

Abstract
Discrimination amongst S-thiazolinyl (STaz), S-benzoxazolyl (SBox), and S-ethyl anomeric leaving group was achieved by fine-tuning of the activation conditions. Preferential glycosidation of a certain leaving group is neither determined by the strength of activating reagents nor the stability of the leaving group itself; instead, the type of activation comes to the fore and plays the key role. The activation conditions established herein were applied to a sequential five-step synthesis of a hexasaccharide using six monosaccharide building blocks equipped with six different leaving groups.
Introduction
Traditional linear approaches to oligosaccharide assembly are often cumbersome and, consequently, the availability of complex glycostructures remains insufficient to address challenges of modern glycosciences.1–3 Recent strategic improvements have significantly shortened the number of synthetic steps required for oligosaccharide assembly.4 In principle, the use of the selective activation concept wherein one leaving group is activated in the presence of another offers flexible oligosaccharide sequencing. This approach does not rely on the nature of the protecting groups, like chemoselective armed-disarmed approach wherein the reactivity of the same leaving group on both glycosyl donor and acceptor counterparts is adjusted by protecting groups.5–8 Instead, it requires a few leaving groups (LGa, LGb, LGc, etc., Scheme 1) that can be sequentially activated. Unfortunately, this relatively simple concept is limited to the number of available leavings groups compatible with the principle of sequential activation.9 In most cases, only two or at most three leaving groups can be aligned for selective activation sequences.
Scheme 1.
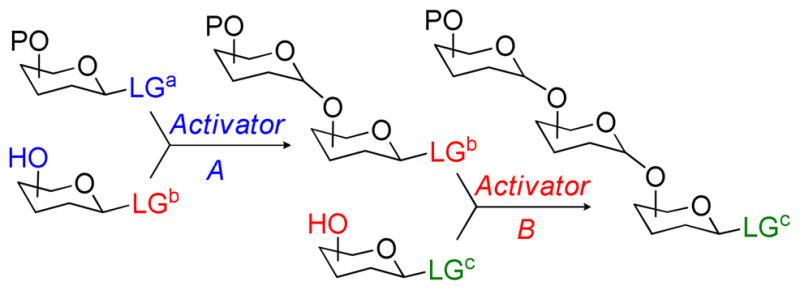
Selective activation-based approach to oligosaccharide synthesis (for orthogonal strategy LGc = LGa)
In part this limitation can be addressed by related semi-orthogonal10,11 and orthogonal12,13 strategies. The only requirement for the orthogonal approach is a set of two orthogonal leaving groups (LGc = LGa, Scheme 1) and a pair of compatible orthogonal activators. Although the orthogonal strategy is an excellent concept for flexible sequencing of oligosaccharides its scope remained narrow. Indeed, only one example involving orthogonal activation of the S-ethyl and fluoride leaving group was known for about a decade since its invention in 1994.12,13 To expand the orthogonal concept to other systems in 2004 we reported that S-thiazolinyl (STaz) and S-alkyl/aryl glycoside combination14,15 is a viable alternative of the original orthogonal concept pioneered by Kanie, Ito, and Ogawa. More recently, Hotha et al.16 introduced a similar concept using propargyl and n-pentenyl glycosides as well as orthoesters thereof.
Very unexpectedly, it has been also determined that structurally similar STaz vs. S-benzoxazolyl (SBox) leaving groups17 also represent a viable orthogonal pair. The uniqueness of this approach is that both leaving groups employed are of essentially the same class, glycosyl thioimidates. While reactions of both SBox and STaz glycosyl donors promoted with MeOTf were very effective, MeI or BnBr were only effective for the STaz glycosyl donors. This discovery created a basis for the development of the STaz-SBox orthogonal strategy, but it also signified the gap in our understanding of the thioimidate activation. From our early mechanistic study,18,19 we already knew that MeOTf-promoted activation of the SBox glycosyl donors proceeds via the anomeric sulfur atom (direct activation). This was confirmed by isolating the departed S-methylated aglycone MeSBox (Scheme 2a).
Scheme 2.

Activation of thioimidates: a study with SBox (a)18,19 and STaz glycosides (b).17
To explain the different activation pattern of the STaz glycosyl donors, we anticipated that their activation proceeds via the nitrogen atom (remote activation). Indeed, this remote activation of STaz showed marginally slower reactions with a powerful promoter, such as MeOTf.17 When weak alkylating reagents are used (MeI, BnBr), a powerful nucleophile is needed to replace the iodine or bromine, respectively. Evidently, this can be achieved with STaz glycosides that bear the reactive nitrogen atom, but not with the SBox glycosides, which can only be activated via the exocyclic sulfur.18,19 The credibility of this working hypothesis was verified by a series of experiments in which we isolated the N-benzylated by-product whose identity was proven by spectral and X-ray methods (Scheme 2b).17
Results and Discussion
With a better understanding of the mechanistic pathways for thioimidate activation, we were well positioned to undertake further studies of the expeditious oligosaccharide assembly via orthogonal and selective activation concepts. Previously, we demonstrated that SBox leaving group in glycosyl donors 1 and 4 can be efficiently activated over glycosyl acceptor 2 equipped with S-alkyl anomeric moiety (Table 1, entries 1 and 2) or O-pentenyl acceptor 6 (entry 3).18,20 These glycosylations were promoted by AgOTf, which is a very powerful activator of thioimidates, but is entirely neutral towards thioglycosides or pentenyl glycosides. Resultantly, disaccharides 3, 5, and 7 were obtained in nearly quantitative yields. Activations of SBox glycosyl donors 8 and 11 over STaz glycosyl acceptors 9 and 12 in the presence of Cu(OTf)2 were also previously reported (entries 4 and 5).15 Although the synthesis of 10 and 13 was successful, our subsequent in depth study indicated that the additional reinforcement of the protecting groups was essential for these couplings. Indeed, in both reactions armed glycosyl donors and disarmed glycosyl acceptors are employed.
Table 1.
Activation of SBox glycosyl donors over glycosyl acceptors bearing SEt, O-pentenyl, STaz or F leaving groups
| Entry | Donor | Acceptor | Conditions | Product | Yield, ratio α/β | Ref |
|---|---|---|---|---|---|---|
| 1 |
 1 |
 2 |
AgOTf |
 3 |
99%, β only | 18 |
| 2 |
 4 |
2 | AgOTf |
 5 |
98%, α only | 20 |
| 3 | 4 |
 6 |
AgOTf |
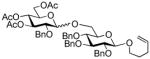 7 |
99%, 8.0/1 | 20 |
| 4 |
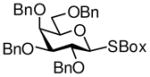 8 |
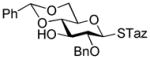 9 |
Cu(OTf)2 |
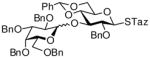 10 |
99%, 7.0/1 | 15 |
| 5 |
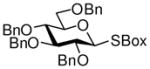 11 |
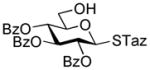 12 |
Cu(OTf)2 |
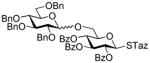 13 |
66%, 1.4/1 | 15 |
| 6 | 1 | 12 | Bi(OTf)3 |
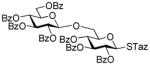 14 |
69%, β only | 17 |
| 7 | 11 | 9 | Cu(OTf)2 |
 15 |
71%, 2.4/1 | |
| 8 | 8 |
 16 |
Cu(OTf)2 |
 17 |
52%, >25/1 | |
| 9 | 1 |
 18 |
MeOTf |
 19 |
95%, β only |
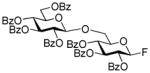
Further studies revealed that indeed SBox can be activated over the STaz group independently of the protecting groups in the presence of Bi(OTf)3 (entry 6).17 This finding created the basis for the development of the thioimidate-only orthogonal strategy for oligosaccharide synthesis because also STaz glycosides can be activated over the SBox moiety under alkylation conditions (vide supra). However, only relatively modest yields in the 70% yield-range could be generally obtained in the presence of Bi(OTf)3. Our further efforts to improve this result did not result in the improved yields. The investigation of secondary glycosyl acceptors 9 and 16 (entries 7 and 8) provided similar glycosylation outcome to that of the primary acceptor 12. Continuing the search of other suitable selective activations, we found that the SBox leaving group can be reliably activated over glycosyl fluorides and a representative example is shown in entry 9. Thus, glycosidation of 1 with fluoride acceptor 18 performed in the presence of MeOTf afforded disaccharide 19 in 95% yield.
Based on results summarized in Table 1, we conclude that SBox glycosides are excellent glycosyl donors that can be selectively activated over a variety of other leaving groups. Particularly high yields have been achieved with the use of thioglycosides, O-pentenyl glycosides and glycosyl fluorides as glycosyl acceptors. However, the activation of the SBox leaving group over glycosyl acceptors equipped with STaz moiety represents a less efficient pathway towards multistep oligosaccharide synthesis.
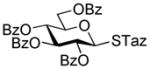
As the continuation of this study, a comprehensive investigation of glycosyl donors equipped with the STaz leaving group appealed to us as an attractive venue. Previously, we determined that thioglycosides 21 and 24 and O-pentenyl glycoside 6 successfully withstand typical reaction conditions required for the activation of STaz glycosides 20 or 23.15 These results are depicted in Table 2 (entries 1–3). As aforementioned, while SBox glycosides are generally more labile than their STaz counterparts towards a variety of acidic or basic reagents, the STaz leaving group can be successfully activated under alkylation conditions in the presence of either BnBr or MeI. SBox glycosides remain completely inert and these reaction conditions; and these selective activations were successfully applied to the building blocks of both the armed and disarmed series (entries 4 and 5, respectively). Selective, as opposed to chemoselective, nature of this activation was ultimately proven by the activation of the disarmed STaz donor 20 over the “armed” SBox acceptor 27. The resulting disaccharide 31 was isolated in 79% yield (entry 6). Glycosylation of secondary glycosyl acceptors 32 and 35 equipped with the SBox moiety was equally successful and the corresponding disaccharides 33 and 36 were obtained in good yields and complete 1,2-cis stereoselectivity (entries 7 and 8). Based on the results summarized in Tables 1 and 2, we believe that if the activation of one thioimidoyl leaving group over another is required, the preferred mode for such activation is the activation of the STaz glycosyl donor over the SBox glycosyl acceptor rather than opposite.
Table 2.
Activation of STaz glycosyl donors over glycosyl acceptors bearing SEt, SPh, O-pentenyl, or SBox leaving groups
| Entry | Donor | Acceptor | Conditions | Product | Yield, ratio α/β | Ref |
|---|---|---|---|---|---|---|
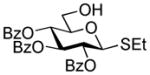
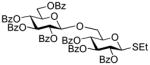
| 1 |
 20 |
 21 |
AgOTf |
 22 |
81%, β only | 15 |
| 2 |
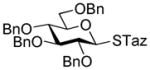 23 |
 24 |
AgOTf |
 25 |
99%, 2.0/1 | 15 |
| 3 | 23 | 6 | AgOTf |
 26 |
91%, 2.0/1 | 15 |
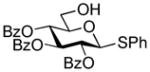
| 4 | 23 |
 27 |
MeI BnBr |
 28 |
82%, 6/1 85%, 3.2/1 |
|
| 5 | 20 |
 29 |
BnBr |
 30 |
76%, β only | |
| 6 | 20 | 27 | BnBr |
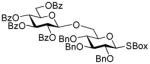 31 |
79%, β only | |
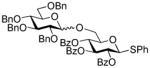
| 7 | 23 |
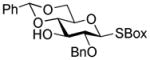 32 |
BnBr |
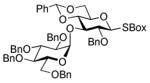 33 |
70%, >25/1 | |
| 8 |
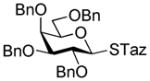 34 |
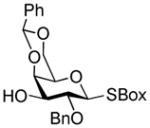 35 |
BnBr |
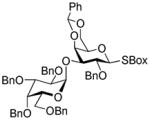 36 |
77%, >25/1 |
Additionally, the investigation of thioglycosides as glycosyl donors in selective activations seemed attractive because in all previous examples thioglycosides were used as glycosyl acceptors. Many reliable reaction conditions for the glycosidation of thioglycosides have been developed21 and some were found compatible with selective activations. For example, NIS/AgOTf-promoted activation of thioglycosides created a basis for the first example of orthogonal activation over glycosyl fluorides.12
MeOTf-promoted activation of thioglycosides22 allows for a very selective activation over O-pentenyl glycosides (Table 3, entries 1 and 2), which created a basis for the semi-orthogonal strategy for oligosaccharide synthesis.10 Thioglycosides (both S-alkyl and S-aryl) can be also activated over STaz glycosides, and this can be accomplished in the presence of NIS and TfOH (entries 3 and 4).14,15 The use of catalytic TfOH is necessary because stoichiometric amount of TfOH would also trigger the activation of the STaz leaving group. Although SEt and STaz leaving groups can be reliably differentiated at the monosaccharide level, it represents a particular difficulty at the advanced stage of the assembly when oligosaccharide donor is used. Thus, we noticed that the efficiency of the orthogonal activation of STaz vs. SEt drops at the stage of tetra- and pentasaccharides.15 Typically, oligosaccharide donor is much less reactive than its respective monosaccharide counterpart, and if additional amounts of TfOH are required to ensure the completion of activation of such S-ethyl donor, this can pose a problem for the acceptor equipped with the STaz moiety that may be also activated in this enhanced acidic environment.
Table 3.
Activation of SEt glycosyl donors over glycosyl acceptors bearing O-pentenyl, STaz or SBox leaving groups
| Entry | Donor | Acceptor | Conditions | Product | Yield, ratioα/β | Ref |
|---|---|---|---|---|---|---|
| 1 |
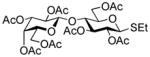 37 |
6 | MeOTf |
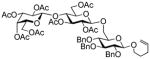 38 |
98%,β only | 10 |
| 2 | 5 | 6 | MeOTf |
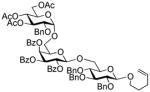 39 |
92%,β only | 20 |
| 3 |
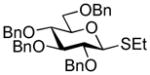 40 |
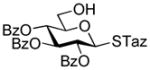 12 |
NIS/TfOH |
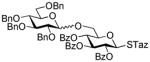 13 |
98%, 1.1/1 | 15 |
| 4 | 40 |
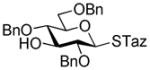 41 |
NIS/TfOH |
42 |
80%, 2.1/1 | 15 |
| 5 | 40 |
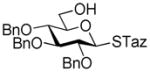 43 |
DMTST |
 44 |
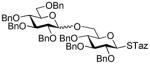
83%, 1.2/1 | |
| 6 |
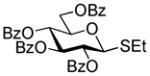 45 |
12 | DMTST |
 14 |
79%, β only | |
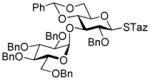
| 7 | 40 | 9 | DMTST |
 15 |
77%, >25/1 | |
| 8 |
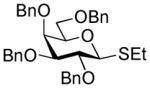 46 |
16 | DMTST |
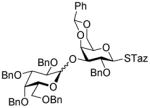 17 |
74%, 15/1 | |
| 9 | 22 | 12 | DMTST |
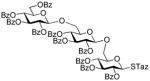 47 |
67%, β only | |
| 10 |
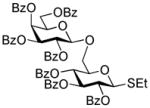 48 |
12 | DMTST |
 49 |
64%, β only | |
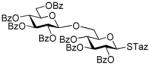
| 11 | 45 | 29 | DMTST |
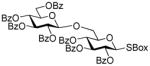 30 |
54%, β only |
Therefore, further search of promoters suitable for more selective activation of thioglycosides over STaz glycosides appealed to us as a useful expansion of this study. Herein, we report that dimethyl (methylthio) sulfonium trifluoromethanesulfonate (DMTST)23 is unable to activate the STaz leaving group, whereas it is a well-known activator for thioglycosides.24 This allowed us to perform reliable activations of various thioglycosides over a broad range of STaz acceptors (entries 5–10, Table 3). We believe that the differential nature of activation of the SEt and STaz moieties is due to the modes by which these two leaving groups are activated: direct for S-alkyl glycosides25 and remote for STaz glycosides.17 Herein the formation of the anomeric sulfonium ion in case of thioglycosides25 is a more likely pathway than the formation of the N-S bond, as it would have to be the case with the STaz glycosides. The yields obtained here are in line with yields obtained in DMTST activations of thioglycosides that typically do not exceed the 80%-range. Therefore, these results do not support the selective nature of this activation that is actually very efficient as no activation of the STaz leaving group was noticed. At this point, these results were deemed suitable for performing selective activations with oligosaccharide building blocks. We believe that further search of suitable promoters to differentiate the reactivity of STaz and SEt leaving groups may help to improve the outcome of these selective couplings. In this context, we noticed that the differentiation between thioglycosides and SBox glycosides can be also achieved in the presence of DMTST (entry 11). However, the efficiency of this selective activation is significantly lower than that of SEt over STaz, because the SBox moiety is activated via the anomeric sulfur like S-ethyl glycosides.
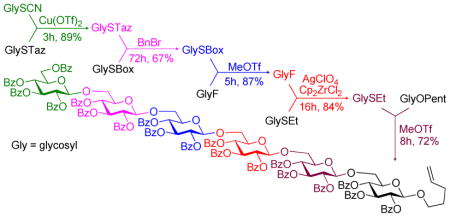
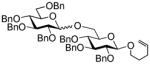
With the study of a variety of selective activations summarized in Tables 1–3, we decided to undertake multi-step sequential selective activations. As a possible LGa for the first stage activation we were initially considering the most common highly reactive glycosyl donors: glycosyl bromides and trichloroacetimidates. In our hands, however, their activation over the STaz leaving group was somewhat inconsistent.15 With the reinvestigation of glycosyl thiocyanates, reactive glycosyl donors introduced by Kochetkov,26–28 we determined that their activation can be reliably achieved with Cu(OTf)2, reaction conditions under which STaz group reacts sluggishly.29 Indeed, Cu(OTf)2-promoted activation of thiocyanate donor 50 over STaz glycosyl acceptor 12 was very efficient and the resulting disaccharide 14 was isolated in 89% yield (Scheme 3). The STaz moiety of disaccharide 14 was then activated over SBox glycosyl acceptor 29 under alkylation conditions to afford the corresponding trisaccharide 51 in 67% yield. Subsequently, the SBox leaving group of 51 was activated over glycosyl fluoride acceptor 18 in the presence of MeOTf. This selective activation resulted in the formation of tetrasaccharide 52 in 87% yield. The activation of glycosyl fluorides over thioglycosides is a well-established protocol dating back to Nicolaou’s study of the two-step activation strategy for oligosaccharide synthesis.30,31 Herein, a protocol used in the original Ogawa’s orthogonal strategy was adopted.12 Thus, activation of glycosyl fluoride tetrasaccharide 52 over thioglycosides acceptor 21 was performed in the presence of AgClO4 and Cp2ZrCl2 and the resulting pentasaccharide 53 was isolated in 84% yield. Finally, thioglycoside 53 was activated over O-pentenyl glycosyl acceptor 54 in the presence of MeOTf. The resulting hexasaccharide 55 was isolated in 72% yield.
Scheme 3.

Synthesis of hexasaccharide 55 via the five-step sequential activation strategy.
It is noteworthy that the use of O-pentenyl moiety at this last stage represents an important practical aspect of oligosaccharide synthesis. On one hand, O-pentenyl can be glycosidated to elongate the sequence further or it can be easily hydrolyzed if the complete deprotection is required. On another hand, O-pentenyl glycoside represents a conjugation-amendable linker that can be either used in thiolene conjugation32 or converted into thiol,33 aldehyde34,35 or carboxylic acid and to effect other common conjugation techniques.36–38
Conclusions
We presented a thorough study of the selective activation of different leaving groups that allowed us to align six different leaving groups to perform five-step synthesis of a linear hexasaccharide from six monosaccharide building blocks. It is to be expected that the same or similar sequential activation would be suitable for the synthesis of other oligosaccharide sequences including those of high biological significance and/or therapeutic relevance.
Experimental part
General
Column chromatography was performed on silica gel 60 (70–230 mesh), reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40 °C. Dichloromethane and 1,2-Dichloroethane were distilled from CaH2 directly prior to application. Methanol was dried by refluxing with magnesium methoxide, distilled and stored under argon. Pyridine was dried by refluxing with CaH2 and then distilled and stored over molecular sieves (3 Å). Molecular sieves (3 Å and 4 Å), used for reactions, were crushed and activated in vacuo at 390 °C during 8 h in the first instance and then for 2–3 h at 390 °C directly prior to application. AgOTf was co-evaporated with toluene (3 × 10 mL) and dried in vacuo for 2–3 h directly prior to application. DMTST was prepared in accordance to previously reported methods. 1H-NMR spectra were recorded at 300 and 500 MHz, 13C-NMR spectra were recorded at 75 and 125 MHz.
2-Thiazolinyl 2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside (16)
A solution of ethyl 3-O-acetyl-2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside39 (0.48 g, 1.08 mmol) and activated molecular sieves (3 Å, 0.54 g) in CH2Cl2 (16 mL) was stirred under argon for 1 h. Freshly prepared solution of Br2 in CH2Cl2 (10 mL, 1/165, v/v) was then added and the reaction mixture was kept for 10 min at rt. After that, the solid was filtered-off and the filtrate was concentrated in vacuo at rt and dried. Crude residue was then treated with NaSTaz (2.16 mmol) and 15-crown-5 (0.21 mmol) in dry acetonitrile (5 mL) under argon for 6 h at rt. Upon completion, the mixture was diluted with dichloromethane, the solid was filtered-off and the residue was washed with dichloromethane (10 mL). The combined filtrate was washed with 1% aq. NaOH (1 × 15 mL) and water (2 × 15 mL). The organic layer was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography to afford 2-thiazolinyl 3-O-acetyl-2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside as white foam in 80% yield (0.44 g, 0.87 mmol). Selected analytical data: Rf = 0.62 (ethyl acetate/hexane, 7/3, v/v); 1H NMR: δ, 2.05 (s, 3H, COOCH3), 3.37 (t, 2H, CH2S), 3.63 (m, 1H, H-5), 4.00 – 4.05 (m, 2H, J2,3 = 9.7 Hz, H-2, 4), 4.17, 4.28 (m, 2H, CH2N), 4.36 (dd, 1H, J6a,6b = 11.3 Hz, H-6b), 4.55 (dd, 1H, J5,6a = 3.5 Hz, H-6a), 4.65 (d, 1H, J2 = 10.9 Hz, ½ CH2Ph), 4.83 (d, 1H, J2 = 10.9 Hz, frac12; CH2Ph), 5.00 (dd, 1H, J3,4 = 3.5 Hz, H-3), 5.43 (d, 1H, J1,2 = 9.9 Hz, H-1), 5.50 (s, 1H, >CHPh), 7.26 – 7.54 (m, 10H, aromatic) ppm. 2-Thiazolinyl 3-O-acetyl-2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside was dissolved in methanol (5 mL) containing 1M NaOCH3 and the resulting mixture was stirred for 1 h at rt. Dowex (H+) was added until neutral pH, the resin was filtered off and washed successively with methanol. The combined filtrate was concentrated in vacuo and the residue was purified by silica gel column chromatography to afford compound the title compound as a white solid in 75% yield (0.30 g, 0.65 mmol). Analytical data for 16: Rf = 0.40 (ethyl acetate); [α]D25 = −15.3° (c = 1, CHCl3); 1H NMR: δ, 2.61 (s, 1H, J = 8.0 Hz), 3.69 (dd, 2H, JCH2S,CH2N = 8.3 Hz, CH2S), 3.61 (m, 1H, H-5), 3.74 (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.85 (m, 1H, H-3), 4.03 (dd, 1H, J6a,6b = 12.6 Hz, H-6a), 4.11 – 4.31 (m, 3H, H-4, CH2N), 4.38 (dd, 1H, H-6b), 4.83 (dd, 2H, J = 11.1 Hz, CH2Ph), 5.34 (d, 1H, J1,2 = 9.8 Hz, H-1), 5.56 (s, 1H, >CHPh), 7.26 – 7.52 (m, 10 H, aromatic) ppm. 13C NMR: δ, 35.3, 64.4, 69.2, 70.4, 74.6, 75.8, 75.9, 78.3, 84.5, 101.6, 126.7 (×2), 128.1, 128.4 (×4), 128.6 (×2), 129.5, 137.7, 138.0, 163.9 ppm; HR-FAB MS [M+Na]+ calcd for C23H25NO5S2Na 482.1072, found 482.1080.
2-Thiazolinyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-galactopyranoside (34)
The solution of ethyl 2,3,4,6-O-benzyl-1-thio-β-D-galactopyranoside (46)40 (1.0 g, 1.71 mmol) and activated molecular sieves (3 Å, 0.85 g) in CH2Cl2 (25 mL) was stirred under argon for 1 h. Freshly prepared solution of Br2 in CH2Cl2 (16 mL, 1/165, v/v) was then added and the reaction mixture was kept for 5 min at rt. After that, the solid was filtered-off and the filtrate was concentrated in vacuo at rt. The crude residue was then treated with NaSTaz (3.4 mmol) and 15-crown-5 (0.34 mmol) in dry acetonitrile (10 mL) under argon for 6 h at rt. Upon completion, the mixture was diluted with dichloromethane (20 mL), the solid was filtered-off and the residue was washed with dichloromethane (10 mL). The combined filtrate was washed with 1% aq. NaOH (1 × 20 mL) and water (2 × 20 mL). The organic layer was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (ethyl acetate-toluene gradient elution) to afford the title compound as a white solid in 75% (0.82 g, 1.27 mmol). Analytical data for 34: Rf = 0.41 (ethyl acetate/hexane, 4/6, v/v); [α]D22 = +16.6° (c = 1, CHCl3); 1H NMR:δ, 3.32 (dd, 2H, JCH2S,CH2N = 8.32 Hz, CH2S), 3.61–3.71 (m, 4H, H-3, 4, 6a, 6b), 3.92 (dd, 1H, J2,3 = 9.5 Hz, H-2), 4.00 (dd, 1H, J4,5 = 2.3 Hz), 4.10 – 4.24 (m, 2H, CH2N), 4.39 – 4.50 (dd, 2H, J = 11.8 Hz, CH2Ph), 4.62 (d, 1H, J2 = 11.6 Hz, ½CH2Ph), 4.69 –4.85 (m, 2H, CH2Ph), 4.95 (d, 1H, J2 = 11.58 Hz, ½ CH2Ph), 5.31 (d, 1H, J1,2 = 9.96 Hz, H-1), 7.27–7.34 (m, 20H, aromatic) ppm; 13C NMR: δ, 35.2, 64.4, 68.5, 72.9, 73.6, 73.7, 74.9, 75.9, 77.8, 84.2, 85.3, 127.7 (×3), 127.8, 127.9, 128.1 (×2), 128.2 (×2), 128.4 (×3), 128.4 (×2), 128.5 (×5), 128.6 (×2), 138.0, 138.1, 138.3, 138.7, 164.2 ppm; HR-FAB MS [M+Na]+ calcd for C37H39NO5S2Na 664.2167, found 664.2164.
2-Benzoxazolyl 2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside (35)
A solution of ethyl 3-O-acetyl-2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside39 (0.4 g, 0.90 mmol) and activated molecular sieves (3 Å, 0.45 g) in CH2Cl2 (14 mL) was stirred under argon for 1 h. Freshly prepared solution of Br2 in CH2Cl2 (9 mL, 1/165, v/v) was then added and the reaction mixture was kept for 10 min at rt. After that, the solid was filtered-off and the filtrate was concentrated in vacuo at rt and dried. Crude residue was then treated with KSBox (1.8 mmol) and 18-crown-6 (0.18 mmol) in dry acetone (5 mL) under argon for 6 h at rt. Upon completion, the mixture was diluted with dichloromethane (10 mL), the solid was filtered-off and the residue was washed with dichloromethane (5 mL). The combined filtrate was washed with 1% aq. NaOH (1 × 15 mL) and water (2 × 15 mL). The organic layer was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography to afford 2-benzoxazolyl 3-O-acetyl-2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside as a colorless foam in 78% yield (0.375 g, 0.70 mmol). Selected analytical data: Rf = 0.75 (ethyl acetate/hexane, 7/3, v/v); 1H NMR: δ, 2.07 (s, 1H, CO2CH3), 3.76 (m. 1H, H-5), 4.03 (dd, 1H, J6a,6b = 12.6 Hz, H-6b), 4.25 (dd, 1H, J2,3 = 9.7 Hz, H-2), 4.34 (dd, 1H, J6a,6b = 12.7 Hz, H-6a), 4.50 (dd, 1H, J4,5 = 2.9 Hz, H-4), 4.72 (d, 1H, J2 = 10.92 Hz, ½ CH2Ph), 4.87 (d, 1H, J2 = 11.6 Hz, ½ CH2Ph), 5.07 (ddd, 1H, J = 3.47, 6.14, 9.63 Hz, H-3), 5.51, (s, 1H, >C HPh), 5.58 (d, 1H, J1,2 = 9.9 Hz, H-1), 7.27 – 7.48 (m, 14H, aromatic) ppm. 2-Benzoxazolyl 3-O-acetyl-2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside was dissolved in methanol (5 mL) containing 1M NaOCH3 and the resulting mixture was stirred for 1 h at rt. Dowex (H+) was added until neutral pH, the resin was filtered off and washed successively with methanol. The combined filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography to obtain the title compound as a white solid in 70% yield (0.25 g, 0.50 mmol). Analytical data for 35: Rf = 0.44 (ethyl acetate/hexane, 7/3, v/v); [α]D21 = −67.1° (c = 1, CHCl3); 1H NMR: δ, 2.67 (d, 1H, J = 8.4 Hz, OH), 3.73 (m, 1H, H-5), 3.93 −3.98 (m, 2H, H-2, 3), 4.05 (dd, 1H, J6a,6b = 12.7 Hz, H-6a), 4.31–4.38 (m, 2H, H-4, 6b), 4.88 (dd, 2H, J = 10.81 Hz, CH2Ph), 5.14 (d, 1H, J1,2 = 9.7 Hz, H-1), 5.57 (s, 1H, >CHPh), 7.24 – 7.80 (m, 14H, aromatic) ppm; 13C NMR: δ, 60.1, 70.5, 74.5, 75.7, 75.9, 78.1, 84.8, 101.5, 110.2, 119.0, 124.4, 124.5, 124.6, 126.6 (×2), 128.0, 128.3 (×2), 128.4, 128.5 (×2), 129.5, 137.6, 137.9, 141.8, 151.9, 162.1 ppm; HR-FAB MS [M+Na]+ calcd for C27H25NO6SNa 514.1300, found 514.1295.
General glycosylation procedures
Method A - Cu(OTf)2 - promoted glycosylation
A mixture the glycosyl donor (0.11 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (4 Å, 200 mg) in (ClCH2)2 (2 mL) was stirred under argon for 1 h at rt. Cu(OTf)2 (0.13 – 0.22 mmol) was added and the reaction mixture was stirred for 3–5 h at rt. Upon completion, the reaction mixture was diluted with CH2Cl2, the solid was filtered-off, and the residue was washed with CH2Cl2. The combined filtrate (30 mL) was washed with 20% aq. NaHCO3 (15 mL) and water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford a di- or oligosaccharide derivative. Anomeric ratios (if applicable) were determined by comparison of the integral intensities of relevant signals in 1H-n.m.r. spectra.
Method B - A typical Bi(OTf)3- promoted glycosylation procedure
A mixture the glycosyl donor (0.11 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (3 Å, 200 mg) in (ClCH2)2 (2 mL) was stirred under argon for 1 h at rt. The reaction mixture was cooled down at 0 °C and then Bi(OTf)3 (0.22 mmol) was added. After that, the reaction mixture was allowed to warm up and was stirred for additional 1–2 h at rt. Upon completion, the reaction mixture was diluted with CH2Cl2, the solid was filtered-off, and the residue was washed with CH2Cl2. The combined filtrate (30 mL) was washed with 20% aq. NaHCO3 (15 mL) and water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford a di- or oligosaccharide derivative. Anomeric ratios (if applicable) were determined by comparison of the integral intensities of relevant signals in 1H-n.m.r. spectra.
Method C – MeOTf-promoted glycosylation procedure
A mixture the glycosyl donor (0.11 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (3 Å, 200 mg) in (ClCH2)2 (2 mL) was stirred under argon for 1 h at rt. MeOTf (0.33 mmol) was added and the reaction mixture was stirred for 3–5 h at rt. Upon completion, the reaction mixture was diluted with CH2Cl2, the solid was filtered-off, and the residue was washed with CH2Cl2. The combined filtrate (30 mL) was washed with 20% aq. NaHCO3 (15 mL) and water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford a di- or oligosaccharide derivative. Anomeric ratios (if applicable) were determined by comparison of the integral intensities of relevant signals in 1H-n.m.r. spectra.
Method D - typical BnBr promoted glycosylation procedure
A mixture of the glycosyl donor (0.11 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (3Å, 200 mg) in (ClCH2)2 (2 mL) was stirred under argon for 1 h. BnBr (0.33–0.99 mol) was added and the reaction mixture was stirred for 24–36 h at 55 °C. Upon completion, the reaction mixture was diluted with CH2Cl2, the solid was filtered-off and the residue was washed with CH2Cl2. The combined filtrate (30 mL) was washed with 20% aq. NaHCO3 (10 mL) and water (3 × 10 mL), the organic phase was separated, dried with MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to allow the corresponding disaccharide. Anomeric ratios (if applicable) were determined by comparison of the integral intensities of relevant signals in 1H-n.m.r. spectra.
Method E - DMTST-promoted glycosylation procedure
A mixture of glycosyl donor (0.11 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (4Å, 200 mg) in (ClCH2)2 (2 mL) was stirred under argon for 1h at rt. The reaction mixture was cooled to 0 °C and then DMTST (0.033 mmol) was added. After that, the reaction mixture was allowed to warm up and was stirred for additional 4–6 h at rt. Upon completion, the reaction mixture was quenched with triethyl amine (1 drop), the solid was filtered off, the filtrate was diluted with CH2Cl2 (30 mL), washed with 1% NaOH (15 mL) and water (3 × 10 mL). The organic layer was separated, dried with MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate – toluene gradient elution) to obtain the corresponding disaccharide. Anomeric ratios (if applicable) were determined by comparison of the integral intensities of relevant signals in 1H-n.m.r. spectra.
Method F - AgClO4/Cp2ZrCl2 promoted glycosylation procedure
A mixture the glycosyl donor (0.11 mmol), glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (4 Å, 200 mg) in (ClCH2)2 (2 mL) was stirred under argon for 1 h at rt. AgClO4 (0.22 mmol) and Cp2ZrCl2 (0.22 mmol) were then added and the reaction mixture was stirred for 3–5 h at rt. Upon completion, the reaction mixture was diluted with CH2Cl2, the solid was filtered-off, and the residue was washed with CH2Cl2. The combined filtrate (30 mL) was washed with 20% aq. NaHCO3 (15 mL) and water (3 × 10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford a di- or oligosaccharide derivative. Anomeric ratios (if applicable) were determined by comparison of the integral intensities of relevant signals in 1H-n.m.r. spectra.
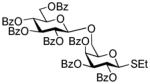
2-Thiazolinyl 2,3,4-tri-O-benzoyl-6-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-1-thio-β-D-glucopyranoside (14)
The title compound was obtained by Method B from benzoxazolyl 2,3,4,6-tetra-O-benzoyl-1-thio-β-D-glucopyranoside (1)19 and 2-thiazolinyl 2,3,4-tri-O-benzoyl-1-thio-β-D-glucopyranoside (12)15 in 69% yield as a white foam. The title compound was also obtained by Method A from 2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl thiocyanate (50)41 and 12 in 89%. Analytical data for 14 was in a good agreement with those reported previously.42
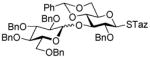
2-Thiazolinyl 2-O-benzyl-3-O-(2,3,4,6-O-tetra-benzyl-α/β-D-glucopyranosyl)-4,6-O-benzylidene-1-thio-β-D-glucopyranoside (15)
This compound was obtained by Method A from 2-benzoxazolyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-glucopyranoside (11)43 and 2-thiazolinyl 2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-glucopyranoside (9)14 in 71% yield (α/β, 2.4/1) as a white foam. In addition, the title compound was also obtained Method E from ethyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-glucopyranoside (40)40 and 2-thiazolinyl 2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-glucopyranoside (9)14 in 77% yield (α/β = >25/1). Analytical data for compound 15 was in a good agreement with those reported previously.14
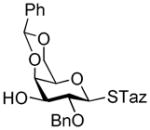
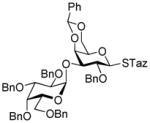
2-Thiazolinyl 2-O-benzyl-3-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl)-4,6-O-benzylidene-1-thio-β-D-galactopyranoside (17)
This compound was obtained by Method A from 2-benzoxazolyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-galactopyranoside (8)44 and 2-thiazolinyl 2-O-benzyl-4,6-O-benzylidene-thio-β-D-galactopyranoside (16) in 52% yield (α/β = >25/1). In addition, this compound was also achieved by Method E from ethyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-galactopyranoside (46)40 and 16 in 74% yield (α/β, 15/1) as a white syrup. Analytical data for 17: Rf = 0.55 (ethyl acetate/toluene, 3/7, v/v); [α]D25 = +48.5° (c = 1, CHCl3); 1H NMR: δ, 3.31 – 3.39 (m, 4H, H-6a′, 6b′, CH2S), 3.55 (dd, 1H, J4′,5′ = 9.7 Hz, J5′,6a′ = 3.0 Hz, H-5′), 3.73 (m, 1H, J5,6a = 2.0 Hz, H-5), 3.87–4.39 (m, 14H, 2×CH2Ph, CH2N, H-2, 3, 4, 6a, 6b, 2′, 3′, 4′), 4.50–4.97 (m, 6H, 3×CH2Ph), 5.30 (d, 1H, J1′,2′ = 3.7 Hz, H-1′), 5.34 (d, 1H, J1,2 = 9.7 Hz, H-1), 5.48 (s, 1H, >CHPh), 7.08–7.60 (m, 30H, aromatic) ppm; 13C NMR: δ, 35.2, 64.4, 69.2, 69.5, 69.7, 70.1, 72.0, 72.1, 73.1, 73.2, 74.9, 75.2, 75.6, 75.7, 76.2, 76.5, 76.7, 78.7, 84.9, 93.02, 101.5, 126.7 (×2), 127.5, 127.6 (×2), 127.7 (×2), 127.7 (×4), 127.8, 127.9 (×2), 128.3 (×3), 128.4 (×7), 128.5 (×4), 129.2, 137.9, 138.2, 138.4, 138.7 (×2), 138.9, 164.1 ppm; HR-FAB MS [M+Na]+ calcd for C57H59NO10S2Na 1004.3478, found 1004.3475.
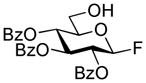
2,3,4-Tri-O-benzoyl-6-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-β–D-glucopyranosyl fluoride (19)
The title compound was obtained by Method C from 2-benzoxazolyl 2,3,4,6-tetra-O-benzoyl-1-thio-β-D-glucopyranoside (1)19 and 2,3,4-tri-O-benzoyl-β-D-glucopyranosyl fluoride (18)45 in 95% yield as a white foam. Analytical data for 19: Rf = 0.51 (ethyl acetate/toluene, 1/9, v/v); [α]D23 = +19.3° (c = 1, CHCl3); 1H NMR: δ,3.97 (dd, 1H, J5′,6a′ = 4.1 Hz, J6a′,6b′ = 8.2 Hz, H-6a′), 4.15 (m, 3H, H-5, 5′, 6b′), 4.47 (dd, 1H, J5,6a = 7.2 Hz, J6a,6b = 10.2 Hz, H-6a), 4.63 (dd, 1H, J5,6b = 3.0 Hz, J6a,6b = 12.2 Hz, H-6b), 5.04 (d, 1H, J1′,2′ = 7.8 Hz, H-1′), 5.29 (d, 1H, J1,2 = 5.9 Hz, H-1), 5.45–5.50 (m, 2H, H-2, 4), 5.56 (dd, 1H, J2′,3′ = 7.8 Hz, H-2′), 5.68 (dd, 1H, J4′,5′ = 9.7 Hz, H-4′), 5.74 (dd, 1H, J3,4 = 8.5 Hz, H-3), 5.94 (dd, 1H, J3′,4′ = 9.7 Hz, H-3′), 7.18–7.95 (m, 35H, aromatic) ppm; 13C NMR: δ, 62.1, 67.6, 68.2, 68.7, 70.4, 70.5 (×2), 70.7, 70.9, 71.5, 71.9, 73.5, 101.1, 104.7, 107.2, 124.5 (×2), 127.4 (×4), 127.5 (×2), 127.6 (×6), 127.7, 127.8, 127.9, 128.0, 128.2, 128.4, 128.7, 128.9 (×5), 129.0 (×4), 129.1 (×2), 132.3, 132.4 (×2), 132.6 (×2), 132.7, 132.8, 137.0, 164.0, 164.3, 164.4 (×2), 164.5, 165.3, 165.4 ppm; HR-FAB MS [M+Na]+ calcd for C61H49FO17Na 1095.2851, found 1095.2871.
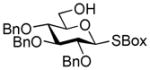
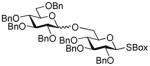
2-Benzoxazolyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranosyl)-1-thio-β-D-glucopyranoside (28)
The title compound was obtained from 2-thiazolinyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-glucopyranoside (23)15 and 2-benzoxazolyl 2,3,4-tri-O-benzyl-1-thio-β-D-glucopyranoside (27)46 by Method D in 85% yield (α/β = 3.2/1) as a colorless syrup. Analytical data for 28: Rf = 0.52 (ethyl acetate/hexane, 3/7, v/v); 1H NMR; δ, 3.28 (dd, 1H, J2,3 = 9.8 Hz, H-2), 3.82 (dd, 1H, J3′,4′ = 9.2 Hz, H-3′), 3.42–3.82 (m, 10H, H-3, 4, 5, 6a, 6b, 2′, 4′, 5′, 6a′, 6b′), 4.43–4.95 (m, 14H, 7×CH2Ph), 5.09 (d, 1H, J1′,2′ = 3.5 Hz, H-1′), 5.38 (d, 1H, J1,2 = 10.1 Hz, H-1), 7.10–7.65 (m, 39H, aromatic) ppm; 13C NMR: δ, 65.5, 68.7, 70.4, 72.3, 73.5, 75.0, 75.3, 75.7, 75.8, 77.4, 79.8, 80.3, 80.8, 81.8, 85.0, 86.7, 97.3, 119.2, 124.4, 124.6, 127.6 (×2), 127.6 (×2), 127.7 (×2), 127.8 (×2), 127.8 (×3), 128.0 (×3), 128.0 (×3), 128.1 (×3), 128.3 (×3), 128.5 (×3), 128.5 (×3), 128.6 (×3), 128.6 (×3), 128.7 (×2), 137.7, 138.2, 138.4, 138.5, 138.7, 138.7, 138.8, 139.0, 142.0, 152.0, 161.7 ppm; HR-FAB MS [M+Na]+ calcd for C68H67NO11SNa 1128.4333, found 1128.4368.
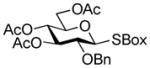
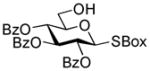
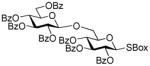
2-Benzoxazolyl 2,3,4-tri-O-benzoyl-6-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-1-thio-β-D-glucopyranoside (30)
The title compound was obtained by Method D from 2-thiazolinyl 2,3,4,6-tetra-O-benzoyl-1-thio-β-D-glucopyranoside (20)15 and 2-benzoxazolyl 2,3,4-tri-O-benzoyl-1-thio-β-D-glucopyranoside (29)43 in 76% yield as a colorless syrup. Analytical data for 30: Rf = 0.45 (ethyl acetate/hexane, 3/7, v/v); [α]D24 = +48.5 ° (c = 1, CHCl3); 1H NMR; δ, 3.53 (dd, 1H, J5,6b = 8.3 Hz, H-6b), 3.86 (dd, 1H, J6a,6b = 4.8 Hz, H-6a), 4.12 (m, 1H, H-5′), 4.15 (m, 1H, H-5), 4.34 (dd, 1H, J5,6b′ = 7.9 Hz, H-6b′), 4.47 (dd, 1H, J6a′,6b′ = 2.6 Hz, H-6a′), 4.74 (m, 1H, H-2), 5.43 (d, 1H, J4′,5′ = 8.4 Hz, H-4′), 5.68 (dd, 2H, H-3, 4), 5.72 (dd, 1H, J3′,4′ = 9.6 Hz, H-3′), 5.90–5.99 (m, 3H, H-1, 1′, 2′), 7.09–8.10 (m, 39H, aromatic) ppm; 13C NMR: δ, 63.3, 64.4, 67.8, 68.8, 79.4, 71.0, 72.3, 74.5, 77.6, 84.3, 110.5, 119.2, 121.4, 124.9, 124.9, 126.9 (×2), 128.7 (×5), 128.8 (×5), 128.9 (×2), 129.0, 129.1, 129.2, 129.5, 129.5, 130.5 (×3), 130.1 (×5), 130.3 (×5), 130.5 (×2), 133.3, 133.7, 133.8 (×3), 133.9, 134.7, 141.9, 152.2, 164.7, 165.5, 165.5, 165.6, 166.1, 166.4 ppm; HR-FAB MS [M+Na]+ calcd for C68H53NO18SNa 1226.2281, found 1226.2283.
2-Benzoxazolyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-β-D-glucopyranosyl)-1-thio-β–D-glucopyranoside (31)
This compound was obtained by Method D from 2-thiazolinyl 2,3,4,6-tetra-O-benzoyl-1-thio-β-D-glucopyranoside (20)14 and 2-benzoxazolyl 2,3,4-tri-O-benzyl-1-thio-β-D-glucopyranoside (27)46 in 79% yield as a colorless syrup. Analytical data for 31: Rf = 0.56 (ethyl acetate/toluene, 2/8, v/v); [α]D22 = −3.4° (c = 0.5, CHCl3); 1H NMR: δ, 3.44–3.73 (m, 6H, H-2, 3, 4, 5, 6a, 6b), 4.10 (m, 1H, H-5′), 4.34 (dd, 1H, J6a′,6b′ = 12.0 Hz, J5,6a′ = 4.9 Hz, H-6a′), 4.38 (dd, 1H, J5′,6b′ = 2.8 Hz, H-6b′), 4.58 (d, 1H, J2 = 10.8 Hz, ½CH2Ph), 4.73–4.92 (m, 6H, H-2′, 2×CH2Ph, ½CH2Ph), 5.41 (dd, 2H, J = 10.3 Hz, H-1, 4′), 5.71 (m, 1H, H-3′), 5.89 (d, 1H, J1′,2′ = 5.3 Hz, H-1′), 7.08–7.92 (m, 39H, aromatic) ppm; 13C NMR: δ, 63.4, 64.2, 67.6, 68.7, 69.3, 72.1, 75.2, 75.7, 75.9, 76.3, 78.6, 81.0, 85.1, 86.7, 97.8, 110.21, 119.1, 121.1, 124.5, 124.6, 126.6, 127.9 (×2), 128.0 (×3), 128.1 (×2), 128.3 (×3), 128.4 (×4), 128.5 (×4), 128.6 (×4), 128.6 (×4), 129.2, 129.4, 129.86 (×3), 130.1 (×2), 130.2 (×2), 133.1, 133.7, 135.1, 137.6, 137.9, 138.4, 141.9, 151.9, 161.7, 164.6, 165.3, 166.2 ppm; HR-FAB MS [M+Na]+ calcd for C68H59NO15SNa 1184.3503, found 1184.3490.
2-Benzoxazolyl 2-O-benzyl-4,6-O-benzylidene-3-O-(2,3,4,6-O-tetra-benzyl-α-D-glucopyranosyl)-1-thio-β-D-glucopyranoside (33)
This compound was obtained by Method D from 2- thiazolinyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-glucopyranoside (23)14 and 2-benzoxazolyl 2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-glucopyranoside (32)43 in 70% yield. Analytical data for 33: Rf = 0.59 (ethyl acetate/toluene, 1/4, v/v); [α]D28 = +37.9° (c = 0.5, CHCl3); 1H NMR: δ,3.30 (m, 2H, H-6a, 6b), 3.51 (dd, 1H, J1′,2′ = 3.6 Hz, J2′,3′ = 6.9 Hz, H-2′), 3.65 (dd, 1H, J4′,5′ = 9.7 Hz, H-4′), 3.75 (m, 2H, J = 5.46 Hz, H-6a′, 6b′), 3.89–4.05 (m, 4H, H-2, 3′, 4, 5), 4.20 (d, 1H, J2 = 12.0 Hz, ½CH2Ph), 4.29 (dd, 1H, J3,4 = 9.0 Hz, H-3), 4.37 (m, 2H, H-5′, ½CH2Ph), 4.50 (d, 1H, J2 = 12.0 Hz, ½CH2Ph), 4.60 (d, 1H, J2 = 12.3 Hz, ½CH2Ph), 4.73–4.91 (m, 5H, 2×CH2Ph, ½CH2Ph), 5.03 (d, 1H, J2 = 10.8 Hz, ½CH2Ph), 5.48 (s, 1H, >CHPh), 5.58 (d, 1H, J1,2 = 9.6 Hz, H-1), 5.70 (d, 1H, J1′,2′ = 3.6 Hz, H-1′), 6.94 – 7.72 (m, 34H, aromatic) ppm; 13C NMR: δ, 68.1, 68.9, 70.1, 70.7, 71.5, 73.5, 75.3, 75.9, 76.5, 78.8, 79.2, 81.9, 82.2, 85.8, 96.4, 102.4, 110.4, 119.4, 124.8, 126.6 (×3), 127.6, 127.7 (×3), 127.8, 128.0 (×3), 128.13 (×3), 128.3 (×3), 128.3 (×3), 128.4 (×3), 128.4 (×3), 128.5, 128.7, 128.8, 129.7, 136.9, 137.0, 137.8, 138.1, 138.9, 139.1, 142.0, 142.1, 142.4, 142.6, 152.1, 160.9 ppm; HR-FAB MS [M+Na]+ calcd for C61H59NO11SNa 1036.3707, found 1036.3716.
2-Benzoxazolyl 2-O-benzyl-4,6-O-benzylidene-3-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl)-1-thio-β-D-galactopyranoside (36)
This compound was obtained by Method D from 2-thiazolinyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-galactopyranoside (34) and 2- benzoxazolyl 2-O-benzyl-4,6-O-benzylidene-1-thio-β-D-galactopyranoside (35) in 77% yield (α/β = >25/1) as a white solid. Analytical data for 36: Rf = 0.34 (ethyl acetate/toluene, 1/4, v/v); [α]D25 = +41.1° (c = 1, CHCl3); 1H NMR: δ,3.33 (dd, 1H, J5,6a = 5.7 Hz, J6a,6b = 9.7 Hz, H-6a), 3.46 (m, 1H, H-5′), 3.43 (m, 1H, H-3′), 3.51 (dd, 1H, J5,6b = 2.8 Hz, H-6b), 3.71 (m, 1H, J5,6b = 2.1 Hz, H-5), 3.92 −3.99 (m, 3H, H-2, 3, 6a′), 4.07 – 4.19 (m, 3H, H-2, 4, 4′), 4.28 (dd, 1H, J6b′,6a′ = 12.7 Hz, H-6b′), 4.33 (s, 2H, CH2Ph), 4.41 (dd, 1H, J3,4 = 7.5 Hz, H-4), 4.50 – 4.66 (m, 4H, 2×CH2Ph), 4.74–4.93 (m, 4H, 2×CH2Ph), 5.31 (d, 1H, J1′,2′ = 3.5 Hz, H-1′), 5.46 (d, 1H, J1,2 = 9.9 Hz, H-1), 5.48 (s, 1H, >CHPh), 7.10 – 7.47 (m, 34H, aromatic) ppm; 13C NMR: δ, 67.7, 69.1, 70.4, 71.9, 72.2, 73.1, 73.3, 75.2, 75.4, 75,7, 76.2, 76.5, 85.4, 92.9, 110.3, 119.1, 124.4, 124.6, 126.7 (×3), 127.6, 127.7 (×4), 127.8 (×3), 127.8 (×3), 127.9 (×4), 128.2 (×4), 128.3 (×3), 128.4 (×2), 128.4 (×2), 128.5 (×4), 128.5 (×4), 128.6 (×3), 129.3, 137.9, 138.0, 138.4, 138.7, 138.9, 142.1, 152.1, 161.1 ppm; HR-FAB MS [M+Na]+ calcd for C67H59NO11SNa 1036.3707, found 1036.3704.
2-Thiazolinyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-D-glucopyranosyl)-1-thio-β–D-glucopyranoside (44)
The title compound was obtained by Method E from ethyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-glucopyranoside (40)40 and 2-thiazolinyl 2,3,4-tri-O-benzyl-1-thio-β-D-glucopyranoside (43)42 in 83% yield (α/β = 1.2/1) as a colorless foam. Analytical data for 44 was in good agreement with those reported previously.42
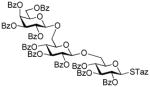
2-Thiazolinyl 2,3,4-tri-O-benzoyl-6-O-(2,3,4-tri-O-benzoyl-6-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-β-D-glucopyranosyl)-1-thio-β–D-glucopyranoside (47)
The title compound was obtained by Method E from ethyl 2,3,4-tri-O-benzoyl-6-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-1-thio-β–D-glucopyranoside (22)47 and 2-thiazolinyl 2,3,4-tri-O-benzoyl-1-thio-β-D-glucopyranoside (12)15 in 67% yield as a white foam. Analytical data for 47: Rf = 0.46 (ethyl acetate/toluene, 1/4, v/v); [α]D21 = +5.8° (c = 1, CHCl3); 1H NMR; δ, 3.29–3.39 (m, 2H, CH2S), 3.59 (m, 1H, H-6a′), 3.85 (m, 2H, H-5′,6b″), 4.00 (m, 1H, 6a″), 4.13–4.29 (m, 2H, CH2N), 4.44 (m, 2H, H-5, 6a′), 4.66 (m, 1H, H-6b), 4.67 (d, 1H, J1″,2″ = 7.8 Hz, H-1″), 5.04 (dd, 1H, J4″,5″ = 9.5 Hz, H-4″), 5.11 (dd, 1H, J2″,3″ = 7.8 Hz, H-2″), 5.21 (d, 1H, J1′,2′ = 7.9, H-1′), 5.27 (dd, 1H, J2′,3′ = 4.0 Hz, H-2′), 5.59–5.62 (m, 2H, H-3″, 4′), 5.65 (dd, 1H, J4,5 = 8.3 Hz, H-4), 5.77 (dd, 1H, J2,3 = 7.1 Hz, H-2), 5.78 (d, 1H, J1,2 = 7.9 Hz, H-1), 5.91 (dd, 1H, J3,4 = 9.6 Hz, H-3), 6.15 (dd, 1H, J3′,4′ = 9.7 Hz, H-3′), 7.15–8.10 (m, 50H, aromatic); 13C NMR; δ, 58.8, 63.5, 64.4, 68.2, 68.4, 69.8, 69.9, 70.6, 70.8, 72.0, 72.3, 72.5, 72.9, 73.0, 74.2, 74.3, 83.5, 100.2, 101.5, 125.5 (×2), 127.2, 127.9, 128.4 (×4), 128.5 (×7), 128.6 (×6), 128.8 (×3), 128.9 (×2), 129.0, 129.1 (×4), 129.2 129.3, 129.4 (×2), 129.5, 129.8, 129.9 (×2), 130.0 (×8), 130.1(×5), 130.2 (×2), 130.3(×4), 133.3 (×2), 133.4 (×2), 133.5 (×2), 133.6 (×2), 165.2, 165.3, 165.4, 165.6 (×2), 166.0 (×2), 166.4 ppm; HR-FAB MS [M+Na]+ calcd for C91H75NO25S2Na 1668.3967, found 1668.4023.
2-Thiazolinyl 2,3,4-tri-O-benzoyl-6-O-(2,3,4-tri-O-benzoyl-6-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-β-D-glucopyranosyl)-1-thio-β–D-glucopyranoside (49)
The title compound was obtained by Method E from ethyl 2,3,4-tri-O-benzoyl-6-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl)-1-thio-β–D-glucopyranoside (48)42 and 2-thiazolinyl 2,3,4-tri-O-benzoyl-1-thio-β-D-glucopyranoside (12)15 in 64% yield as a white foam. Analytical data for 49: Rf = 0.44 (ethyl acetate/toluene, 1/4, v/v); [α]D24 = +12.9° (c = 1, CHCl3); 1H NMR: δ, 3.28 (m, 1H), 3.37 (m, 1H), 3.54 (dd, 1H, J = 6.5 Hz), 3.88 (m, 2H), 3.97 (m, 2H, J = 8.7 Hz), 4.06 (d, 1H, J = 10.5 Hz), 4.11 (m, 1H), 4.26 (m, 1H), 4.43 (ddd, 1H, J = 5.8, 10.9 Hz), 4.54 (dd, 1H, J = 5.9 Hz), 4.59 (m, 1H), 4.65 (d, 1H, J = 7.7 Hz), 5.04 (dd, 1H, J = 9.4 Hz), 5.15 (m, 2H, J = 8.1, 9.3 Hz), 5.55 (dd, 1H, J = 9.4 Hz), 5.64 (dd, 1H, J = 9.58 Hz), 5.69 (dd, 1H, J = 9.8 Hz), 5.77–5.83 (m, 2H), 5.86 – 5.92 (m, 2H), 6.02 (d, 1H, J = 2.0 Hz), 7.20–8.20 (m, 50H, aromatic) ppm; 13C NMR: δ, 57.7, 58.8, 63.5, 64.4, 68.3, 68.8, 69.9, 70.6, 71.7, 71.6, 72.3, 72.5, 72.9, 73.1, 74.3, 74.8, 78.4, 83.5, 100.2, 102.2, 125.5 (×2), 127.3, 127.9 (×4), 128.4 (×2), 128.5 (×3), 128.5, 128.6 (×4), 128.7 (×4), 128.8, 128.9 (×3), 129.0 (×3), 129.1 (×4), 129.2 129.3, 129.4 (×2), 129.5, 129.8 (×2), 129.9, 130.0 (×2), 130.1, 130.2 (×2), 130.3 (×2), 133.4 (×4), 133.5 (×4), 133.6 (×4), 136.3 (×2), 163.0, 165.2, 165.3 (×2), 165.5 (×2), 165.7 (×2), 165.9, 166.2 ppm; HR-FAB MS [M+Na]+ calcd for C91H75NO25S2Na 1668.3967, found 1668.3940.
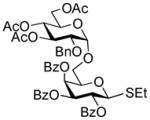
2-Benzoxazolyl 2,3,4-tri-O-benzoyl-6-O-(2,3,4-tri-O-benzoyl-6-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-β-D-glucopyranosyl)-1-thio-β–D-glucopyranoside (51)
The title compound was obtained by Method D from 14 and 2-benzoxazolyl 2,3,4-tri-O-benzoyl-1-thio-β-D-glucopyranoside (29)43 in 67% yield as a colorless syrup. Analytical data for 51: Rf = 0.48 (ethyl acetate/toluene, 1.5/8.5, v/v); [α]D28 = +47.4° (c = 1, CHCl3); 1H NMR: δ, 3.43–3.45 (m, 2H, H-6a, 6b), 3.66 (dd, 1H, J5′,6a′ = 5.5 Hz, J6a′,6b′ = 11.5 Hz, H-6a′), 3.84 (m, 1H, H-5′), 4.06–4.16 (m, 3H, H-5, 5″, 6b′), 4.30 (dd, 1H, J5″,6a″ = 5.1 Hz, J6a″,6b″ = 12.1 Hz, H-6a″), 4.54–4.58 (m, 2H, H-4, 6b″), 4.95 (d, 1H, J1′,2′ = 11.0 Hz, H-1′), 5.22 (dd, 1H, H-4′), 5.50–5.55 (m, 2H, H-2′, 3′), 5.60–5.69 (m, 2H, H-3″, 4″), 5.71 (dd, 1H, J3,4 = 9.6 Hz, H-3), 5.77 (d, 1H, J1″,2″ = 5.3 Hz, H-1″), 5.86 (dd, 1H, J2,3 = 9.7 Hz, H-2), 5.95 (d, 1H, J2″,3″ = 9.5 Hz), 5.96 (d, 1H, J1,2 = 10.3 Hz, H-1), 7.15–8.10 (m, 54H, aromatic) ppm; 13C NMR: δ, 60.6, 63.0, 63.3, 68.1, 68.6, 69.3, 69.5, 70.0, 70.8, 71.7, 72.4, 72.4, 73.2, 74.3, 76.7, 77.6, 77.7, 84.1, 97.8, 102.0, 110.3, 119.1, 121.1, 124.6, 124.8, 126.6, 128.2 (×2), 128.4 (×2), 128.4 (×2), 128.5 (×4), 128.6 (×4), 128.8, 128.9, 129.0, 129.1, 129.1, 129.2, 129.4 (×3), 129.7, 129.8 (×2), 129.9 (×4), 130.0 (×4), 130.0 (×4), 130.0 (×4), 130.10 (×2), 130.3, 133.1, 133.2, 133.3 (×2), 133.4, 133.5, 133.6 (×2), 133.6, 134.4, 141.7, 152.0, 161.4, 164.5, 165.1, 165.2, 165.2, 165.4, 165.9, 166.0, 166.3 ppm; HR-FAB MS [M+Na]+ calcd for C95H75NO26SNa 1700.4196, found 1700.4182.
O-(2,3,4-Tri-O-benzoyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-benzoyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzoyl-β-D-glucopyranosyl fluoride (52)
The title compound was obtained by Method C from 51 and 2,3,4-tri-O-benzoyl-β-D-glucopyranosyl fluoride (18)45 in 87% yield as a colorless syrup. Analytical data for 52: Rf = 0.42 (ethyl acetate/toluene, 1/9, v/v); [α]D25 = −2.0° (c = 1, CHCl3); 1H NMR: δ, 3.54 (m, 1H), 3.59 (m, 1H), 3.86 (m, 3H), 4.00–4.08 (m, 3H), 4.14 (dd, 1H, J = 12.1 Hz), 4.34 (m, 1H), 4.46 (dd, 1H, J = 4.9, 11.4 Hz), 4.60 (dd, 1H, J = 11.8 Hz), 4.76 (d, 1H, J =7.7 Hz), 4.82 (d, 1H, J = 7.9 Hz), 5.11 (d, 1H, J = 7.8 Hz), 5.21 (dd, 1H, J = 9.8), 5.28 (dd, 1H, J = 9.3 Hz), 5.34 (d, 1H, J = 6.2 Hz), 5.40 (dd, 1H, J = 9.7 Hz), 5.46 (m, 1H), 5.53 (dd, 1H, J = 9.5 Hz), 5.62–5.70 (m, 3H), 5.75 (dd, 1H, J = 9.6 Hz), 5.79–5.85 (m, 2H), 6.16 (dd, 1H, J = 9.6 Hz), 7.22–8.10 (m, 65H, aromatic) ppm; 13C NMR: δ, 63.4, 68.1, 68.7, 68.9, 69.1, 69.7, 69.8, 70.2, 71.7, 72.0 (×3), 72.4, 72.5, 72.8, 72.9, 73.0, 73.8, 74.2, 74.7, 100.9, 101.5, 101.7, 106.2, 128.4 (×4), 128.5 (×6), 128.6 (×6), 128.7 (×6), 128.8 (×4), 128.9 (×4), 129.0 (×2), 129.1 (×4), 129.2 (×4), 129.5 (×2), 129.7 (×2), 129.8 (×2), 129.9 (×3), 130.0 (×6), 130.1 (×6), 130.2 (×2), 133.2 (×2), 133.3 (×3), 133.4 (×2), 133.5 (×3), 133.6, 133.7, 133.8 (×2), 134.1, 165.0, 165.2, 165.3 (×2), 165.4 (×3), 165.5, 165.7, 165.9 (×2), 166.0, 166.3 ppm; HR-FAB MS [M+Na]+ calcd for C115H93FO33Na 2043.5481, found 2043.5452.
Ethyl O-(2,3,4-tri-O-benzoyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-benzoyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-benzoyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzoyl-1-thio-β-D-glucopyranoside (53)
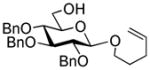
The compound was obtained by Method F from 52 and ethyl 2,3,4-tri-O-benzoyl-thio-β-D-glucopyranoside (21)48 in 84% yield as white solid. Analytical data for 53: Rf = 0.46 (ethyl acetate/toluene, 1.5/8.5, v/v;[α]D26 = −9.3 ° (c = 1, CHCl3); 1H NMR: δ,1.14 (t, SCH2CH3), 2.68 (m, 2H, SCH2CH3), 3.58 (m, 1H, J = 5.5, 6.2 Hz), 3.67–3.75 (m, 3H), 3.85 – 3.92 (m, 6H), 4.02 (dd, 1H, J = 10.8 Hz), 4.12 (m, 1H), 4.35 (m, 1H), 4.48 (dd, 1H, J = 5.5, 12.1 Hz), 4.57 (dd, 1H, J = 11.0 Hz), 4.70 (d, 1H, J = 10.0 Hz), 4.76 (d, 1H, J = 7.7 Hz), 4.86 (m, 2H), 5.16 (d, 1H, J = 7.9 Hz), 5.25 (dd, 1H, J =9.6 Hz), 5.33 (dd, 1H, J = 9.3 Hz), 5.39 (dd, 1H, J = 9.6 Hz), 5.43–5.56 (m, 7H), 5.70 (dd, 1H, J = 9.7 Hz), 5.76–5.87 (m, 2H), 5.90–5.95 (m, 2H), 6.23 (dd, 1H, J = 9.7 Hz), 6.95–8.90 (m, 80H, aromatic) ppm; 13C NMR: δ, 14.9, 31.1, 63.5, 68.0 (×2), 69.0, 69.3, 69.7, 69.9, 70.1, 70.2, 70.7, 71.0, 72.2, 72.3, 72.4, 72.5 (×2), 72.7, 72.8, 73.0 (×2), 73.8, 74.2, 74.7, 78.4, 83.7, 101.2, 101.3, 101.4, 101.8, 128.2 (×3), 128.3 (×5), 128.4 (×6), 128.5 (×4), 128.6 (×3), 128.7 (×6), 128.8 (×3), 128.9 (×4), 129.0 (×4), 129.1 (×3), 129.2 (×4), 129.3 (×2), 129.5 (×2), 129.7 (×4), 129.8 (×7), 129.9 (×4), 130.0 (×9), 130.1 (×10), 133.1 (×2), 133.2 (×2), 133.3 (×2), 133.4 (×3), 133.5 (×2), 133.6, 133.8, 165.2 (×2), 165.3 (×3), 165.4 (×2), 165.6 (×2), 165.8, 165.9 (×4), 166.0, 166.3 ppm; HR-FAB MS [M+Na]+ calcd for C144H120O41SNa 2559.6923, found 2559.6938.
4-Pentenyl O-(2,3,4-tri-O-benzoyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-benzoyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-benzoyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-benzoyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzoyl-β-D-glucopyranoside (55)
This compound was obtained by Method C from 53 and 4-pentenyl 2,3,4-tri-O-benzoyl-β-D-glucopyranoside (54)16 in 72% yield as a white solid. Analytical data for 53 Rf = 0.50 (ethyl acetate/toluene, 2/8, v/v); [α]D22 = −10.5 ° (c = 1, CHCl3), 1H NMR; δ, 1.41–1.58 (m, 4H), 1.89 (m, 2H), 3.28 (m, 1H), 3.55 (m, 2H), 3.71 (dd, 1H, J = 11.5 Hz), 3.75–3.81 (m, 5H), 3.90 – 4.07 (m, 7H), 4.12 (m, 1H), 4.31 (m, 1H), 4.47 (dd, 1H, J = 5.4, 12.1 Hz), 4.56 (dd, 1H, J = 2.6, 9.3 Hz), 4.64 (d, 1H, J = 7.9 Hz), 4.75 (d, 1H, J = 1.6 Hz), 4.79–4.87 (m, 6H), 5.12 (d, 1H, J = 7.8 Hz), 5.24 (dd, 1H, J = 9.7 Hz), 5.31 (dd, 1H, J = 1.8, 9.6 Hz), 5.38 – 5.55 (m, 9H), 5.58–5.63 (m, 1H), 5.68 (dd, 1H, J = 9.7 Hz), 5.67 (dd, 1H, J = 9.7 Hz), 5.86 (dd, 1H, J = 9.6 Hz), 5.90 −5.97 (m, 2H, J = 9.6 Hz), 6.06 (dd, 1H, J = 9.6 Hz), 6.16 (dd, 1H, J = 9.7 Hz), 6.90–8.00 (m, 95H, aromatic) ppm; 13C NMR: δ, 28.7, 29.9, 30.0 (×2), 31.1, 63.0, 63.4, 67.6, 68.2, 69.8, 69.9, 70.1, 70.5 (×2), 70.9 (×2), 72.1 (×2), 72.3 (×2), 72.4 (×3), 72.8 (×2), 72.9, 73.0 (×2), 74.5, 89.9 (×2), 101.2, 101.3 (×3), 101.4, 101.6, 115.0, 128.2 (×5), 128.3 (×5), 128.4 (×5), 128.5 (×7), 128.6 (×5), 128.7 (×6), 128.8 (×7), 129.00 (×10), 129.1 (×6), 129.2 (×6), 129.3 (×4), 129.4 (×4), 129.7 (×4), 129.8 (×8), 129.9 (×6), 130.0 (×12), 130.1 (×12), 130.2 (×6), 165.2 (×3), 165.3, 165.4 (×3), 165.5 (×2), 165.6 (×2), 165.7, 165.9 (×4), 166.0, 166.3 ppm; HR-FAB MS [M+Na]+ calcd for C174H146O50Na 3057.8780, found 3057.8748.
Supplementary Material
Acknowledgments
This work was supported by awards from NIGMS (GM077170) and NIAID (AI067494). Dr. Winter and Mr. Kramer (UM – St. Louis) are thanked for HRMS determinations.
Footnotes
Supporting Information Available: 1H and 13C NMR spectra for all new compounds, this material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Seeberger PH, Werz DB. Nature. 2007;446:1046–1051. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]
- 2.Galonic DP, Gin DY. Nature. 2007;446:1000–1007. doi: 10.1038/nature05813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prescher JA, Bertozzi CR. Nature Chem Bio. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 4.Smoot JT, Demchenko AV. Adv Carbohydr Chem Biochem. 2009;62:161–250. doi: 10.1016/S0065-2318(09)00005-5. [DOI] [PubMed] [Google Scholar]
- 5.Fraser-Reid B, Wu Z, Udodong UE, Ottosson H. J Org Chem. 1990;55:6068–6070. [Google Scholar]
- 6.Fraser-Reid B, Udodong UE, Wu ZF, Ottosson H, Merritt JR, Rao CS, Roberts C, Madsen R. Synlett. 1992:927–942. and references therein. [Google Scholar]
- 7.Douglas NL, Ley SV, Lucking U, Warriner SL. J Chem Soc, Perkin Trans. 1998;1:51–65. [Google Scholar]
- 8.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 9.Kaeothip S, Demchenko AV. Carbohydr Res. 2011;346:1371–1388. doi: 10.1016/j.carres.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demchenko AV, De Meo C. Tetrahedron Lett. 2002;43:8819–8822. [Google Scholar]
- 11.Lopez JC, Uriel C, Guillamon-Martin A, Valverde S, Gomez AM. Org Lett. 2007;9:2759–2762. doi: 10.1021/ol070753r. [DOI] [PubMed] [Google Scholar]
- 12.Kanie O, Ito Y, Ogawa T. J Am Chem Soc. 1994;116:12073–12074. [Google Scholar]
- 13.Ito Y, Kanie O, Ogawa T. Angew Chem Int Ed. 1996;35:2510–2512. [Google Scholar]
- 14.Demchenko AV, Pornsuriyasak P, De Meo C, Malysheva NN. Angew Chem Int Ed. 2004;43:3069–3072. doi: 10.1002/anie.200454047. [DOI] [PubMed] [Google Scholar]
- 15.Pornsuriyasak P, Demchenko AV. Chem Eur J. 2006;12:6630–6646. doi: 10.1002/chem.200600262. [DOI] [PubMed] [Google Scholar]
- 16.Vidadala SR, Thadke SA, Hotha S. J Org Chem. 2009;74:9233–9236. doi: 10.1021/jo901837z. [DOI] [PubMed] [Google Scholar]
- 17.Kaeothip S, Pornsuriyasak P, Rath NP, Demchenko AV. Org Lett. 2009;11:799–802. doi: 10.1021/ol802740b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamat MN, De Meo C, Demchenko AV. J Org Chem. 2007;72:6947–6955. doi: 10.1021/jo071191s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamat MN, Rath NP, Demchenko AV. J Org Chem. 2007;72:6938–6946. doi: 10.1021/jo0711844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demchenko AV, Malysheva NN, De Meo C. Org Lett. 2003;5:455–458. doi: 10.1021/ol0273452. [DOI] [PubMed] [Google Scholar]
- 21.Zhong W, Boons G-J. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinheim, Germany: 2008. pp. 261–303. [Google Scholar]
- 22.Lonn H. J Carbohydr Chem. 1987;6:301–306. [Google Scholar]
- 23.Ravenscroft M, Roberts RMG, Tillett JG. J Chem Soc Perkin Trans. 1982;2:1569–1972. [Google Scholar]
- 24.Andersson F, Fugedi P, Garegg PJ, Nashed M. Tetrahedron Lett. 1986;27:3919–3922. [Google Scholar]
- 25.Mydock LK, Kamat MN, Demchenko AV. Org Lett. 2011;13:2928–2931. doi: 10.1021/ol2009818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kochetkov NK, Klimov EM, Malysheva NN. Tetrahedron Lett. 1989;30:5459–5462. [Google Scholar]
- 27.Kochetkov NK, Klimov EM, Malysheva NN, Demchenko AV. Carbohydr Res. 1991;212:77–91. doi: 10.1016/0008-6215(91)84047-i. [DOI] [PubMed] [Google Scholar]
- 28.Kochetkov NK, Klimov EM, Malysheva NN, Demchenko AV. Carbohydr Res. 1992;232:C1–C5. doi: 10.1016/s0008-6215(00)91007-3. [DOI] [PubMed] [Google Scholar]
- 29.Kaeothip S, Akins SJ, Demchenko AV. Carbohydr Res. 2010;345:2146–2150. doi: 10.1016/j.carres.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 30.Nicolaou KC, Dolle RE, Papahatjis DP, Randall JL. J Am Chem Soc. 1984;106:4189–4192. [Google Scholar]
- 31.Nicolaou KC, Ueno H. In: Preparative Carbohydrate Chemistry. Hanessian S, editor. Marcel Dekker, Inc; New York: 1997. pp. 313–338. [Google Scholar]
- 32.Hoyle CE, Bowman CN. Angew Chem Int Ed. 2010;49:1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 33.Noti C, Paz JL, Polito L, Seeberger PH. Chem Eur J. 2006;12:8664–8686. doi: 10.1002/chem.200601103. [DOI] [PubMed] [Google Scholar]
- 34.Rele SM, Iyer SS, Baskaran S, Chaikof EL. J Org Chem. 2004;69:9159–9170. doi: 10.1021/jo049092r. [DOI] [PubMed] [Google Scholar]
- 35.Jeon I, Iyer K, Danishefsky SJ. J Org Chem. 2009;74:8452–8455. doi: 10.1021/jo901682p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Payne RJ, Wong CH. Chem Commun. 2010;46:21–43. doi: 10.1039/b913845e. [DOI] [PubMed] [Google Scholar]
- 37.Gamblin DP, Scanlan EM, Davis BG. Chem Rev. 2009;109:131–163. doi: 10.1021/cr078291i. [DOI] [PubMed] [Google Scholar]
- 38.Pozsgay V, Kubler-Kielb J. ACS Symp Ser. 2008:989. [Google Scholar]; Carbohydrate-Based Vaccines. :36–70. [Google Scholar]
- 39.Garegg PJ, Oscarson S. Carbohydr Res. 1985;136:207–213. [Google Scholar]
- 40.Weïwer M, Sherwood T, Linhardt RJ. J Carbohydr Chem. 2008;27:420–427. [Google Scholar]
- 41.Ranade SC, Kaeothip S, Demchenko AV. Org Lett. 2010;12:5628–5631. doi: 10.1021/ol1023079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pornsuriyasak P, Gangadharmath UB, Rath NP, Demchenko AV. Org Lett. 2004;6:4515–4518. doi: 10.1021/ol048043y. [DOI] [PubMed] [Google Scholar]
- 43.Kamat MN, Demchenko AV. Org Lett. 2005;7:3215–3218. doi: 10.1021/ol050969y. [DOI] [PubMed] [Google Scholar]
- 44.Mydock LK, Demchenko AV. Org Lett. 2008;10:2103–2106. doi: 10.1021/ol800345j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Konda Y, Toida T, Kaji E, Takeda K, Harigaya Y. Carbohydr Res. 1997;301:123–143. [Google Scholar]
- 46.Mydock LK, Demchenko AV. Org Lett. 2008;10:2107–2110. doi: 10.1021/ol800648d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pornsuriyasak P, Demchenko AV. Tetrahedron: Asymmetry. 2005;16:433–439. [Google Scholar]
- 48.Agoston K, Kroeger L, Dekany G, Thiem J. J Carbohydr Chem. 2007;26:513–525. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.