

Abstract
Little is known about the transcription factors that mediate lineage commitment of multipotent hematopoietic precursors. One candidate is the Ets family transcription factor PU.1, which is expressed in myeloid and B cells and is required for the development of both these lineages. We show here that the factor specifically instructs transformed multipotent hematopoietic progenitors to differentiate along the myeloid lineage. This involves not only the up-regulation of myeloid-specific cell surface antigens and the acquisition of myeloid growth-factor dependence but also the down-regulation of progenitor/thrombocyte-specific cell-surface markers and GATA-1. Both effects require an intact PU.1 transactivation domain. Whereas sustained activation of an inducible form of the factor leads to myeloid lineage commitment, short-term activation leads to the formation of immature eosinophils, indicating the existence of a bilineage intermediate. Our results suggest that PU.1 induces myeloid lineage commitment by the suppression of a master regulator of nonmyeloid genes (such as GATA-1) and the concomitant activation of multiple myeloid genes.
Keywords: differentiation, eosinophil, GATA-1, transcription factor
The differentiation of hematopoietic stem cells into specialized blood cells involves global changes in gene expression, resulting in expression of a characteristic set of genes in each mature cell type. Although a number of transcription factors that regulate the expression of cell type-specific genes are well characterized, little is known about the transcriptional events that lead to lineage commitment of multipotent progenitors. Gene inactivation studies in mice are of somewhat limited value in addressing this question because of commonly encountered gene redundancies and difficulties in defining the earliest committed progenitors. On the other hand, in vitro work with isolated multipotent progenitors is not practical, as they represent only a small fraction of hematopoietic tissues and are not easily maintained in culture. One approach to circumvent these difficulties relies on the use of hematopoietic cell lines with multilineage potential, with the disadvantage that cell lines are often aberrant in their properties. An alternative in vitro system, developed in our laboratory, is based on the generation of primary transformed hematopoietic cells. This can be achieved by infecting chicken blastoderm cell suspensions with the Gag–Myb–Ets-encoding E26 leukemia virus, leading to the outgrowth of proliferating primary colonies in semisolid medium. 50%–80% of these colonies consist of cells with properties of multipotent progenitors, the rest contain myeloblasts and occasional erythroid cells and eosinophils (Graf et al. 1992). The Myb–Ets-transformed multipotent progenitors (termed MEPs) express cell-surface antigens characteristic of normal multipotent hematopoietic progenitors and thrombocytes (McNagny et al. 1992, 1997). They can be induced to differentiate into thrombocytes when v-Myb is inactivated (Frampton et al. 1995), into erythrocytes when v-Ets is deleted or inactivated (Kraut et al. 1994; Rossi et al. 1996a), and into myeloblasts and eosinophils when the Ras or PKC pathways are activated (Graf et al. 1992). The differentiation of MEPs into myeloid cells and eosinophils involves the up-regulation of the transcription factors PU.1 and C/EBP and the down-regulation of GATA-1, factors that are known to be expressed in normal cells of the corresponding lineages (Graf et al. 1992; Kulessa et al. 1995; McNagny et al. 1998).
It is widely assumed that lineage-specific gene expression programs are established and maintained by both positive and negative interactions between transcription factors that are expressed in different compartments of the hematopoietic system (for review, see Ness and Engel 1995). Thus, whereas myeloid cells appear to be specified by a combination of PU.1 and C/EBP proteins (for review, see Tenen et al. 1997), eosinophils (at least in chickens) require a combination of C/EBP and moderate levels of GATA-1 (Kulessa et al. 1995; McNagny et al. 1998). In all cases, promoters of lineage-specific genes contain binding sites for the corresponding proteins and these sites are important for function. The lineage-restricted factors in turn cooperate with more ubiquitously expressed regulators, such as c-Myb and AML-1, to activate their cognate promoters (for review, see Tenen et al. 1997). However, the absence of a factor may be as important as its presence. Thus, forced expression of GATA-1 in chicken myeloid cells leads to a suppression of myeloid gene expression followed by up-regulation of either eosinophil- or progenitor-specific genes (Kulessa et al. 1995). In both MEPs and eosinophils, PU.1 expression is extinguished, whereas C/EBP is down-regulated in MEPs but not eosinophils (Kulessa et al. 1995; McNagny et al. 1998). These findings raised the possibility that PU.1 and C/EBP themselves induce multipotent progenitors to differentiate along the myeloid and eosinophil lineages, respectively. We have addressed this question by introducing PU.1 (this paper) and C/EBP (Nerlov et al. 1998) into MEPs using recombinant versions of the E26 retrovirus that express these factors.
PU.1 is a transcription factor with a winged helix–turn–helix-type DNA-binding domain that is a member of the Ets family of proteins and is expressed specifically in myeloid and B-lymphoid cells of the hematopoietic system (Klemsz et al. 1990). In mice in which the PU.1 gene has been inactivated, these lineages either completely fail to develop (Scott et al. 1994) or are delayed and highly aberrant (McKercher et al. 1996). Experiments in chimeric animals have shown that the factor is required in a cell-autonomous manner and appears to affect the differentiation of multipotent myeloid/B-cell progenitors (Scott et al. 1997). These observations raised the intriguing possibility that PU.1 is involved at the level of commitment of multipotent progenitors and is not simply required for late functions of myeloid and B cells.
Here we show that PU.1 instructs MEPs to exit the multipotent state and to differentiate into myeloblasts. This process becomes irreversible within 2–3 days of forced expression of an active PU.1 form and is preceded by the down-regulation of GATA-1, a factor incompatible with myeloid gene expression. Thus, PU.1 is the first transcription factor shown to mediate commitment of multipotent progenitors to the myeloid lineage.
Results
Hematopoietic cells transformed by a PU.1-expressing E26 virus are exclusively myeloid
To assess the effect of PU.1 on MEPs we constructed a recombinant version of the E26 virus expressing human PU.1 from an internal ribosomal entry site (E26-PU.1; Fig. 1A). Proviral DNA corresponding to this construct, as well as that of wild-type E26 virus (E26–WT), were used to transiently transfect the Q2bn-packaging cell line to produce virus. These cells were then cocultivated with suspensions of 2-day chicken blastoderms (which contain large numbers of E26 target cells in their yolk sac) and the infected cells plated in semisolid medium for 2 weeks at 37°C. Approximately 100–200 transformed colonies per plate were obtained with each construct. These were pooled and analyzed for expression of lineage-specific cell-surface antigens. (MEP21 detects MEPs, EOS47 detects eosinophils, and MYL51/2 detects myeloblasts). As shown in Figure 1B, E26–WT-transformed colonies consisted of ∼50% MEPs, ∼8% eosinophils, and ∼40% myeloblasts (which arise spontaneously from MEPs under these culture conditions; Graf et al. 1992); in contrast, the presence of PU.1 in the E26 virus led to a dramatic increase in the proportion of myeloid cells at the expense of MEPs and eosinophils, which were reduced to almost background levels. Similar results were obtained for other MEP (MEP26) and myeloid (1C3, MHC II) markers (data not shown). Clonal expansion of individual E26–PU.1-transformed colonies yielded only myeloid populations, and we observed no expression of erythroid, B-, or T-cell markers (data not shown). These results suggest that PU.1 accelerates the differentiation of MEPs into myeloid cells.
Figure 1.


Phenotype of colonies transformed by E26–PU.1 virus. (A) Structure of the E26–WT and E26–PU.1 proviruses. In E26–PU.1, the PU.1 cDNA is translated from a bicistronic genomic RNA via an internal ribosomal entry site (IRES). (LTR) Long terminal repeat. (B) Distribution of cell types in colonies transformed by E26–WT and E26–PU-1. (Light-shaded bars) MEP21; (dark-shaded bars) EOS47; (solid bars) MYL51/2. Transformed colonies were pooled and analyzed by FACS for cell surface antigen expression. The average number of cells positive for each antigen, as determined in two independent experiments, is shown (no value deviated >4% from the mean). In the case of the E26–PU.1-transformed populations ∼15% weakly MYL51/2-positive cells were scored as negative. Staining with MEP26 (another MEP/thrombocyte marker) and anti-MHC II or 1C3 (myeloid lineage markers) antibodies gave similar results as for MEP21 and MYL51/2, respectively; surface markers for the erythroid, B-, and T-cell lineages could not be detected.
Activation of a conditional PU.1 allele in MEPs induces myeloid differentiation
To ensure that the predominance of myeloid cells in E26-PU.1-transformed colonies was the result of an instructive effect and not to the selective survival of myeloid cells, we made a conditional allele of PU.1 by fusing the hormone-binding domain of the human estrogen receptor (hER) to the carboxyl terminus of human PU.1 (Fig. 2A). The inducibility of this PU.1–estrogen receptor chimera (PUER) was tested by transient transfection with a reporter construct containing three PU.1 binding sites upstream of the herpes simplex virus (HSV) thymidine kinase (TK) minimal promoter driving the luciferase gene (Fig. 2A), in fibroblasts maintained in the presence or absence of β-estradiol (βE). As seen in Figure 2B, PUER activity was inducible by βE from background levels to a level indistinguishable from that of wild-type PU.1. Next, we constructed an E26 derivative containing PUER (E26–PUER; analogous to E26–PU.1 in Fig. 1A) and used this virus to transform blastoderm cultures. Individual clones were isolated and those that contained no myeloid cells or eosinophils were then selected for further studies. Such E26–PUER-transformed MEP clones were treated with βE, and after 2, 4, and 6 days, their phenotype was analyzed. As illustrated in Figure 2C, we observed a rapid down-regulation of MEP21 as well as MEP26 antigen (another MEP/thrombocyte restricted marker; McNagny et al. 1992) and an up-regulation of the myeloid markers 1C3 and MYL51/2, whereas EOS47 antigen was not detectable at all. Almost identical time courses of antigenic changes were observed with four clones examined, whereas another three exhibited slower kinetics of both down- and up-regulation of antigens. No effect whatsoever of βE was observed on E26–WT-transformed MEPs.
Figure 2.
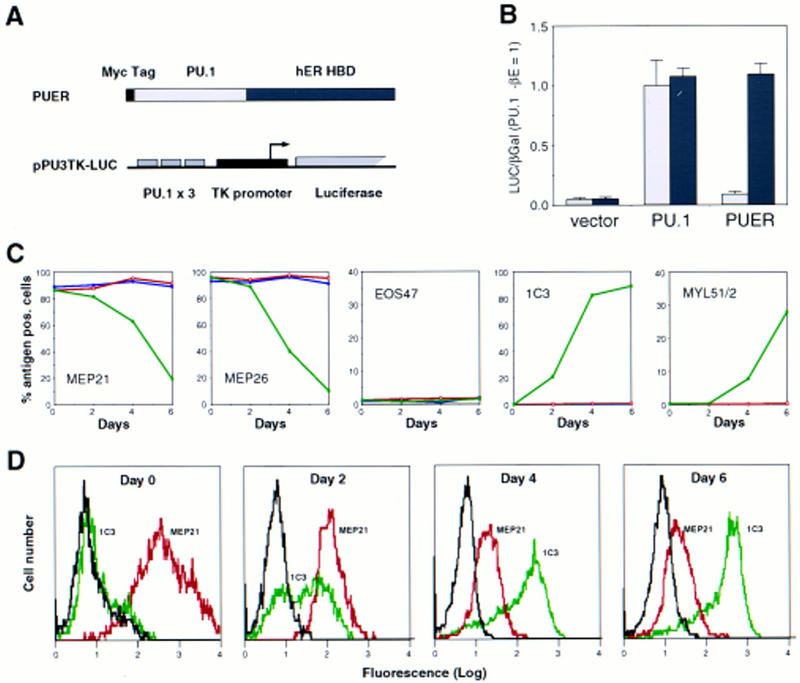
Induction of myeloid differentiation by an inducible PU.1 allele. (A) Structure of the inducible PU.1 allele and of the PU.1 reporter construct. The hormone-binding domain (HBD) of the human estrogen receptor (hER) was fused to the carboxyl terminus of the PU.1 protein, generating PUER. The reporter pPU3TK–LUC contains three PU.1-binding sites upstream of the HSV thymidine kinase minimal promoter (B) Cotransfection of PU.1/PUER with the reporter pPU3TK–LUC and pRSV–βGal control plasmid in the presence (solid bar) or absence (shaded bar) of βE. The luciferase/β-galactosidase ratios were expressed relative to those obtained with the PU.1 expression vector in the absence of βE. The average values of four determinations are shown; (error bars) standard deviations. (C) Effect of βE on the phenotype of MEPs transformed by E26–PUER. MEP clones transformed by E26–PUER were treated with βE (green graphs) or left untreated (red graphs), and the expression of MEP (MEP21, MEP26), eosinophil (EOS47), and myeloid (1C3, MYL51/2) specific antigens determined at the indicated timepoints. MHC II surface antigen was induced more slowly and only in a subset of the clones (data not shown). βE treatment had no detectable effect on E26–WT MEPs (blue graphs). No decrease in cell viability caused by PUER activation was observed. (D) FACS profiles of βE-treated E26–PUER–MEPs. MEP21 antigen (red graphs) and 1C3 antigen (green graphs) expression was determined at 2, 4, and 6 days after addition of βE. (c-Src) Control antibody (black graphs). Data are displayed as cell number (linear scale) plotted against fluorescence intensity (logarithmic scale).
Although the observed changes in antigen expression are consistent with an instructive effect of the factor, they could also be the result of PUER-mediated growth arrest or death of E26–PUER MEPs followed by the outgrowth of a previously undetected subpopulation of myeloid cells. If this was the case, one would expect the sudden emergence of a subpopulation of cells with surface markers characteristic of myeloblasts (e.g., MEP21−/1C3+). If, on the other hand, cells in the population are reprogrammed by PU.1, one would expect a gradual decrease in MEP antigen expression in the whole population, together with a gradual increase in the intensity of expression of myeloid markers. Therefore, we reanalyzed our FACS data (Fig. 2D) and found that treatment with βE induced a gradual change in the entire MEP population, with cells exhibiting intermediate levels of both MEP21 and 1C3 antigens at day 2 and cells expressing high levels of 1C3 antigen and low levels of MEP21 at day 6. There was also a significant decrease in MEP21 fluorescence intensity already after a 2-day βE pulse, which is not apparent from the results plotted in Figure 2C, in which all cells above a certain threshold scored as positive. We conclude from these results that selection is not likely to play a role in the phenotypic changes observed after PUER activation in MEP cells and that these changes are the result of an instructive role of the factor.
PU.1 activation in MEPs results in down-regulation of GATA-1
Our previous results showed that expression of GATA-1 is incompatible with the myeloid gene expression program, as forced expression in myeloid cell lines led to their conversion into either eosinophils or MEPs (Kulessa et al. 1995). Therefore, we analyzed the effect of PUER activation on GATA-1 protein expression in MEPs. As shown by the Western blot in Figure 3A, treatment with βE of E26–PUER MEPs for an increasing number of days led to a decrease of GATA-1 expression, with a 6-day treatment resulting in complete GATA-1 down-regulation. To determine whether the disappearance of GATA-1 protein was caused by the down-regulation of GATA-1 mRNA levels, four E26–PUER MEP clones were pooled and induced with βE. Total RNA was isolated and GATA-1 mRNA levels analyzed after 2 and 4 days of induction (Fig. 3B). This analysis showed a significant down-regulation of GATA-1 mRNA on PUER activation compared with uninduced control cells. As a control, GAPDH mRNA was not down-regulated (but rather up-regulated) during this time course (cf. with 28S rRNA for RNA loading).
Figure 3.


Effect of PUER activation on GATA-1 protein and mRNA expression. (A) E26–PUER transformed MEPs (from a single clone) were treated with βE, and aliquots subjected to Western analysis at different time points with anti-GATA-1 antibody. (B) Pooled MEP clones (four clones) were treated with βE and RNA isolated after 2 and 4 days of induction, as well as from uninduced cells. Sequential Northern blot analysis was performed with chicken GATA-1 and GAPDH cDNAs. The 28S rRNA is shown as a control for total RNA loading.
PU.1 induces irreversible myeloid lineage commitment in MEPs and reveals the formation of EOS47 antigen-positive cells
To correlate the observed PU-1-induced down-regulation of GATA-1 expression with the ability of PU.1 to induce myeloid commitment of MEPs, we determined the time required for irreversible commitment to myeloid differentiation. For this purpose, E26–PUER-transformed MEPs were exposed to βE pulses of 1–4 days, the inducer was removed by thorough washing, and the cells were tested for marker expression on day 8 (Fig. 4A). The data in Figure 4, B–D, show that following a 1-day βE pulse, the majority of the cells down-regulated their MEP markers and ∼10% expressed myeloid markers instead. Interestingly, another subpopulation of the cells (>50%) were positive for EOS47 antigen, a marker that is exclusively expressed on immature and mature eosinophils within the chick bone marrow (McNagny et al. 1992, 1996). After a 3-day pulse, essentially all E26–PUER–MEPs were committed to myeloid differentiation, expressing three different myeloid markers, but no MEP or eosinophil markers, and had the morphology of myeloblasts, in that they exhibited a large, faintly stained cytoplasm and excentric nuclei (Fig. 5A). Furthermore, in contrast to the parental MEP cells, their growth was dependent on chicken myelomonocytic growth factor (Fig. 5B), a hallmark of normal as well as E26-transformed myeloid cells (Leutz et al. 1984). Finally, they had completely lost GATA-1 expression and acquired C/EBPβ protein instead (Fig. 5C).
Figure 4.
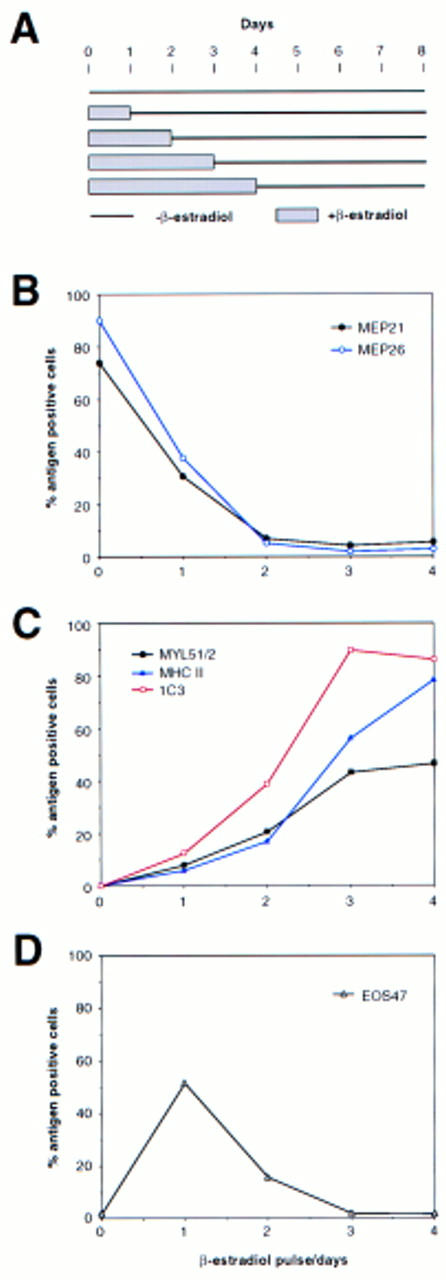
Induction of myeloid differentiation by transient PUER activation. (A) Experimental protocol. E26–PUER MEPs were subjected to a βE pulse of 1–4 days, washed extensively, incubated for a total of 8 days, and assayed for the lineage-specific markers described below. (B) Expression of MEP-specific [(•, black) MEP21; (○, blue) MEP26], (C) myeloid-specific [(▪, black) MYL51/2; (▵, blue) MHC 11; ( , red) 1C3], and (D) eosinophil-specific (EOS47) antigens. Results are representative of five independent clones. E26–WT MEPs showed no change in antigen expression (data not shown).
Figure 5.
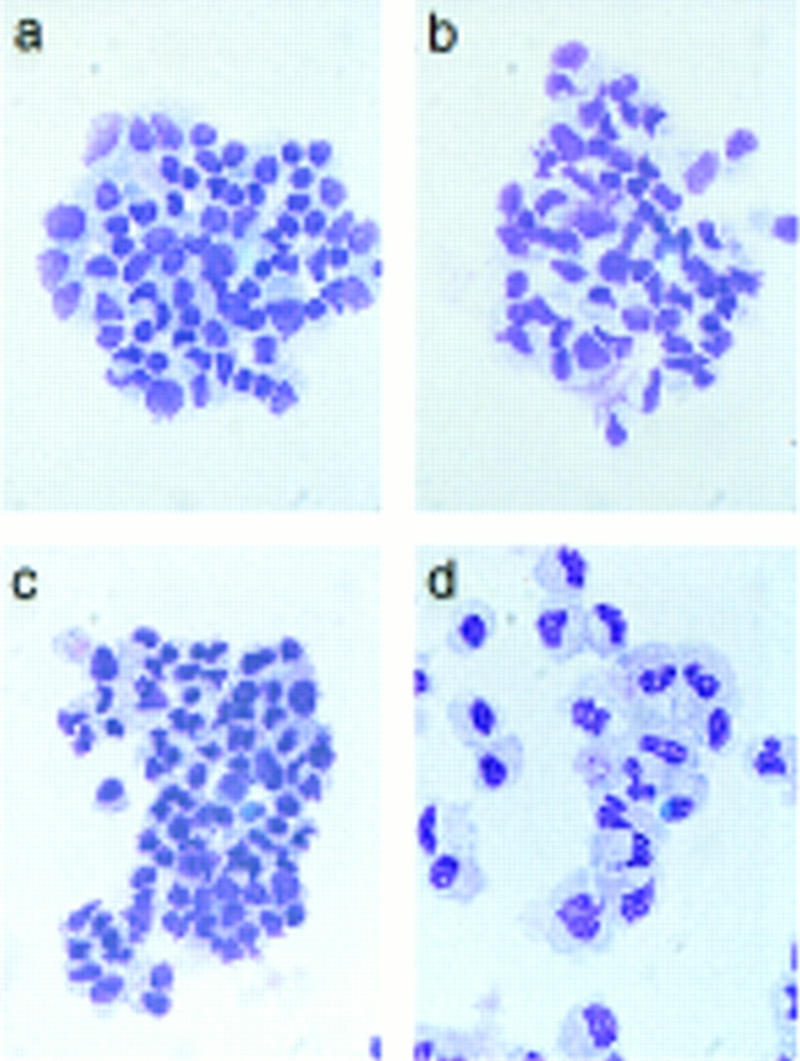


Phenotype of E26–PUER-transformed MEPs pulse treated for 3 days with βE. (A) May–Gruenwald–Giemsa staining of E26–WT cells untreated (a) or subjected to a 3-day βE pulse (b), and E26–PUER MEPs untreated (c), or βE treated (d). (B) cMGF dependence of E26–PUER cells βE untreated or treated as indicated. Cells were incubated under low serum conditions with or without recombinant cMGF (rcMGF) for 2 days and the number of cells in S phase determined by short-term [3H]thymidine incorporation. Average values of triplicate determinations are shown; (error bars) standard deviations. (C) E26–WT and E26–PUER MEPs untreated or pulsed with βE for 4 days were expanded in liquid culture for 10 days and subjected to Western blot analyses with anti-chicken GATA-1 (top) and anti-chicken C/EBPβ antisera (bottom).
The formation of MEP21-negative, EOS47-positive cells following a 1-day treatment of E26–PUER cells with βE raised the question of whether they expressed other eosinophil markers such as peroxidase (which is restricted to mature avian eosinophils; Brune and Spitznagel 1973). Therefore, we compared the hormone-induced cells with eosinophils obtained from MEPs after activation of a C/EBPβ–ER chimera (Nerlov et al. 1998). As can be seen from the FACS profiles of Figure 6A, the βE-induced E26–PUER cells expressed slightly lower levels of EOS47 antigen than the βE-induced control C/EBPβ–ER cells. In addition, they exhibited no granules and, unlike the C/EBPβ–ER cells, were peroxidase negative (Fig. 6B). These results therefore show that a short-term activation of PUER in MEPs induces the formation of cells with properties of immature eosinophils.
Figure 6.
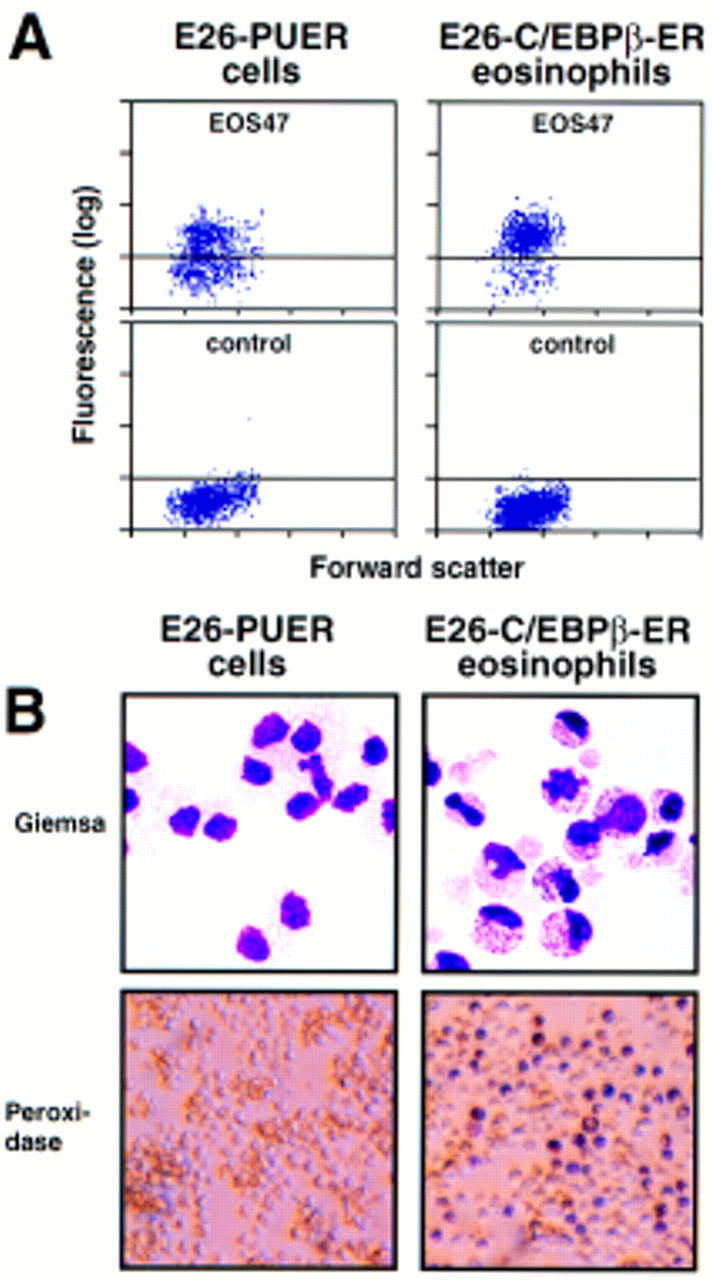
Phenotype of E26–PUER-transformed MEPs pulse treated for 1 day with β-estradiol. (A) EOS47 antigen expression of 1-day βE pulse-treated E26–PUER MEPs (left) and 3-day pulse-treated control E26–C/EBPβ–ER MEPs (right). The fluorescence intensities of cells stained with EOS47 (top) and anti-c-Src (control) antibodies (bottom) are shown. Fluorescence intensity (y-axis; logarithmic scale) is plotted against forward scatter (x-axis; linear scale). (B) May–Gruenwald–Giemsa (top) and peroxidase staining (bottom) of the above cells.
The PU.1 transactivation domain is required for induction of myeloid differentiation
To study the role of the transactivation domain of PU.1, we constructed a series of amino-terminal deletion mutants. The transactivation domain is located at the amino terminus of the protein, the middle contains a proline-, glutamic acid-, serine-, and threonine-rich (PEST) domain, whereas the DNA-binding domain is located at the carboxyl terminus. (Fig. 7A; Klemsz and Maki 1996). The various mutants were cotransfected with the pPU3TK–LUC reporter construct (Fig. 2A) into Q2bn fibroblasts to test their transcription activation potential. As shown in Figure 7B, deleting the first 23 amino acids of PU.1 had little effect on transactivation, deleting 56 or 80 amino acids severely reduced activity, whereas deleting 100 amino acids led to an almost complete loss of activity. We then fused various parts of the PU.1 protein to the GAL4 DNA-binding domain and tested the activity of the chimeric proteins on a GAL4 site-containing reporter construct (Fig. 7C). This showed strong activation by PU.1 amino acids 1–55, some activity of amino acids 56–99, and no activity of amino acids 100–170 (Fig. 7D), consistent with the above results, as well as previous reports (Hagemeier et al. 1993; Klemsz and Maki 1996). The two activating regions synergized (G4-PU 1–99), whereas the PEST domain-containing region, although inactive by itself, had a negative effect (cf. G4 PU 1–99 with G4 PU 1–170; G4 PU 56–99 with G4 PU 56–170). We conclude that the major transactivation domain of PU.1 resides in the amino-terminal 55 amino acids and a minor domain between amino acids 56 and 99.
Figure 7.




Requirement of the PU.1 transactivation domain for induction of myeloid differentiation. (A) Maps of full-length PU.1 and amino-terminal deletion mutants (the number indicates the first amino acid still present). The positions of structural and functional domains (Klemsz and Maki 1996) are indicated above. (B) Transactivation potential of PU.1 alleles (see Fig. 2B legend). The activity of PU.1 WT was arbitrarily assigned a value of 1. The averages of three determinations are shown; (error bars) standard deviation. (C) Maps of PU.1 constructs fused to the GAL4 DNA-binding domain. (D) Transactivation potential of the GAL4–PU.1 fusions on the pG5B–Luc reporter. The normalized luciferase activity is expressed as fold activation above the value obtained with control vector (pcDNAI).
Next, we inserted the PU.1 amino-terminal deletion mutants into the E26 virus and tested the resulting recombinant viruses for their transforming specificities. As shown in Figure 8A, the induced decrease in MEP-type colonies relative to myeloid colonies was strictly dependent on the transactivation domain. Thus, strong PU.1 alleles (PU.1 WT, PUDN23) induced a virtually complete MEP to myeloid cell conversion, whereas transactivation-deficient PU.1 alleles (PUDN100, PUDN123, PUDN143) were ineffective. The latter even led to a partial suppression of myeloid differentiation, suggesting a dominant-negative effect, most likely caused by competition with endogenous PU.1 for PU.1 DNA-binding sites. That these effects were not a reflection of different protein expression levels was shown by Western blots of extracts from cells transformed by the various viruses with a monoclonal antibody against the Myc epitope used to tag the PU.1 proteins (Fig. 8B). Moreover, the effect on differentiation was highly sensitive to PU.1 activity, as seen by the close correlation between the ratio of myeloid cells to MEPs (plotted on a logarithmic scale) and the transactivation strength (plotted on a linear scale) of the PU.1 alleles tested (Fig. 8C). Therefore, we conclude that the ability of PU.1 to induce myeloid differentiation in E26-transformed multipotent progenitors depends on the integrity of its transactivation domain.
Figure 8.
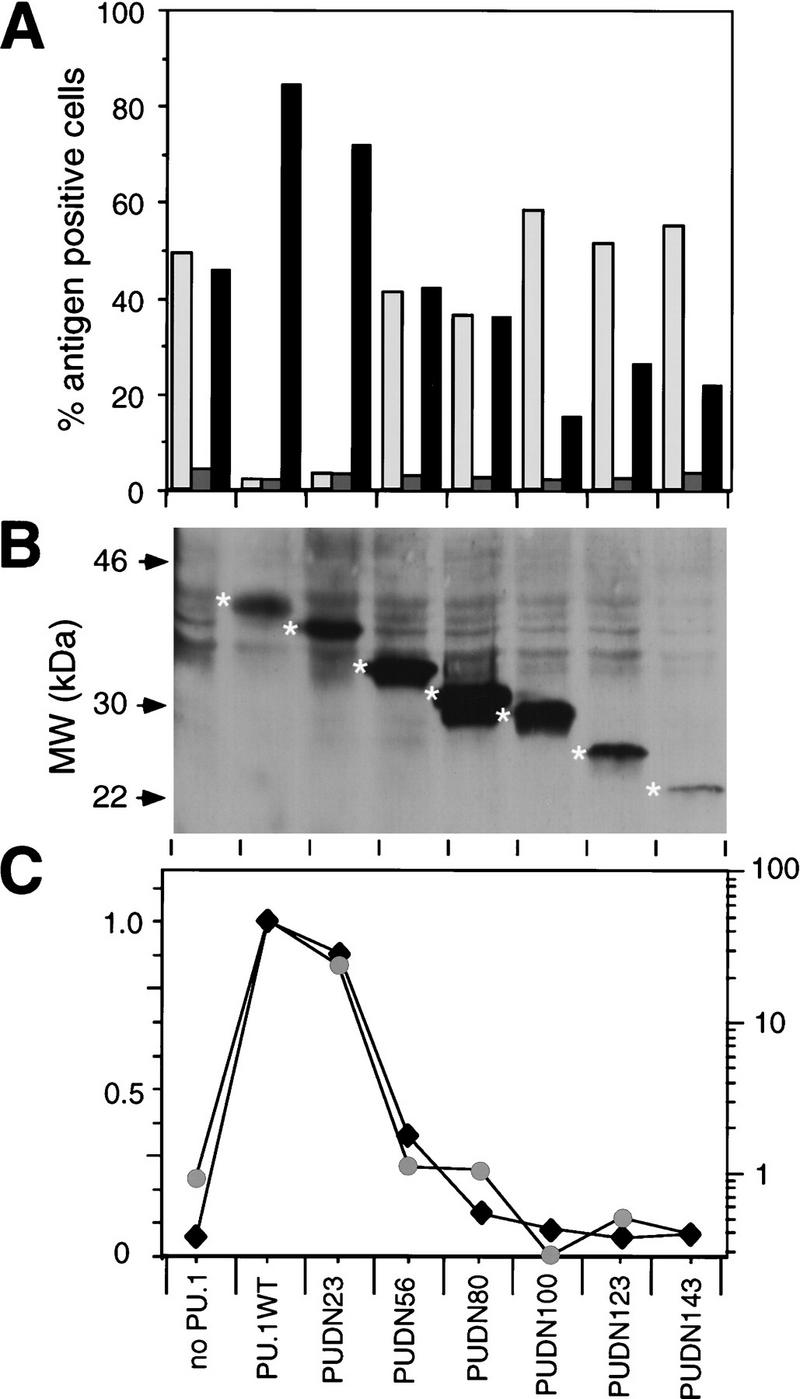
(A) Biological activity of PU.1 transactivation domain deletions. E26 derivatives containing PU.1 amino-terminal deletion mutants were constructed, and viruses were produced and used to infect blastoderm cultures, along with E26–WT control virus. Proportions of MEPs, eosinophils and myeloid cells were determined as in Fig. 1. (Light-shaded box) MEP21; (dark-shaded box) EOS47; (solid box) MYL51/2. Average values of three independent transformation assays are shown. (B) Equal amounts of protein from pooled colonies transformed by E26 viruses encoding the indicated PU.1 alleles were subjected to Western blotting with the 9E10 monoclonal antibody (directed against the amino-terminal epitope tag of the PU.1 proteins). The positions of molecular weight markers are shown at left; specific bands representing PU.1 proteins are indicated with asterisks. (C) Correlation between the ratio of MYL51/2-positive and MEP21-positive cells transformed by E26–PUDN viruses and the transactivation potential of the corresponding PUDN constructs. (Black diamonds) Luciferase activity; (shaded circles) MYL/MEP.
Discussion
PU.1-induced myeloid lineage commitment in multipotent hematopoietic precursors
Here we have demonstrated that PU.1 is able to induce myeloid lineage commitment in primary cultures of MEPs transformed by the E26 virus in a manner strictly dependent on the PU.1 transactivation domain. The cells obtained expressed cell-surface antigens specific for the myeloid lineage and required cMGF for their growth, a characteristic of both normal and E26-transformed myeloid cells (Leutz et al. 1984). Furthermore, they expressed C/EBPβ, a myeloid/eosinophil-specific transcription factor, but not GATA-1. Our results, therefore, provide a first example of a transcription factor capable of selectively instructing multipotent progenitors to differentiate along the myeloid lineage. Thus, they suggest a role for PU.1 in the earliest steps of myelopoiesis, and may explain the impairment of myeloid development in PU.1 mutant mice (Scott et al. 1994, 1997; McKercher et al. 1996). That the changes induced by PU.1 are the result of its capacity to reprogram multipotent progenitors rather than to simply induce ectopic gene expression, is indicated by the finding that subjecting E26–PUER-transformed MEPs to a 3-day pulse of βE is sufficient to induce the entire progenitor population to differentiate into myeloblasts. Once committed, the cells are probably locked in the myeloid compartment because of the ability of PU.1 to regulate its own expression (Chen et al. 1995). The activation in MEPs of the Ras or PKC pathway likewise leads to the differentiation of myeloid cells (Graf et al. 1992). This suggests that induction of PU.1 expression is downstream of a signaling pathway that emanates from an as yet unknown extracellular stimulus. However, because the biological effects described here occurred in the absence of such stimuli, it is not likely that PU.1 protein needs to be additionally modified by the signal transduction machinery, at least in the context studied.
It cannot be ruled out that the presence of Ets domains in both PU.1 and the Gag–Myb–Ets oncoprotein leads to functional interference between the two proteins. However, the observation that the PU.1 Ets domain by itself does not promote (but rather inhibits) myeloid differentiation argues against this possibility. To settle this point, it will be necessary to test the ability of PU.1 to direct the lineage choice of untransformed cells, a question we are currently addressing using ES cell in vitro differentiation.
PU.1-mediated suppression of GATA factors in myeloid lineage commitment
We have demonstrated previously that maintenance of the myeloid phenotype requires the absence of GATA-1, because intermediate levels of ectopic GATA-1 expression in myeloid cells resulted in a reprogramming into eosinophils, whereas higher levels of GATA-1 led to cells expressing MEP/thrombocyte markers (Kulessa et al. 1995). During both processes, PU.1 expression was extinguished (McNagny et al. 1998). Conversely, several lines of evidence suggest that the mechanism by which PU.1 induces myeloid (and eosinophil) commitment in MEP cells involves the down-regulation of GATA-1. Thus, the level of GATA-1 protein expression observed at different times after PUER activation correlated with the percentage of cells that became irreversibly committed after hormone withdrawal, with predominantly eosinophil commitment obtained after an approximately two- to threefold down-regulation of GATA-1 and myeloid commitment after further reduction. Consistent with this observation, the difference in ectopic GATA-1 levels required for the generation of either eosinophils or MEPs from myeloblasts was only two- to threefold (Kulessa et al. 1995). Our results thus support the notion that the capacity of PU.1 to reprogram MEP cells can, at least in part, be explained by its ability to down-regulate GATA-1 expression, most likely at the level of transcription, and suggest that GATA-1 and PU.1 have antagonistic roles in hematopoietic lineage decisions at the branch point between erythroid/megakaryocytic and myeloid precursors. This conclusion is in accordance with observations made with chimeras between PU.1 null and wild-type mice, which showed a complete lack of mutant-derived myeloid/B cells, whereas the erythroid/megakaryocytic lineages were unimpaired (Scott et al. 1997).
The observations that sustained activation of PUER in E26–PUER cells induces myeloid differentiation, whereas a 1-day pulse induces the formation of immature eosinophils, indicate that myeloid differentiation proceeds via a transient myeloid/eosinophil precursor. A possible explanation is that an incomplete down-regulation of GATA-1 induces the expression of endogenous C/EBP to levels that are sufficient to drive the cells into the eosinophil lineage but that are insufficient to induce their maturation. Evidence for this type of mechanism is provided in Nerlov et al. (1998).
The proposed role of GATA factors in the maintenance of multipotent progenitors is supported by the finding that a GATA-1 promoter construct driving a temperature-sensitive SV40 large T antigen (LTAg) can transform murine cells with myeloid/erythroid potential (Cairns et al. 1994). Consistent with our observations, these cells down-regulate GATA-1 (and LTAg) expression when induced to differentiate along the myeloid lineage. It should be noted, however, that other GATA family members (such as GATA-2) are also expressed in SV40 LTAg-transformed progenitors, enriched human pluripotent stem cells, and MEP cells, but not in myeloblasts (Cairns et al. 1994; Orlic et al. 1995; C. Nerlov and T. Graf, unpubl.). Whether these are also down-regulated by PU.1 remains to be investigated.
It is interesting that ectopic activation of PU.1 (through retroviral insertion) in GATA-1 expressing murine erythroleukemia (MEL) cells results in an induction of proliferation and arrest of differentiation, whereas GATA-1 remains expressed (Baron and Farrington 1994). When these cells are induced to mature into erythrocytes, PU.1 is down-regulated, and this is a prerequisite for differentiation to occur (Rao et al. 1997). The peaceful coexistence of GATA -1 and PU.1 in MEL cells could be caused by the existence of different regulatory elements that control GATA-1 expression in multipotent progenitors and in early erythroid cells. A similar mechanism could be operative in mast cells in which PU.1 and GATA-1 are also known to coexist (Henkel and Brown 1994).
Specificity of the PU.1-induced myeloid differentiation
The specificity of the myeloid-instructive capacity of PU.1 is illustrated by the fact that other lineage-restricted transcription factors do not show this effect. Thus, C/EBPα, which is normally expressed in myeloid cells and in eosinophils, induces MEPs to differentiate exclusively into eosinophils. C/EBPβ, although predominantly inducing eosinophil differentiation, can induce myeloid differentiation in addition (Nerlov et al. 1998). Furthermore, MafB, whose expression is restricted to myeloid cells of the hematopoietic system (Sieweke et al. 1996), does not induce myeloid differentiation of MEPs (M. Sieweke and T. Graf, unpubl.). We were unable to observe the formation of either B or T cells following the expression of PU.1 in MEP cells, in spite of the known requirement of PU.1 for the development of these two cell types in vivo (Scott et al. 1994, 1997; McKercher et al. 1996). This may reflect the fact that the differentiation potential of MEPs is restricted to nonlymphoid cell types.
Materials and methods
Cell culture and FACS analysis
Infection of 2-day chicken blastoderm by cocultivation with Q2bn packaging cells transfected with E26 proviral DNA has been described (Rossi et al. 1996b). For analysis of bulk cultures, cells were plated in methylcellulose and phenotyped by indirect immunofluorescence (IIF) on a Becton–Dickinson FACScan 2 weeks after infection (Graf et al. 1992). The MEP21, MEP26, EOS47, MYL51/2, 1C3, and c1a (anti-MHC II) monoclonal antibodies have been described (Kornfeld et al. 1983; Ewert et al. 1984; Mandi et al. 1987; Graf et al. 1992; McNagny et al. 1992). For analysis of individual MEP clones, colonies were picked and expanded, analyzed by IIF, and clones selected that were positive for MEP21 and MEP26 antigen expression and negative for lineage-specific markers. For induction, cells were cultured in the presence of 0.5 μm βE. In pulse-chase experiments, cells were washed three times in medium after the indicated period of βE treatment and replated in the absence of inducer. Phenotypes were analyzed by IIF as above. [3H]thymidine incorporation assays for growth factor dependence were performed as described (Rossi et al. 1996b), except that recombinant, rather than crude, cMGF was used (Leutz et al. 1989). Peroxidase activity was detected as described (Graf et al. 1992), and morphological analyses were performed on cytospins fixed with methanol and May–Gruenwald–Giemsa stained (Diff-Quik, Harleco).
Transfection and Western blot analysis
Transient transfection into Q2bn fibroblasts was done with 0.5 μg of PU.1 expression vectors, 1 μg of the pPU3–TKLUC reporter (obtained from Dr. N. Kraut, Fred Hutchinson Cancer Research Center, Seattle, WA), or 1 μg of pG5B–Luc reporter, and 0.25 μg of pRSV–β-Gal internal control plasmid as described (Frampton et al. 1996). Cell extracts were made and Western blotting performed as described (Frampton et al. 1996), with the anti-Myc 9E10 monoclonal antibody (Evan et al. 1985), anti-GATA-1 antiserum (Kulessa et al. 1995; generously provided by Dr. G. Goodwin, The Institute of Cancer Research, Sutton, Surrey, UK), and anti-C/EBPβ antiserum (Katz et al. 1993; kindly provided by Dr. A. Leutz).
RNA extraction and Northern blotting
Total cellular RNA was prepared according to Chomczynski and Sacchi (1987). After electrophoresis through a 1.2% formaldehyde–agarose gel, RNA was transferred to Duralose (Stratagene) by capillary blotting and sequentially probed with chicken GATA-1 (Yamamoto et al. 1990) and GAPDH cDNA (Dugaiczyk 1983) labeled by random priming.
DNA constructs
Human PU.1 cDNA (Ray et al. 1990) and PCR-generated deletion constructs were amino-terminally tagged with a Myc epitope and cloned into pcDNAI (Invitrogen) for transient transfection analysis, and the E26 proviral vector pMI3–IRES (behind an EMCV IRES element (Rossi et al. 1996b) for MEP infection. The human estrogen receptor hormone-binding domain (amino acids 282–595), derived from pMV–7MER (Eilers et al. 1989), was fused to the carboxyl terminus of PU.1, generating PUER. This was cloned into pcDNAI and pMI3–IRES as above, generating pCMV–PUER and pMI3–IR–PUER (which contains the E26–PUER provirus). The pG5B–LUC reporter was constructed by cloning a SacI–XhoI fragment of pG5B–CAT (containing five GAL4 binding sites upstream of the E1b minimal promoter; Lillie and Green 1989) into the same sites in pGL3-basic (Promega), the E1b promoter transcribing toward the luciferase gene. GAL4 fusions were constructed by cloning a HindIII–EcoRI fragment from pGBT9 (Clontech) into the same sites in pcDNAI, generating pCMV-GD. Fragments of the PU.1-coding sequence were flanked by EcoRI and XhoI by PCR and cloned into the same sites in pCMV–GD.
Acknowledgments
We thank Dr. J. Ghysdael for PU.1 cDNA, Drs. P. Orban, G. Goodwin, and A. Leutz for antibodies, members of the Graf laboratory for helpful discussions, Dr. K. McNagny for critically reading the report, S. Leillard for secretarial assistance, and G. Döderlein for excellent technical assistance. C.N. is a fellow of the Danish Medical Research Council. This work was partially funded by the Deutsche Forschungs Gemeinschaft, SFB 229.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL graf@embl-heidelberg.de; FAX 49 6221 387 516.
References
- Baron MH, Farrington SM. Positive regulators of the lineage-specific transcription factor GATA-1 in differentiating erythroid cells. Mol Cell Biol. 1994;14:3108–3114. doi: 10.1128/mcb.14.5.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune K, Spitznagel JK. Peroxidaseless chicken leukocytes: Isolation and characterization of antibacterial granules. J Infect Dis. 1973;127:84–94. doi: 10.1093/infdis/127.1.84. [DOI] [PubMed] [Google Scholar]
- Cairns LA, Crotta S, Minuzzo M, Moroni E, Granucci F, Nicolis S, Schiro R, Pozzi L, Giglioni B, Riccardi-Castagnoli P, Ottolenghi S. Immortalization of multipotent growth-factor dependent hematopoietic progenitors from mice transgenic for GATA-1 driven SV40 tsA58 gene. EMBO J. 1994;13:4577–4586. doi: 10.1002/j.1460-2075.1994.tb06779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-M, Ray-Gallet D, Zhang P, Hetherington CJ, Gonzalez DA, Zhang D-E, Moreau-Gachelin F, Tenen DG. PU.1 (Spi-1) autoregulates its expression in myeloid cells. Oncogene. 1995;11:1549–1560. [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single step isolation of RNA by acid guanidinium isothiocyanate method. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Dugaiczyk A. Cloning and sequencing of a deoxyribonucleic acid copy of glyceraldehyde-3-phosphate dehydrogenase messenger ribonucleic acid isolated from chicken muscle. Biochemistry. 1983;22:1605–1613. doi: 10.1021/bi00276a013. [DOI] [PubMed] [Google Scholar]
- Eilers M, Picard D, Yamamoto KR, Bishop JM. Chimeras of Myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature. 1989;340:66–68. doi: 10.1038/340066a0. [DOI] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for the human c-myc oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewert DL, Munchus MS, Chen CL, Cooper MD. Analysis of structural properties and cellular distribution of avian Ia antigen by using monoclonal antibody to monomorphic determinants. J Immunol. 1984;132:2524–2530. [PubMed] [Google Scholar]
- Frampton J, McNagny KM, Sieweke M, Philip A, Smith G, Graf T. v-Myb DNA binding is required to block thrombocytic differentiation of Myb-Ets-transformed multipotent hematopoietic progenitors. EMBO J. 1995;14:2866–2875. doi: 10.1002/j.1460-2075.1995.tb07286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton J, Ramqvist T, Graf T. v-Myb of E26 leukemia virus up-regulates bcl-2and suppresses apoptosis in myeloid cells. Genes & Dev. 1996;10:2720–2731. doi: 10.1101/gad.10.21.2720. [DOI] [PubMed] [Google Scholar]
- Graf T, McNagny KM, Brady G, Frampton J. Chicken “erythroid” cells transformed by the Gag-Myb-Ets encoding E26 leukemia virus are multipotent. Cell. 1992;70:201–213. doi: 10.1016/0092-8674(92)90096-u. [DOI] [PubMed] [Google Scholar]
- Hagemeier C, Cook A, Kouzarides T. The retinoblastoma protein binds E2F residues required for activation in vivo and TBP binding in vitro. Nucleic Acids Res. 1993;21:4998–5004. doi: 10.1093/nar/21.22.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel G, Brown MA. PU.1 and GATA: Components of a mast cell-specific interleukin 4 intronic enhancer. Proc Natl Acad Sci. 1994;91:7737–7741. doi: 10.1073/pnas.91.16.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz S, Kowenz-Leutz E, Müller C, Meese K, Ness SA, Leutz A. The NF-M transcription factor is related to C/EBPβ and plays a role in signal transduction, differentiation and leukemogenesis of avian myelomonocytic cells. EMBO J. 1993;12:1321–1332. doi: 10.1002/j.1460-2075.1993.tb05777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemsz MJ, Maki RA. Activation of transcription by PU.1 requires both acidic and glutamine residues. Mol Cell Biol. 1996;16:390–397. doi: 10.1128/mcb.16.1.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemsz MJ, McKersher SR, Celada A, Van Beveren C, Maki RA. The macrophage and B cell-specific transcription factor PU.1 is related to the etsoncogene. Cell. 1990;61:113–124. doi: 10.1016/0092-8674(90)90219-5. [DOI] [PubMed] [Google Scholar]
- Kornfeld S, Beug H, Doederlein G, Graf T. Detection of avian hematopoietic cell surface antigens with monoclonal antibodies to myeloid cells. Exp Cell Res. 1983;143:383–394. doi: 10.1016/0014-4827(83)90065-4. [DOI] [PubMed] [Google Scholar]
- Kraut N, Frampton J, McNagny KM, Graf T. A functional Ets DNA-binding domain is required to maintain multipotency of hematopoietic progenitors transformed by Myb–Ets. Genes & Dev. 1994;8:33–44. doi: 10.1101/gad.8.1.33. [DOI] [PubMed] [Google Scholar]
- Kulessa H, Frampton J, Graf T. GATA-1 reprograms avian myelomonocytic cell lines into eosinophils, thromboblasts, and erythroblasts. Genes & Dev. 1995;9:1250–1262. doi: 10.1101/gad.9.10.1250. [DOI] [PubMed] [Google Scholar]
- Leutz A, Beug H, Graf T. Purification and characterization of cMGF, a novel chicken myelomonocytic growth factor. EMBO J. 1984;3:3191–3197. doi: 10.1002/j.1460-2075.1984.tb02278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutz A, Damm K, Sterneck E, Kowenz E, Ness S, Frank R, Gausepohl H, Pan Y-CE, Smart J, Hayman M, Graf T. Molecular cloning of the chicken myelomonocytic growth factor (cMGF) reveals relationship to interleukin 6 and granulocyte colony stimulating factor. EMBO J. 1989;8:175–181. doi: 10.1002/j.1460-2075.1989.tb03362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie JW, Green MR. Transcription activation by the adenovirus E1a protein. Nature. 1989;338:39–44. doi: 10.1038/338039a0. [DOI] [PubMed] [Google Scholar]
- Mandi Y, Pusztai R, Baranji K, Seprenyi G, Tarodi B, Bakay M, Beladi I. The role of interferon in the adenovirus-induced augmentation of granulocyte-mediated cytotoxicity in chicken. Immunobiology. 1987;174:210–220. doi: 10.1016/S0171-2985(87)80040-2. [DOI] [PubMed] [Google Scholar]
- McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, Maki RA. Targeted disruption of the PU.1gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15:5647–5658. [PMC free article] [PubMed] [Google Scholar]
- McNagny KM, Lim F, Grieser S, Graf T. Cell surface proteins of chicken hematopoietic progenitors, thrombocytes and eosinophils detected by novel monoclonal antibodies. Leukemia. 1992;6:975–984. [PubMed] [Google Scholar]
- McNagny KM, Rossi F, Smith G, Graf T. The eosinophil-specific cell surface antegen, EOS47, is a chicken homologue of the oncofetal antigen melanotransferrin. Blood. 1996;87:1343–1352. [PubMed] [Google Scholar]
- McNagny KM, Pettersson I, Rossi F, Flamme I, Shevchenko A, Mann M, Graf T. Thrombomucin, a novel cell surface antigen that defines thrombocytes and multipotent hematopoietic progenitors. J Cell Biol. 1997;138:1395–1407. doi: 10.1083/jcb.138.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNagny KM, Sieweke M, Döderlein G, Graf T, Nerlov C. Regulation of eosinophil-specific gene expression by a c/EBP-Ets complex and GATA-1. EMBO J. 1998;17:3669–3680. doi: 10.1093/emboj/17.13.3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlov, C., K.M. McNagny, G. Döderlein, E. Kowenz-Leutz, and T. Graf. 1998. Distinct C/EBP functions are required for eosinophil lineage commitment and maturation. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Ness SA, Engel JD. Vintage reds and whites: Combinatorial transcription factor utilization in hematopoietic differentiation. Curr Opin Genet Dev. 1995;4:718–724. doi: 10.1016/0959-437x(94)90139-t. [DOI] [PubMed] [Google Scholar]
- Orlic D, Anderson S, Biesecker LG, Sorrentino BP, Bodine DM. Pluripotent hematopoietic stem cells contain high levels of mRNA for c-kit, GATA-2, p45 NF-E2, and c-myb and low levels or no mRNA for c-fms and the receptors for granulocyte colony-stimulating factor and interleukins 5 and 7. Proc Natl Acad Sci. 1995;92:4601–4605. doi: 10.1073/pnas.92.10.4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao G, Rekthman N, Cheng G, Krasikov T, Skoultchi AI. Deregulated expression of the PU.1 transcription factor blocks murine erythroleukemia cell terminal differentiation. Oncogene. 1997;14:123–131. doi: 10.1038/sj.onc.1200807. [DOI] [PubMed] [Google Scholar]
- Ray D, Culine S, Tavitian A, Moreau-Gachelin F. The human homologue of the putative proto-oncogene Spi-1: Characterization and expression in tumors. Oncogene. 1990;5:663–668. [PubMed] [Google Scholar]
- Rossi F, McNagny KM, Logie C, Stewart AF, Graf T. Excision of Ets by an inducible site-specific recombinase causes differentiation of differentiation of Myb-Ets-transformed hematopoietic progenitors. Curr Biol. 1996a;6:866–872. doi: 10.1016/s0960-9822(02)00610-3. [DOI] [PubMed] [Google Scholar]
- Rossi F, McNagny KM, Smith G, Frampton J, Graf T. Lineage commitment of transformed haematopoietic progenitors is determined by the level of PKC activity. EMBO J. 1996b;15:1984–1901. [PMC free article] [PubMed] [Google Scholar]
- Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- Scott EW, Fisher RC, Olson MC, Kehrli EW, Simon MC, Singh H. PU.1 functions in a cell-autonomous manner to control the differentiation of multipotential lymphoid-myeloid progenitors. Immunity. 1997;6:437–447. doi: 10.1016/s1074-7613(00)80287-3. [DOI] [PubMed] [Google Scholar]
- Sieweke MH, Tekotte H, Frampton J, Graf T. MafB is an interaction partner and repressor of Ets-1 that inhibits erythroid differentiation. Cell. 1996;85:49–60. doi: 10.1016/s0092-8674(00)81081-8. [DOI] [PubMed] [Google Scholar]
- Tenen DG, Hromas R, Licht JD, Zhang D-E. Transcription factors, normal myeloid development, and leukemia. Blood. 1997;90:489–519. [PubMed] [Google Scholar]
- Yamamoto M, Ko LJ, Leonard MW, Beug H, Orkin SH, Engel JD. Activity and tissue-specific expression of the transcription factor NF-E1 multigene family. Genes & Dev. 1990;4:1650–1662. doi: 10.1101/gad.4.10.1650. [DOI] [PubMed] [Google Scholar]