Sigma) for 72 hr, and either harvested for analysis of protein or cultured for an additional 96 hr and stained with crystal violet as described previously (Qin et al. 1992).
The WT-LP, C706F-LP, PSM.7-LP, PSM.9I-RB, and CMV-NEO vectors have been described previously (Knudsen and Wang 1997). The puromycin-resistance plasmid, pBABE-PURO was kindly provided by Dr. H. Land (Imperial Cancer Research Fund, London, UK). The p16ink4a expression plasmid has been described previously (Knudsen and Wang 1997). The cyclin E and cyclin A plasmids have been described previously (Knudsen and Wang 1996, 1997). The T-Ag expression plasmids were obtained from Dr. J. DeCaprio (Dana-Farber Cancer Institute, Boston, MA) and Dr. S. Subramani (UCSD). The GFP-expression plasmid, Green Lantern, is commercially available (GIBCO-BRL).
Microinjection, immunofluorescence, and flow cytometry
Cells were microinjected with glass capillary needles, made with a Kopf vertical pipette puller. The plasmids were coinjected at the concentration of 100 ng/μl GFP and 0.4–200 ng/μl of the effector plasmid. Injections were carried out with an Eppendorf microinjector. All plasmids utilized for microinjection were doubly purified to remove any impurities. Plasmids were originally purified either by cesium chloride banding or Qiagen Maxi-Prep kits. The resulting plasmid DNA (50 μg) was then repurified over a Qiagen Midi-column. Plasmid concentration was determined photometrically, and the integrity of all plasmids was determined by visual analysis following agarose gel electrophoresis. Injected cells were grown on acid-washed glass coverslips. The approximate number of plasmid molecules introduced into the injected cells was determined on the basis of previously reported injection volumes (Capecchi 1980; Huang et al. 1996). For the plasmids utilized in this study the approximate number of plasmid molecules per injected cell is given as follow: 0.4 ng/μl = 1–4 plasmid molecules per injected cells; 2.0 ng/μl = 5–20 plasmid molecules per injected cell; 10 ng/μl = 25–100 plasmid molecules per injected cell; 50 ng/μl = 125–500 plasmid molecules per injected cell; 100 ng/μl = 250–1000 plasmid molecules per injected cell; 200 ng/μl = 500–2000 plasmid molecules per injected cells.
Several different culture conditions were used for the injection of S-phase cells. First, Rat-1 cells that had been cultured for 24 hr in aphidicolin-containing media were used. The injected cells were cultured for an additional 16 hr in aphidicolin to allow for the accumulation of the plasmid-encoded protein. Cells were washed extensively to remove the aphidicolin and BrdU was added immediately to the medium. Cells were then fixed following a 6 hr labeling period. Alternatively, Rat-1 cells were rendered quiescent via culture in DMM for 72 hr. Cells were stimulated with serum for 14 hr and then cultured in the presence of aphidicolin for and additional 6 hr. These S-phase cells were subjected to microinjection and then cultured an additional 14 hr in aphidicolin. These cells were then released from the aphidicolin arrest in the presence of BrdU, fixed, and stained following 6 hr of labeling. Finally, quiescent cells that had been serum stimulated for 12 hr were subjected to microinjection, and BrdU was added to the cells 6 hr post-injection. Cells were fixed after 6 hr of labeling and then stained for the incorporation of BrdU.
For the injection of early G1 cells, Rat-1 cells that had been rendered quiescent by culture in DMM for 72 hr were released into medium containing 10% FBS for 2 hr and then microinjected. BrdU was added immediately following injection. Cells were then fixed after 18 hr in the presence of BrdU.
Following microinjection and labeling with BrdU to measure DNA synthesis, cells were fixed in 3.7% formaldehyde in PBS for 15 min and then permeabilized in 0.3% Triton X-100 in PBS for 10 min. Primary antibody staining (Accurate Scientific, 1:500 dilution) was carried out in PBS supplemented with 5 mg/ml BSA and 0.5% NP-40, 10 mm MgCl2, and 100 U/μl DNase 1 for 1 hr at 37°C in a humidified chamber. The coverslips were then washed in PBS. Secondary antibody (Jackson Laboratories, 1:100 dilution) was diluted and incubated in PBS supplemented with 5mg/ml BSA and 0.5% NP-40. Coverslips were washed again in PBS, and mounted on glass slides.
Fluorescent microscopy was performed with a Zeiss Axiophot Microscope with either 40× or 63× objective lenses. Photographs of stained cells were recorded with a Hamamatsu CCD camera. Digital images were printed on a Mitsubishi color sublimation printer.
For flow cytometry, cells were transfected with a CD20 cell surface marker and the indicated expression plasmids. Forty-eight hours post-transfection, cells were harvested by trypsinization (trypsin does not effect the staining for CD20). Cells were stained for the expression of CD20 with an FITC conjugated anti-CD20 antibody (PharMingen). These cells were then fixed in 80% ethanol and stained with propidium iodide. The DNA content of CD20-positive and negative cells was determined by use of a FACSCAN cytometer equipped with cellfit software (Becton-Dickinson).
Quantitative RT–PCR
Total cellular RNA was isolated with Trizol reagent (GIBCO-BRL) according to manufacturer’s instruction. First-strand cDNAs were synthesized from 2 μg of DNase I-treated total RNA by use of SuperScript II reverse transcriptase (Life Technology, GIBCO-BRL) and random antisense primers according to the manufacturer’s instruction. Diluted cDNAs were subjected to PCR amplifications with two sets of primers specific for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (GGTCATCAATGGGAAACCCA TCAC and TGATGGCATGGACTGTGGTCATGA) and cyclin A (AGACCCTGCATTTGGCTGTG and ACAAACTCTGCTACTTCTGG). In all PCR reactions, the GAPDH primers were added to the reaction mixture four cycles later than the cyclin A primers. PCR-amplified products were separated on a 1% agarose gel and analyzed by Southern blotting with 32P-labeled GAPDH and cyclin A cDNAs as probes. The expression levels of GAPDH and cyclin A were quantitated with a PhosphoImager (Molecular Dynamics) from five PCR reactions that were performed in the linear range. The relative expression levels of cyclin A were normalized to the expression levels of GAPDH from the given PCR reaction, and the cyclin A levels from asynchronous growing cells were arbitrarily set to 100%.
Antibodies, immunoblotting, and kinase reactions
Cyclin D1 was detected with a polyclonal antibody kindly provided by Dr. S.I. Reed (Scripps Research Institute, La Jolla, CA). Cyclin E was detected with monoclonal antibody from PharMingen. Cyclin A was detected with a polyclonal antibody kindly provided by Dr. T. Hunter (Salk Institute, La Jolla, CA). Cyclin B was detected with a monoclonal antibody from PharMingen. Cdk2 protein was detected with polyclonal antibody provided by Dr. M. Pagano (Mitotix, Cambridge, MA) or from Santa Cruz Scientific. The Cdc2 and RB antibodies have been previously described (Welch and Wang 1993). p107 and p130 antibodies were obtained from Santa Cruz Scientific. The p21cip1 antibody was from PharMingen, and p27kip1 antibody was kindly provided by Dr. J. Roberts (Fred Hutchinson Cancer Research Center). Immunoblotting was carried out by use of standard procedures and Immobilon P membranes.
Kinase reactions were carried out essentially as described (Knudsen and Wang 1996), with the exception that only 20 μg of total cell lysate from the indicated cells was utilized per kinase reaction. The kinase complexes were isolated via immunoprecipitation with the indicated antibodies and protein A–Sepharose and then used to phosphorylate histone H1 as a substrate. The nonspecific rabbit anti-mouse antibody was obtained from Cappell.
Acknowledgments
We thank Dr. Karen E. Knudsen for critical reading of the manuscript and also thank all the members of the Wang and Feramisco laboratories for thoughtful discussions and/or technical assistance. We are also grateful to Dr. Tony Hunter, Dr. James A. DeCaprio, Dr. Steven I. Reed, Dr. Jim Roberts, Dr. Webster Cavenee, and Dr. Doug Green for helpful discussion and/or provision of reagents. This work was supported by a grant to J.Y.J.W. (CA58320) from the National Institutes of Health (NIH), and a grant to J.R.F. from the California Tobacco Related Disease Research Program. E.S.K. is supported by a training grant to the UCSD Cancer Center from National Cancer Institute–NIH (T32CA09290).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jywang@ucsd.edu FAX (619) 534-2821.
References
- Akiyama T, Ohuchi T, Sumida S, Matsumoto K, Toyoshima K. Phosphorylation of the retinoblastoma protein by cdk2. Proc Natl Acad Sci. 1992;89:7900–7904. doi: 10.1073/pnas.89.17.7900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alevizopoulos K, Vlach J, Hennecke S, Amati B. Cyclin E and c-Myc promote cell proliferation in the presence of p16(INK4a) and of hypophosphorylated retinoblastoma family proteins. EMBO J. 1997;16:5322–5333. doi: 10.1093/emboj/16.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway in cell cycle control and cancer. Exp Cell Res. 1997;237:1–6. doi: 10.1006/excr.1997.3776. [DOI] [PubMed] [Google Scholar]
- Beijersbergen RL, Bernards R. Cell cycle regulation by the retinoblastoma family of growth inhibitor proteins. Biochim Biophys Acta. 1996;1287:103–120. doi: 10.1016/0304-419x(96)00002-9. [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Capecchi MR. High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell. 1980;2:479–488. doi: 10.1016/0092-8674(80)90358-x. [DOI] [PubMed] [Google Scholar]
- Chen PL, Scully P, Shew JY, Wang JYJ, Lee WH. Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell. 1988;58:1193–1198. doi: 10.1016/0092-8674(89)90517-5. [DOI] [PubMed] [Google Scholar]
- Chen Y, Knudsen ES, Wang JYJ. Cells arrested in G1 by the v-Abl tyrosine kinase do not express cyclin A despite the hyperphosphorylation of RB. J Biol Chem. 1996;271:19637–19640. doi: 10.1074/jbc.271.33.19637. [DOI] [PubMed] [Google Scholar]
- Connell-Crowley L, Harper JW, Goodrich DW. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol Biol Cell. 1997;8:287–301. doi: 10.1091/mbc.8.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, Marsilio E, Paucha E, Livingston DM. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–283. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DelSal G, Loda M, Pagano M. Cell cycle and cancer: Critical events at the G1 restriction point. Crit Rev Oncogenesis. 1996;7:127–142. doi: 10.1615/critrevoncog.v7.i1-2.80. [DOI] [PubMed] [Google Scholar]
- Geng Y, Eaton EN, Picón M, Roberts JM, Lundberg AS, Gifford A, Sardet C, Weinberg RA. Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene. 1996;12:1173–1180. [PubMed] [Google Scholar]
- Gill RM, Hamel PA, Zhe J, Zacksenhaus E, Gallie BL, Phillips RA. Characterization of the human RB1 promoter and of elements involved in transcriptional regulation. Cell Growth Differ. 1994;5:467–474. [PubMed] [Google Scholar]
- Goodrich DW, Wang NP, Qian YW, Lee EYHP, Lee WH. The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell. 1991;67:293–302. doi: 10.1016/0092-8674(91)90181-w. [DOI] [PubMed] [Google Scholar]
- Hamel PA, Gill RM, Phillips RA, Gallie BL. Regions controlling hyperphosphorylation and conformation of the retinoblastoma gene product are independent of domains required for transcriptional repression. Oncogene. 1992;7:693–701. [PubMed] [Google Scholar]
- Hamel PA, Phillips RA, Muncaster M, Gallie BL. Speculations on the roles of RB1 in tissue-specific differentiation, tumor initiation, and tumor progression. FASEB J. 1993;7:846–854. doi: 10.1096/fasebj.7.10.8344484. [DOI] [PubMed] [Google Scholar]
- Herrera RE, Mäkelä TP, Weinberg RA. TGF beta-induced growth inhibition in primary fibroblasts requires the retinoblastoma protein. Mol Biol Cell. 1996a;7:1335–1342. doi: 10.1091/mbc.7.9.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera RE, Sah VP, Williams BO, Mäkelä TP, Weinberg RA, Jacks T. Altered cell cycle kinetics, gene expression, and G1 restriction point regulation in Rb-deficient fibroblasts. Mol Cell Biol. 1996b;16:2402–2407. doi: 10.1128/mcb.16.5.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LC, Clarkin KC, Wahl GM. Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc Natl Acad Sci. 1996;93:4827–4832. doi: 10.1073/pnas.93.10.4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T, Pines J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell. 1994;79:573–582. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- Hurford RK, Jr, Cobrinik D, Lee MH, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes & Dev. 1997;11:1447–1463. doi: 10.1101/gad.11.11.1447. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr Recent insights into the functions of the retinoblastoma susceptibility gene product. Cancer Invest. 1997;15:243–254. doi: 10.3109/07357909709039722. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Wang JYJ. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271:8313–8320. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- ————— Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17:5771–5783. doi: 10.1128/mcb.17.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh J, Enders GH, Dynlacht BD, Harlow E. Tumour-derived p16 alleles encoding proteins defective in cell-cycle inhibition. Nature. 1995;375:506–510. doi: 10.1038/375506a0. [DOI] [PubMed] [Google Scholar]
- Kowalik TF, DeGregori J, Schwarz JK, Nevins JR. E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J Virol. 1995;69:2491–2500. doi: 10.1128/jvi.69.4.2491-2500.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JA, Buchkovich KJ, Marshak DR, Anderson CW, Harlow E. The retinoblastoma protein is phosphorylated on multiple sites by human cdc2. EMBO J. 1991;10:4279–4290. doi: 10.1002/j.1460-2075.1991.tb05006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin BT, Gruenwald S, Morla AO, Lee WH, Wang JYJ. Retinoblastoma cancer suppressor gene product is a substrate of the cell cycle regulator cdc2 kinase. EMBO J. 1991;10:857–864. doi: 10.1002/j.1460-2075.1991.tb08018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M, Peters G, Bartek J. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature. 1995;375:503–506. doi: 10.1038/375503a0. [DOI] [PubMed] [Google Scholar]
- Lukas J, Herzinger T, Hansen K, Moroni MC, Resnitzky D, Helin K, Reed SI, Bartek J. Cyclin E-induced S-phase without activation of the pRb/E2F pathway. Genes & Dev. 1997;11:1479–1492. doi: 10.1101/gad.11.11.1479. [DOI] [PubMed] [Google Scholar]
- Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18:753–761. doi: 10.1128/mcb.18.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema RH, Herrera RE, Lam F, Weinberg RA. Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc Natl Acad Sci. 1995;92:6289–6293. doi: 10.1073/pnas.92.14.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994;14:2077–2086. doi: 10.1128/mcb.14.3.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Leone G, Herendeen DR, Kelly TJ, Nevins JR. Expression of the HsOrc1 gene, a human ORC1 homolog, is regulated by cell proliferation via the E2F transcription factor. Mol Cell Biol. 1996;16:6977–6984. doi: 10.1128/mcb.16.12.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992;11:961–971. doi: 10.1002/j.1460-2075.1992.tb05135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmero I, Peters G. Perturbation of cell cycle regulators in human cancer. Cancer Surv. 1996;27:351–367. [PubMed] [Google Scholar]
- Qin XQ, Chittenden T, Livingston DM, Kaelin WG., Jr Identification of a growth suppression domain within the retinoblastoma gene product. Genes & Dev. 1992;6:953–964. doi: 10.1101/gad.6.6.953. [DOI] [PubMed] [Google Scholar]
- Qin XQ, Livingston DM, Kaelin WG, Jr, Adams PD. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnitzky D, Reed SI. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol Cell Biol. 1995;15:3463–3469. doi: 10.1128/mcb.15.7.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnitzky D, Hengst L, Reed SI. Cyclin A-associated kinase activity is rate limiting for entrance into S-phase and is negatively regulated in G1 by p27Kip1. Mol Cell Biol. 1995;15:4347–4352. doi: 10.1128/mcb.15.8.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, Lee WH. Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol Cell Biol. 1994;14:8166–8173. doi: 10.1128/mcb.14.12.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Sidle A, Palaty C, Dirks P, Wiggan O, Kiess M, Gill RM, Wong AK, Hamel PA. Activity of the retinoblastoma family proteins, pRB, p107, and p130, during cellular proliferation and differentiation. Crit Rev Biochem Mol Biol. 1996;31:237–271. doi: 10.3109/10409239609106585. [DOI] [PubMed] [Google Scholar]
- Slansky JE, Farnham PJ. Introduction to the E2F family: Protein structure and gene regulation. Curr Top Microbiol Immunol. 1996;208:1–30. doi: 10.1007/978-3-642-79910-5_1. [DOI] [PubMed] [Google Scholar]
- Strausfeld UP, Howell M, Rempel R, Maller JL, Hunt T, Blow JJ. Cip1 blocks the initiation of DNA replication in Xenopus extracts by inhibition of cyclin-dependent kinases. Curr Biol. 1994;4:876–883. doi: 10.1016/s0960-9822(00)00196-2. [DOI] [PubMed] [Google Scholar]
- Wang JYJ, Knudsen ES, Welch PJ. The retinoblastoma tumor suppressor protein. Adv Cancer Res. 1994;64:25–85. doi: 10.1016/s0065-230x(08)60834-9. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Welch PJ, Wang JYJ. A C-terminal protein-binding domain in the retinoblastoma protein regulates nuclear c-Abl tyrosine kinase in the cell cycle. Cell. 1993;75:779–790. doi: 10.1016/0092-8674(93)90497-e. [DOI] [PubMed] [Google Scholar]
- Zalvide J, Stubdal H, DeCaprio JA. The J domain of simian virus 40 large T antigen is required to functionally inactivate RB family proteins. Mol Cell Biol. 1998;18:1408–1415. doi: 10.1128/mcb.18.3.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G(1)/S cyclin-dependent kinases. J Biol Chem. 1997;272:12738–12746. doi: 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]