

Abstract
Background
Expression of the inducible nitric oxide synthase (iNOS) is commonly induced in inflammation, an important risk factor of cancer. Nitric oxide (NO) and related reactive nitrogen species can directly cause DNA damage to increase DNA mutation. They can also indirectly affect DNA mutation by modulation of DNA repair proteins, in particular through protein S-nitrosylation, a key regulatory mechanism of NO.
Scope of Review
Here we review protein targets, molecular mechanisms, and potential roles of NO in the regulation of DNA repair, with a focus on S-nitrosylation of DNA repair proteins by endogenous NO synthase activity.
Major Conclusions
Recent studies have identified a number of key DNA repair proteins as targets of S-nitrosylation, including O6-alkylguanine-DNA-alkyltransferase (AGT), 8-oxoguanine glycosylase, apurinic-apyrimidinic endonuclease 1, and DNA-dependent protein kinase catalytic subunit. S-nitrosylation has been shown to modulate the activity, stability, and cellular localization of DNA repair proteins. The level of protein S-nitrosylation depends both on NO synthesis by NO synthases and on denitrosylation by a major denitrosylase, S-nitrosoglutathione reductase (GSNOR). Dysregulated S-nitrosylation of AGT due to GSNOR deficiency inactivates AGT-dependent DNA repair and appears to contribute critically to hepatocarcinogenesis.
General Significance
Studies on the S-nitrosylation of DNA repair proteins have started to reveal molecular mechanisms for the contribution of inflammation to mutagenesis and carcinogenesis. The modulation of protein S-nitrosylation to affect the activity of DNA repair proteins may provide a therapeutic strategy to prevent DNA damage and mutation frequently associated with chronic inflammation and to sensitize cancer cells to DNA-damaging drugs.
Keywords: Nitric oxide, S-nitrosylation, DNA repair proteins, Inflammation, Nitrosative stress, Carcinogenesis
Introduction
DNA is subjected to mutagenic modification by environmental agents and during DNA replication. DNA damage, if not repaired, can cause significant pathologies including disruption of nervous, immune, and reproductive functions as well as premature aging and carcinogenesis [1]. Recent cancer genome studies highlight the mutational signatures and prominent roles of DNA repair in cancer biology [2, 3]. To ensure genomic stability and fidelity, several complex and exquisitely controlled pathways have evolved to repair specific DNA lesions. These include base excision repair (BER) for single base modifications and damage, nucleotide excision repair (NER) for bulky adducts, and homologous recombination or non-homologous end-joining for single- and double-strand breaks (DSB) (Figure 1) [4]. The function of key DNA repair proteins is subject to modulation by multiple posttranslational modifications [1], including modification by NO.
Figure 1.
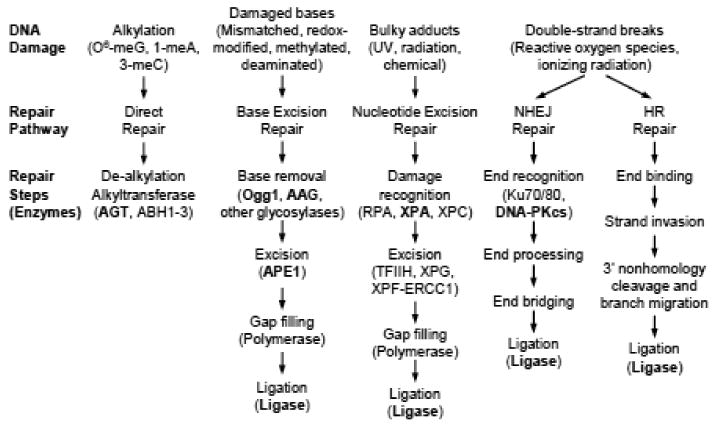
Schematic of the major DNA repair pathways. Proteins highlighted in bold have been shown in vivo or in vitro to be susceptible to modulation by S-nitrosylation (see text for details). Abbreviations: O6-meG, O6-methylguanine; 1-meA, 1-methyladenine; 3-meC, 3-methylcytosine; NHEJ, non-homologous end-joining; HR, homologous recombination.
S-Nitrosylation, the covalent modification of cysteine thiols by NO, plays significant roles in various physiologic and pathophysiologic processes including the regulation of vascular tone, apoptosis, and cancer, among others [5-7]. S-Nitrosylation affects functions of a wide range of proteins and is central to the ubiquitous influence of NO in biological systems [8]. S-Nitrosylation occurs at selective cysteine residues of a protein through redox-based mechanisms by NO or related reactive nitrogen species (RNS) originating from NO synthases (NOS). S-Nitrosylation of proteins and peptides in cells however is not only influenced by NOS activities, but is also prominently regulated by S-nitrosoglutathione reductase (GSNOR) [9], a major denitrosylase [9-11]. GSNOR is the primary means of cells for degrading the main non-protein S-nitrosothiol (SNO), S-nitrosoglutathione (GSNO) [9, 10]. GSNO is believed to be the major source of SNO bioactivity in the cytosol and extracellular fluids [12] and is in equilibrium with protein SNOs in cells. Thus, GSNOR controls the cellular concentration of protein SNOs [9, 10, 13, 14]. Protein S-nitrosylation regulated by GSNOR has been demonstrated to play critical roles in many biological processes including DNA repair and carcinogenesis [9-11, 14-16]. Protein S-nitrosylation has also been shown to be regulated by the denitrosylase activity of the thioredoxin system [17]. Numerous proteins are susceptible to S-nitrosylating agents in vitro, but for most of the proteins, S-nitrosylation by endogenous NO bioactivity in vivo and the biological effect of the S-nitrosylation remain to be fully established.
Extensive studies have focused on the role of NO in DNA damage, repair, and carcinogenesis. NO and related RNS at high levels can cause DNA damage, including alkylation and subsequent deamination, which can lead to GC to AT, GC to CG, and GC to TA mutations [6, 18-23]. RNS can also indirectly increase DNA mutation by inhibition of the DNA repair system. NO can be produced at high levels by iNOS, which is commonly induced in various inflammatory conditions. It has been shown that iNOS activation can inhibit DNA repair pathways including direct repair, BER, and NER [16, 24-27]. DNA repair proteins have been shown to be modulated through S-nitrosylation in vitro by exogenous NO or RNS and in vivo by iNOS induction during inflammation [16, 28-30]. Here we review the findings of the regulation of DNA repair by NO with a focus on S-nitrosylation of DNA repair proteins from endogenous NOS activity.
S-Nitrosylation of DNA Repair Proteins
O6-Alkylguanine-DNA-Alkyltransferase
In direct DNA repair, the key DNA repair protein O6-alkylguanine-DNA alkyltransferase (AGT; also known as O6-Methylguanine-DNA-Methyltransferase, MGMT) repairs highly mutagenic and cytotoxic O6-alkylguanines by transferring the alkyl group from DNA to the enzyme active site cysteine, resulting in irreversible inactivation of AGT and the restoration of guanine [31]. O6-Alkylguanines are produced by alkylating N-nitroso compounds, including dialkylnitrosamines, which are widely present in the environment and can also be formed endogenously [32-34]. O6-Alkylguanines are mispaired by DNA polymerases to thymine during DNA replication, and the O6-alkylguanine:T mispairing, through a further round of DNA replication, can result in GC to AT mutations, DNA double strand breaks, and cell death [35-37]. In vivo, AGT is critical for protecting against dialkylnitrosamine-induced genotoxicity and hepatocellular carcinoma (HCC) [38-41].
Laval and Wink first showed that the alkyltransferase activity in purified AGT and in rat H4 cells was inhibited by diethylamine NONOate treatment and that inhibition of purified AGT by NO was partially reversed by subsequent treatment with dithiothreitol (DTT), suggesting possible involvement of S-nitrosylation in AGT inhibition [42]. Liu et al showed later that in CHO cells transfected with human AGT, treatment with S-nitroso-N-acetylpenicillamine or GSNO led to proteasomal degradation of the AGT protein, reducing half-life of the protein from over 24 h to about 1.3 h [43]. S-Nitroso-N-acetylpenicillamine-induced degradation of AGT was abolished by mutation of the cysteine in the enzyme active site (Cys145Ala), suggesting that AGT degradation may depend on NO modification, potentially via Cys145 S-nitrosylation. Mass spectrum analysis provided more direct evidence for S-nitrosylation at Cys145 of purified AGT following treatment with S-nitroso-N-acetylpenicillamine. These studies showed that AGT in vitro is susceptible to S-nitrosylation at Cys145 by exogenously provided RNS.
More recently, Wei et al demonstrated AGT S-nitrosylation in vivo and its associated hepatocarcinogenic effects in GSNOR-deficient (GSNOR-/-) mice (Figure 2) [16]. They showed that during inflammatory responses following intraperitoneal injection of diethylnitrosamine (DEN) or lipopolysaccharide (LPS), GSNOR deficiency resulted in ubiquitination, proteasomal degradation, and marked depletion of AGT in livers of GSNOR-/- mice. When proteasomal degradation was pharmacologically inhibited, the livers of GSNOR-/- mice showed accumulation of endogenously S-nitrosylated AGT. The proteosome inhibition study suggested that as in cell culture study [43], AGT S-nitrosylation may reduce the half-life of AGT to less than a few hours in vivo, leading to rapid loss of AGT. As a result of AGT depletion, repair of carcinogenic O6-ethylguanines in the livers of DEN-challenged GSNOR-/- mice was significantly impaired. Importantly, the authors showed that GSNOR-/- mice were very susceptible to both spontaneous and DEN-induced HCC. Strikingly, S-nitrosylation and depletion of AGT, accumulation of O6-ethylguanines, and predisposition to HCC due to GSNOR deficiency were all abolished by concurrent deletion of iNOS in GSNOR-/-iNOS-/- mice. Thus the study demonstrated for the first time that AGT is a direct target for S-nitrosylation by iNOS-derived NO bioactivity in vivo and that dysregulated S-nitrosylation of AGT from GSNOR deficiency inactivates the DNA repair system and may contribute critically to hepatocarcinogenesis. It has been further demonstrated most recently that hepatocyte-specific deletion of GSNOR caused nitrosative inactivation of liver AGT and increased mortality from DEN [44], underscoring the importance of the control of S-nitrosylation by GSNOR in liver parenchymal cells.
Figure 2.
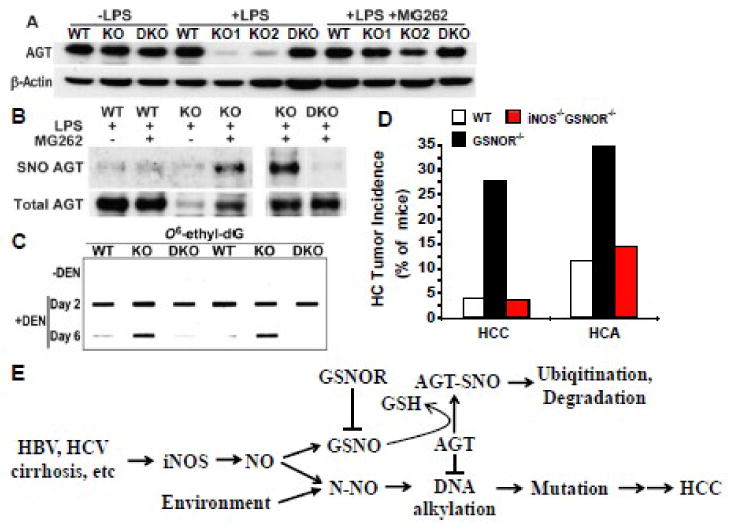
Nitrosative inactivation of O6-alkylguanine-DNA alkyltransferase (AGT) and hepatocarcinogenesis caused by deficiency of S-nitrosoglutathione reductase (GSNOR). A, GSNOR deficiency results in depletion of AGT following induction of iNOS by lipopolysaccharide (LPS). Wild-type (WT), GSNOR-/- (KO), and GSNOR-/-iNOS-/- (DKO) mice were intraperitoneally injected with saline (-LPS) or LPS (+LPS), and liver samples were collected 2 days later for immunoblot analysis of AGT and β-actin. Four hours before tissue collection, some of the LPS-challenged mice were intraperitoneally injected with the proteasome inhibitor MG262 (+LPS +MG262) to inhibit proteasomal degradation of AGT. B, S-Nitrosylation of AGT in livers of LPS-challenged GSNOR-/- mice. The mice were treated as in A and S-nitrosylated AGT (SNO AGT) in liver was detected by the biotin switch assay. C, Impaired repair of O6-ethyldeoxyguanosine (O6-ethyl-dG) in livers of diethylnitrosamine (DEN)-challenged GSNOR-/- mice. Liver DNA from mice before (-DEN) or 2 and 6 days after DEN treatment is probed in immuno-slot blot with monoclonal antibodies against O6-ethyl-dG. D, Increased incidence of spontaneous hepatocellular carcinoma (HCC) and adenoma (HCA) in GSNOR-/- but not GSNOR-/-iNOS-/- mice. E, Model of how iNOS, GSNOR, and AGT interact in HCC oncogenesis: GSNO S-nitrosylates AGT and promotes its degradation via proteasome. (A-D reproduced and adapted from [16], courtesy of AAAS).
The human GSNOR gene (ADH5) is located at approximately 4q23, a region in which chromosomal deletion occurs most frequently in HCC [45-48]. Wei et al showed that the abundance and activity of GSNOR were significantly decreased in cancer samples from ∼50% of patients with HCC [16]. In several human HCC samples that were deficient in GSNOR activity, AGT protein levels were also decreased, suggesting possible protection of AGT by GSNOR in humans [16]. Elevated expression of iNOS has been described in both HCC cells and in the hepatocytes of patients with liver diseases predisposing to HCC [49-54]. Patients with GSNOR deficiency and concurrent iNOS overexpression in the liver may suffer S-nitrosylation and depletion of AGT, and may thus be at an increased risk of HCC. Indeed, gene-expression profiling of liver tissue adjacent to HCC (following resection) showed that both GSNOR deficiency and iNOS overexpression are closely associated with HCC development and a poor prognosis in HCC patients [55]. Inhibition of iNOS-derived S-nitrosylation in the patients may provide a therapeutic strategy to prevent HCC.
8-Oxoguanine Glycosylase
The BER pathway is responsible for the recognition and repair of small, single base modifications like DNA alkylation or oxidation. The BER machinery generally consists of a DNA glycosylase, an apurinic-apyrimidinic (AP)-endonuclease, a DNA polymerase, and a DNA ligase [56]. 8-Oxoguanine glycosylase (Ogg1) is a major DNA glycosylase in the BER pathway with specificity for 8-oxoguanine (8-oxoG), a dominant and highly mutagenic oxidative DNA lesion that favors GC to TA transversions [57, 58]. Targeted deletion of the murine Ogg1 gene greatly reduced 8-oxoG cleavage, suggesting a major role for Ogg1 in the clearance of oxidative DNA damage [59]. Ogg1 relies on critical thiol moieties for catalytic activity and contains a Cys2His2-type zinc (Zn) finger DNA binding motif that could be susceptible to redox modification [60].
NO regulation of Ogg1 was first shown in the purified bacterial ortholog of Ogg1 and in H4 rat hepatoma cells where diethylamine NONOate-treatment irreversibly inhibited enzyme activity [61]. Jaiswal, et al subsequently showed in KMBC cells overexpressing hemagglutinin-tagged Ogg1-1a that 8-oxoG specific BER activity of Ogg1 was inhibited by cytokine-induced iNOS expression and that Ogg1 inhibition was abolished by the pharmacological inhibition of iNOS [28]. Induction of iNOS in these cells resulted in S-nitrosylation of hemagglutinin-tagged Ogg1, and the S-nitrosylation was abolished by iNOS inhibitor 1400W. In addition, NO from iNOS transfected into KMBC cells inhibited endogenous Ogg1 activity [62]. The inhibition of Ogg1 in the iNOS-overexpressing cells was not reversed by peroxynitrite scavengers, indicating that despite an increase in detectable nitrotyrosine, inhibition of 8-oxoG repair is not mediated by peroxynitrite. Ogg1 inhibition was blocked by treatment of these cells with DTT. This may suggest inactivation by DTT of redox-sensitive GSNO or other intermediates that S-nitrosylate Ogg1. DTT treatment of the lysate from iNOS-overexpressing KMBC cells did not restore the Ogg1 activity, suggesting that S-nitrosylation leads to irreversible disruption of the Zn finger motif of Ogg1 [62]. Indeed, treatment of purified hemagglutinin-tagged Ogg1 with S-nitrosylating agents resulted in the release of Zn from the protein [28], although S-nitrosylation of the Zn finger motif or other selective cysteine residues of Ogg1 remains to be formally demonstrated.
Inflammatory cytokine-induction of iNOS expression in three cholangiocarcinoma cell lines led to a NO-dependent increase in oxidative DNA damage and DNA strand breaks [24]. Furthermore, recent evidence suggests that hepatocyte expression of hepatitis C virus core protein in transgenic mice induces iNOS expression and subsequent NO-dependent inhibition of oxidative DNA repair and accumulation of 8-oxoG [27]. In addition, immunohistochemical staining of liver samples from 30 primary sclerosing cholangitis cases revealed de novo expression of iNOS and 8-oxoG in biliary epithelia [62], suggesting a possible role for iNOS-derived NO in DNA damage in vivo. Taken together, these findings suggest that iNOS-derived NO in inflammatory conditions inactivates Ogg1, possibly through S-nitrosylation, and increases oxidative DNA damage, which may provide a mechanism for GC to TA mutations frequently detected in various tumors [2, 3].
Apurinic-Apyrimidinic Endonuclease 1
Apurinic-apyrimidinic endonuclease 1 (APE1; also known as redox factor 1, Ref-1) is the main AP endonuclease in mammals that is critical for the BER pathway of DNA repair. In addition to its role in DNA repair, APE1 also plays roles in regulating transcription and cellular redox responses. APE1 activity is controlled by multiple stimuli, many of which are redox mediated, as well as several post-translational modifications including phosphorylation, acetylation, redox, proteolysis, and S-nitrosylation [63].
Qu et al showed that in HEK-293 cells transfected with hemagglutinin-tagged APE1, GSNO treatment resulted in S-nitrosylation of the overexpressed APE1 protein and its export from the nucleus to the cytoplasm [29]. GSNO-induced S-nitrosylation of APE1 was abolished either by treatment of the GSNO-challenged cells with reducing agents or by the combined mutations of Cys93 and Cys310 to Ser. Inhibition of APE1 S-nitrosylation in both cases prevented GSNO-induced nuclear export of APE1, indicating that S-nitrosylation of APE1 is sufficient for its nuclear export following GSNO treatment. H2O2 treatment of the cells does not lead to APE1 cytoplasmic translocation, suggesting specific effects of S-nitrosylation in the control of APE1 subcellular localization. S-Nitrosylation-dependent regulation of APE1 cytoplasmic localization depends both on active nuclear export and inhibition of importin-mediated nuclear import. It remains to be determined whether S-nitrosylation and subsequent cytoplasmic localization of APE-1 can result from NO bioactivity endogenously generated by NOS. Currently, it is also unknown whether S-nitrosylation of APE1 can regulate its role in DNA repair or its other cellular functions.
Recent studies suggest that glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a mediator of S-nitrosylation of nuclear proteins [30], may potentially also transnitrosylate APE1 in the nucleus and promote its cytoplasmic translocation. GAPDH knockdown in HCT116 cells reduced AP endonuclease activity, increased AP site accumulation, and increased sensitivity to BER-directed genotoxic agents [64], suggesting an important role for GAPDH in the modulation of APE1 activity. GAPDH was also shown to physically interact with APE1 and regulate its function through reduction of oxidized APE1 to restore its endonuclease activity, which may also be regulated through S-nitrosylation-mediated nuclear export of APE1 [29]. It is interesting to note that the same cysteine residue of GAPDH involved in reducing APE1 is also important in transnitrosylation. Taken together, these data suggest that APE1 may be a potential target for S-nitrosylation by SNO-GAPDH. Moreover, thioredoxin, a protein important for the control of S-nitrosylation [17, 65], can translocate to the nucleus upon phorbol myristate acetate stimulation and interact with APE1 [66], and thus could also potentially play a role in APE1 S-nitrosylation.
Notably, whereas APE1 is generally found in the nucleus in most cells, cytoplasmic distribution of APE1 has been described in colon adenomas, breast cancer, and hepatocellular carcinomas [67, 68] and was associated with poor prognosis in breast cancer and HCC [69-71]. S-nitrosylation of APE1, through its regulation of APE1 subcellular localization, may play an important role in the modulation of DNA repair and tumorigenesis and therefore deserves further investigation.
DNA-Dependent Protein-Kinase Catalytic Subunit
DNA-dependent protein-kinase catalytic subunit (DNA-PKcs) is a key double-strand DNA break repair enzyme involved in non-homologous end-joining, a major repair pathway for ionizing radiation-induced DSBs in mammalian cells [4]. DNA-PKcs, along with Ku70/80, mediates the early stage of DSB detection and tethering, which protects DNA from nuclease cleavage [72, 73].
Forrester et al found in a proteomic study that DNA-PKcs is a target for S-nitrosylation in S-nitrosocysteine-treated HEK-293 cells [74]. Subsequently, Kornberg et al showed that in HEK-293 cells overexpressing nNOS, overexpression of GAPDH results in increased DNA-PKcs S-nitrosylation. Conversely, knockdown of GAPDH expression by shRNA reduced S-nitrosylation of DNA-PKcs in GSNO-treated HEK-293 cells [30]. DNA-PKcs S-nitrosylation was dependent on nuclear localization of SNO-GAPDH mediated through binding to Siah1, which contains a nuclear localization sequence. Thus, nuclear DNA-PKcs is targeted by GAPDH for transnitrosylation. It remains to be determined whether S-nitrosylation plays an important role in the regulation of DNA repair by DNA-PKcs. NO has also been reported to increase transcriptional expression and activity of DNA-PKcs, which reduced DNA damage and cell death induced by various DNA damaging agents [75]. NO may be important in the regulation of DNA-PKcs in DNA repair through S-nitrosylation and transcriptional regulation.
Other Potential Targets for S-Nitrosylation in DNA Repair
NO has also been implicated in the modulation of additional proteins important for DNA repair. 3-Alkyladenine DNA glycosylase (AAG) is a broad-specificity DNA glycosylase in BER [76]. Recently, a study using purified recombinant AAG showed that peroxynitrite treatment resulted in tyrosine nitration and inhibition of its activity, whereas NO donor treatment led to a modest increase in the enzyme activity possibly through S-nitrosylation [67]. DNA ligases catalyze the ligation of nicked DNA as the last step of essentially all DNA repair pathways [77]. DNA ligase activities of both purified T4 DNA ligase and cell extracts from CHO cells were reduced after incubation with diethylamine NONOate, which can be blocked by cysteine, suggesting that RNS-mediated S-nitrosylation may be the mechanism for inhibition [78, 79]. Xeroderma Pigmentosum, Complementation Group A (XPA) is a critical component of NER and is involved in the early recognition of helix distorting lesions. Using a synthetic peptide of XPA Cys 4-type Zn finger, Smirnova et al showed that the Zn finger can be disrupted by GSNO treatment, suggesting that XPA may be susceptible to inactivation by GSNO [80]. NO has also been implicated in the arsenite- and LPS-induced inhibition of NER [25, 26, 81, 82]. Whether NO modification of these proteins occurs and plays a role in DNA repair in vivo remains to be determined.
Conclusions
Recent studies have demonstrated that a number of important proteins in the DNA repair system are modified by NO through S-nitrosylation, which can affect their activity, stability, and subcellular localization. The emerging data suggest that the control of S-nitrosylation of DNA repair proteins may depend on both NO synthesis by NOS and degradation by denitrosylases, including in particular, GSNOR. The modulation of protein S-nitrosylation to affect the activity of DNA repair proteins may thus provide a therapeutic strategy to prevent DNA damage and mutation frequently associated with chronic inflammation and to sensitize cancer cells to DNA-damaging drugs.
Highlights.
NO/RNS can directly or indirectly cause DNA mutation
Key DNA repair proteins AGT, Ogg1, APE1, and DNA-PK are targets of S-nitrosylation
S-nitrosylation modulates DNA repair protein activity, stability, and localization
S-nitrosylation levels depend on NO synthesis by NOS and denitrosylation by GSNOR
GSNOR deficiency leads to nitrosative inactivation of AGT and hepatocarcinogenesis
Additional DNA repair proteins may be regulate by S-nitrosylation
Acknowledgments
This work was supported by the National Institutes of Health (R01CA55578, R01CA122359, and P01CA123328 to L.L.).
Abbreviations
- 8-oxoG
8-oxoguanine
- AGT
O6-alkylguanine-DNA-alkyltransferase
- AP
apurinic-apyrimidinic
- APE1
AP endonuclease 1
- BER
base excision repair
- DEN
diethylnitrosamine
- DNA-PKcs
DNA-dependent protein kinase catalytic subunit
- DSB
double-strand break
- DTT
dithiothreitol
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GSNO
S-nitrosoglutathione
- GSNOR
GSNO reductase
- HCC
hepatocellular carcinoma
- LPS
lipopolysaccharide
- NER
nucleotide excision repair
- NO
nitric oxide
- NOS
NO synthase
- Ogg1
8-oxoG glycosylase
- RNS
reactive nitrogen species
- SNO
S-nitrosothiol
- Zn
zinc
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, Beare D, Butler A, Carter RJ, Chen L, Cox AJ, Edkins S, Kokko-Gonzales PI, Gormley NA, Grocock RJ, Haudenschild CD, Hims MM, James T, Jia M, Kingsbury Z, Leroy C, Marshall J, Menzies A, Mudie LJ, Ning Z, Royce T, Schulz-Trieglaff OB, Spiridou A, Stebbings LA, Szajkowski L, Teague J, Williamson D, Chin L, Ross MT, Campbell PJ, Bentley DR, Futreal PA, Stratton MR. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pleasance ED, Stephens PJ, O'Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C, Varela I, Nik-Zainal S, Davies HR, Ordonez GR, Mudie LJ, Latimer C, Edkins S, Stebbings L, Chen L, Jia M, Leroy C, Marshall J, Menzies A, Butler A, Teague JW, Mangion J, Sun YA, McLaughlin SF, Peckham HE, Tsung EF, Costa GL, Lee CC, Minna JD, Gazdar A, Birney E, Rhodes MD, McKernan KJ, Stratton MR, Futreal PA, Campbell PJ. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 5.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 6.Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 8.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 10.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 11.Yang Z, Wang ZE, Doulias PT, Wei W, Ischiropoulos H, Locksley RM, Liu L. Lymphocyte development requires S-nitrosoglutathione reductase. J Immunol. 2010;185:6664–6669. doi: 10.4049/jimmunol.1000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaston B, Reilly J, Drazen JM, Fackler J, Ramdev P, Arnelle D, Mullins ME, Sugarbaker DJ, Chee C, Singel DJ, et al. Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc Natl Acad Sci U S A. 1993;90:10957–10961. doi: 10.1073/pnas.90.23.10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feechan A, Kwon E, Yun BW, Wang Y, Pallas JA, Loake GJ. A central role for S-nitrosothiols in plant disease resistance. Proc Natl Acad Sci U S A. 2005;102:8054–8059. doi: 10.1073/pnas.0501456102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Que LG, Liu L, Yan Y, Whitehead GS, Gavett SH, Schwartz DA, Stamler JS. Protection from experimental asthma by an endogenous bronchodilator. Science. 2005;308:1618–1621. doi: 10.1126/science.1108228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, Messina E, Bowles D, Kontos CD, Hare JM, Stamler JS, Rockman HA. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A. 2009;106:6297–6302. doi: 10.1073/pnas.0901043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei W, Li B, Hanes MA, Kakar S, Chen X, Liu L. S-nitrosylation from GSNOR deficiency impairs DNA repair and promotes hepatocarcinogenesis. Sci Transl Med. 2010;2:19ra13. doi: 10.1126/scitranslmed.3000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burney S, Caulfield JL, Niles JC, Wishnok JS, Tannenbaum SR. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat Res. 1999;424:37–49. doi: 10.1016/s0027-5107(99)00006-8. [DOI] [PubMed] [Google Scholar]
- 19.Hanafy KA, Krumenacker JS, Murad F. NO, nitrotyrosine, and cyclic GMP in signal transduction. Med Sci Monit. 2001;7:801–819. [PubMed] [Google Scholar]
- 20.Hofseth LJ, Hussain SP, Wogan GN, Harris CC. Nitric oxide in cancer and chemoprevention. Free Radic Biol Med. 2003;34:955–968. doi: 10.1016/s0891-5849(02)01363-1. [DOI] [PubMed] [Google Scholar]
- 21.Marnett LJ, Riggins JN, West JD. Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J Clin Invest. 2003;111:583–593. doi: 10.1172/JCI18022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wink DA, Vodovotz Y, Laval J, Laval F, Dewhirst MW, Mitchell JB. The multifaceted roles of nitric oxide in cancer. Carcinogenesis. 1998;19:711–721. doi: 10.1093/carcin/19.5.711. [DOI] [PubMed] [Google Scholar]
- 23.Felley-Bosco E. Role of nitric oxide in genotoxicity: implication for carcinogenesis. Cancer Metastasis Rev. 1998;17:25–37. doi: 10.1023/a:1005948420548. [DOI] [PubMed] [Google Scholar]
- 24.Jaiswal M, LaRusso NF, Burgart LJ, Gores GJ. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000;60:184–190. [PubMed] [Google Scholar]
- 25.Bau DT, Gurr JR, Jan KY. Nitric oxide is involved in arsenite inhibition of pyrimidine dimer excision. Carcinogenesis. 2001;22:709–716. doi: 10.1093/carcin/22.5.709. [DOI] [PubMed] [Google Scholar]
- 26.Cavallo P, Cianciulli A, Mitolo V, Panaro MA. Lipopolysaccharide (LPS) of Helicobacter modulates cellular DNA repair systems in intestinal cells. Clin Exp Med. 2010 doi: 10.1007/s10238-010-0118-1. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 27.Machida K, Tsukamoto H, Liu JC, Han YP, Govindarajan S, Lai MM, Akira S, Ou JH. c-Jun mediates hepatitis C virus hepatocarcinogenesis through signal transducer and activator of transcription 3 and nitric oxide-dependent impairment of oxidative DNA repair. Hepatology. 2010;52:480–492. doi: 10.1002/hep.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaiswal M, LaRusso NF, Nishioka N, Nakabeppu Y, Gores GJ. Human Ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001;61:6388–6393. [PubMed] [Google Scholar]
- 29.Qu J, Liu GH, Huang B, Chen C. Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res. 2007;35:2522–2532. doi: 10.1093/nar/gkl1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol. 2010;12:1094–1100. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pegg AE. Repair of O(6)-alkylguanine by alkyltransferases. Mutat Res. 2000;462:83–100. doi: 10.1016/s1383-5742(00)00017-x. [DOI] [PubMed] [Google Scholar]
- 32.Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer. 2004;4:296–307. doi: 10.1038/nrc1319. [DOI] [PubMed] [Google Scholar]
- 33.Liu RH, Baldwin B, Tennant BC, Hotchkiss JH. Elevated formation of nitrate and N-nitrosodimethylamine in woodchucks (Marmota monax) associated with chronic woodchuck hepatitis virus infection. Cancer Res. 1991;51:3925–3929. [PubMed] [Google Scholar]
- 34.Lundberg JO, Weitzberg E, Cole JA, Benjamin N. Nitrate, bacteria and human health. Nat Rev Microbiol. 2004;2:593–602. doi: 10.1038/nrmicro929. [DOI] [PubMed] [Google Scholar]
- 35.Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst) 2007;6:1079–1099. doi: 10.1016/j.dnarep.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 36.Quiros S, Roos WP, Kaina B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle. 2010;9:168–178. doi: 10.4161/cc.9.1.10363. [DOI] [PubMed] [Google Scholar]
- 37.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glassner BJ, Weeda G, Allan JM, Broekhof JL, Carls NH, Donker I, Engelward BP, Hampson RJ, Hersmus R, Hickman MJ, Roth RB, Warren HB, Wu MM, Hoeijmakers JH, Samson LD. DNA repair methyltransferase (Mgmt) knockout mice are sensitive to the lethal effects of chemotherapeutic alkylating agents. Mutagenesis. 1999;14:339–347. doi: 10.1093/mutage/14.3.339. [DOI] [PubMed] [Google Scholar]
- 39.Iwakuma T, Sakumi K, Nakatsuru Y, Kawate H, Igarashi H, Shiraishi A, Tsuzuki T, Ishikawa T, Sekiguchi M. High incidence of nitrosamine-induced tumorigenesis in mice lacking DNA repair methyltransferase. Carcinogenesis. 1997;18:1631–1635. doi: 10.1093/carcin/18.8.1631. [DOI] [PubMed] [Google Scholar]
- 40.Kawate H, Sakumi K, Tsuzuki T, Nakatsuru Y, Ishikawa T, Takahashi S, Takano H, Noda T, Sekiguchi M. Separation of killing and tumorigenic effects of an alkylating agent in mice defective in two of the DNA repair genes. Proc Natl Acad Sci U S A. 1998;95:5116–5120. doi: 10.1073/pnas.95.9.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsuzuki T, Sakumi K, Shiraishi A, Kawate H, Igarashi H, Iwakuma T, Tominaga Y, Zhang S, Shimizu S, Ishikawa T, et al. Targeted disruption of the DNA repair methyltransferase gene renders mice hypersensitive to alkylating agent. Carcinogenesis. 1996;17:1215–1220. doi: 10.1093/carcin/17.6.1215. [DOI] [PubMed] [Google Scholar]
- 42.Laval F, Wink DA. Inhibition by nitric oxide of the repair protein, O6-methylguanine-DNA-methyltransferase. Carcinogenesis. 1994;15:443–447. doi: 10.1093/carcin/15.3.443. [DOI] [PubMed] [Google Scholar]
- 43.Liu L, Xu-Welliver M, Kanugula S, Pegg AE. Inactivation and degradation of O(6)-alkylguanine-DNA alkyltransferase after reaction with nitric oxide. Cancer Res. 2002;62:3037–3043. [PubMed] [Google Scholar]
- 44.Wei W, Yang Z, Tang CH, Liu L. Targeted deletion of GSNOR in hepatocytes of mice causes nitrosative inactivation of O6-alkylguanine-DNA alkyltransferase and increased sensitivity to genotoxic diethylnitrosamine. Carcinogenesis. 2011 doi: 10.1093/carcin/bgr041. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagai H, Pineau P, Tiollais P, Buendia MA, Dejean A. Comprehensive allelotyping of human hepatocellular carcinoma. Oncogene. 1997;14:2927–2933. doi: 10.1038/sj.onc.1201136. [DOI] [PubMed] [Google Scholar]
- 46.Patil MA, Gutgemann I, Zhang J, Ho C, Cheung ST, Ginzinger D, Li R, Dykema KJ, So S, Fan ST, Kakar S, Furge KA, Buttner R, Chen X. Array-based comparative genomic hybridization reveals recurrent chromosomal aberrations and Jab1 as a potential target for 8q gain in hepatocellular carcinoma. Carcinogenesis. 2005;26:2050–2057. doi: 10.1093/carcin/bgi178. [DOI] [PubMed] [Google Scholar]
- 47.Wong N, Lai P, Lee SW, Fan S, Pang E, Liew CT, Sheng Z, Lau JW, Johnson PJ. Assessment of genetic changes in hepatocellular carcinoma by comparative genomic hybridization analysis: relationship to disease stage, tumor size, and cirrhosis. Am J Pathol. 1999;154:37–43. doi: 10.1016/S0002-9440(10)65248-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeh SH, Chen PJ, Shau WY, Chen YW, Lee PH, Chen JT, Chen DS. Chromosomal allelic imbalance evolving from liver cirrhosis to hepatocellular carcinoma. Gastroenterology. 2001;121:699–709. doi: 10.1053/gast.2001.27211. [DOI] [PubMed] [Google Scholar]
- 49.Kane JM, 3rd, Shears LL, 2nd, Hierholzer C, Ambs S, Billiar TR, Posner MC. Chronic hepatitis C virus infection in humans: induction of hepatic nitric oxide synthase and proposed mechanisms for carcinogenesis. J Surg Res. 1997;69:321–324. doi: 10.1006/jsre.1997.5057. [DOI] [PubMed] [Google Scholar]
- 50.Majano PL, Garcia-Monzon C, Lopez-Cabrera M, Lara-Pezzi E, Fernandez-Ruiz E, Garcia-Iglesias C, Borque MJ, Moreno-Otero R. Inducible nitric oxide synthase expression in chronic viral hepatitis. Evidence for a virus-induced gene upregulation. J Clin Invest. 1998;101:1343–1352. doi: 10.1172/JCI774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rahman MA, Dhar DK, Yamaguchi E, Maruyama S, Sato T, Hayashi H, Ono T, Yamanoi A, Kohno H, Nagasue N. Coexpression of inducible nitric oxide synthase and COX-2 in hepatocellular carcinoma and surrounding liver: possible involvement of COX-2 in the angiogenesis of hepatitis C virus-positive cases. Clin Cancer Res. 2001;7:1325–1332. [PubMed] [Google Scholar]
- 52.Hussain SP, Raja K, Amstad PA, Sawyer M, Trudel LJ, Wogan GN, Hofseth LJ, Shields PG, Billiar TR, Trautwein C, Hohler T, Galle PR, Phillips DH, Markin R, Marrogi AJ, Harris CC. Increased p53 mutation load in nontumorous human liver of wilson disease and hemochromatosis: oxyradical overload diseases. Proc Natl Acad Sci U S A. 2000;97:12770–12775. doi: 10.1073/pnas.220416097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McNaughton L, Puttagunta L, Martinez-Cuesta MA, Kneteman N, Mayers I, Moqbel R, Hamid Q, Radomski MW. Distribution of nitric oxide synthase in normal and cirrhotic human liver. Proc Natl Acad Sci U S A. 2002;99:17161–17166. doi: 10.1073/pnas.0134112100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ikeguchi M, Ueta T, Yamane Y, Hirooka Y, Kaibara N. Inducible nitric oxide synthase and survivin messenger RNA expression in hepatocellular carcinoma. Clin Cancer Res. 2002;8:3131–3136. [PubMed] [Google Scholar]
- 55.Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, Gupta S, Moore J, Wrobel MJ, Lerner J, Reich M, Chan JA, Glickman JN, Ikeda K, Hashimoto M, Watanabe G, Daidone MG, Roayaie S, Schwartz M, Thung S, Salvesen HB, Gabriel S, Mazzaferro V, Bruix J, Friedman SL, Kumada H, Llovet JM, Golub TR. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions. J Biol Chem. 1992;267:166–172. [PubMed] [Google Scholar]
- 58.Wood ML, Dizdaroglu M, Gajewski E, Essigmann JM. Mechanistic studies of ionizing radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry. 1990;29:7024–7032. doi: 10.1021/bi00482a011. [DOI] [PubMed] [Google Scholar]
- 59.Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci U S A. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tani M, Shinmura K, Kohno T, Shiroishi T, Wakana S, Kim SR, Nohmi T, Kasai H, Takenoshita S, Nagamachi Y, Yokota J. Genomic structure and chromosomal localization of the mouse Ogg1 gene that is involved in the repair of 8-hydroxyguanine in DNA damage. Mamm Genome. 1998;9:32–37. doi: 10.1007/s003359900675. [DOI] [PubMed] [Google Scholar]
- 61.Wink DA, Laval J. The Fpg protein, a DNA repair enzyme, is inhibited by the biomediator nitric oxide in vitro and in vivo. Carcinogenesis. 1994;15:2125–2129. doi: 10.1093/carcin/15.10.2125. [DOI] [PubMed] [Google Scholar]
- 62.Jaiswal M, LaRusso NF, Shapiro RA, Billiar TR, Gores GJ. Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterology. 2001;120:190–199. doi: 10.1053/gast.2001.20875. [DOI] [PubMed] [Google Scholar]
- 63.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal. 2009;11:601–620. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Azam S, Jouvet N, Jilani A, Vongsamphanh R, Yang X, Yang S, Ramotar D. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J Biol Chem. 2008;283:30632–30641. doi: 10.1074/jbc.M801401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 66.Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci U S A. 1997;94:3633–3638. doi: 10.1073/pnas.94.8.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jones LE, Jr, Ying L, Hofseth AB, Jelezcova E, Sobol RW, Ambs S, Harris CC, Espey MG, Hofseth LJ, Wyatt MD. Differential effects of reactive nitrogen species on DNA base excision repair initiated by the alkyladenine DNA glycosylase. Carcinogenesis. 2009;30:2123–2129. doi: 10.1093/carcin/bgp256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 69.Di Maso V, Avellini C, Croce LS, Rosso N, Quadrifoglio F, Cesaratto L, Codarin E, Bedogni G, Beltrami CA, Tell G, Tiribelli C. Subcellular localization of APE1/Ref-1 in human hepatocellular carcinoma: possible prognostic significance. Mol Med. 2007;13:89–96. doi: 10.2119/2006-00084.DiMaso. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kakolyris S, Kaklamanis L, Engels K, Fox SB, Taylor M, Hickson ID, Gatter KC, Harris AL. Human AP endonuclease 1 (HAP1) protein expression in breast cancer correlates with lymph node status and angiogenesis. Br J Cancer. 1998;77:1169–1173. doi: 10.1038/bjc.1998.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Puglisi F, Barbone F, Tell G, Aprile G, Pertoldi B, Raiti C, Kelley MR, Damante G, Sobrero A, Beltrami CA, Di Loreto C. Prognostic role of Ape/Ref-1 subcellular expression in stage I-III breast carcinomas. Oncol Rep. 2002;9:11–17. [PubMed] [Google Scholar]
- 72.Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung CS, Peterson SR, Chen DJ. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol Cell Biol. 1999;19:3877–3884. doi: 10.1128/mcb.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kienker LJ, Shin EK, Meek K. Both V(D)J recombination and radioresistance require DNA-PK kinase activity, though minimal levels suffice for V(D)J recombination. Nucleic Acids Res. 2000;28:2752–2761. doi: 10.1093/nar/28.14.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27:557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu W, Liu L, Smith GC, Charles G. Nitric oxide upregulates expression of DNA-PKcs to protect cells from DNA-damaging anti-tumour agents. Nat Cell Biol. 2000;2:339–345. doi: 10.1038/35014028. [DOI] [PubMed] [Google Scholar]
- 76.Asaeda A, Ide H, Asagoshi K, Matsuyama S, Tano K, Murakami A, Takamori Y, Kubo K. Substrate specificity of human methylpurine DNA N-glycosylase. Biochemistry. 2000;39:1959–1965. doi: 10.1021/bi9917075. [DOI] [PubMed] [Google Scholar]
- 77.Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem. 2008;77:313–338. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wink DA, Nims RW, Darbyshire JF, Christodoulou D, Hanbauer I, Cox GW, Laval F, Laval J, Cook JA, Krishna MC, et al. Reaction kinetics for nitrosation of cysteine and glutathione in aerobic nitric oxide solutions at neutral pH. Insights into the fate and physiological effects of intermediates generated in the NO/O2 reaction. Chem Res Toxicol. 1994;7:519–525. doi: 10.1021/tx00040a007. [DOI] [PubMed] [Google Scholar]
- 79.Graziewicz M, Wink DA, Laval F. Nitric oxide inhibits DNA ligase activity: potential mechanisms for NO-mediated DNA damage. Carcinogenesis. 1996;17:2501–2505. doi: 10.1093/carcin/17.11.2501. [DOI] [PubMed] [Google Scholar]
- 80.Smirnova J, Zhukova L, Witkiewicz-Kucharczyk A, Kopera E, Oledzki J, Wyslouch-Cieszynska A, Palumaa P, Hartwig A, Bal W. Reaction of the XPA zinc finger with S-nitrosoglutathione. Chem Res Toxicol. 2008;21:386–392. doi: 10.1021/tx700297f. [DOI] [PubMed] [Google Scholar]
- 81.Chien YH, Bau DT, Jan KY. Nitric oxide inhibits DNA-adduct excision in nucleotide excision repair. Free Radic Biol Med. 2004;36:1011–1017. doi: 10.1016/j.freeradbiomed.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 82.Ding W, Hudson LG, Sun X, Feng C, Liu KJ. As(III) inhibits ultraviolet radiation-induced cyclobutane pyrimidine dimer repair via generation of nitric oxide in human keratinocytes. Free Radic Biol Med. 2008;45:1065–1072. doi: 10.1016/j.freeradbiomed.2008.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]