

Abstract
Dorsal closure in the Drosophila embryo occurs during the later stages of embryogenesis and involves changes in cell shape leading to the juxtaposition and subsequent adherence of the lateral epidermal primordia over the amnioserosa. Dorsal closure requires the activation of a conserved c-jun amino-terminal kinase (JNK) mitogen-activated protein kinase (MAPK) module, as it is blocked by null mutations in JNK kinase [hemipterous (hep)] and JNK [basket (bsk)]. Drosophila JNK (DJNK) functions by phosphorylating and activating DJun, which in turn induces the transcription of decapentaplegic (dpp). We provide biochemical and genetic evidence that a Ste20-related kinase, misshapen (msn), functions upstream of hep and bsk to stimulate dorsal closure in the Drosophila embryo. Mammalian (NCK-interacting kinase [NIK]) and Caenorhabditis elegans (mig-15) homologs of msn have been identified; mig-15 is necessary for several developmental processes in C. elegans. These data suggest that msn, mig-15, and NIK are components of a signaling pathway that is conserved among flies, worms, and mammals to control developmentally regulated pathways.
Keywords: Drosophila, Ste20 kinase, NIK, misshapen, dorsal closure, JNK
Multiple mitogen-activated protein kinase (MAPK) modules have been identified as components of signal transduction pathways, including at least six in yeast and three in mammalian cells (Herskowitz 1995; Kyriakis and Avruch 1996). All MAPK modules are composed of three sequentially acting protein kinases: a MAP kinase, an enzyme that activates MAP kinase [known as MAP kinase kinase (MKK), MAP extracellular signal-related kinase (ERK) kinase (MEK), or Ste7-related kinase], and an enzyme that activates MKK [known as a MAP kinase kinase kinase (MKKK), MAP ERK kinase kinase (MEKK), or Ste11-related kinase] (Davis 1994; Cobb and Goldsmith 1995; Herskowitz 1995). The defining characteristic of these MAP kinase modules is the MAP kinase itself, and in mammalian cells the MAP kinases described thus far include the p42 and p44 MAP kinases (also known as ERK1 and ERK2), c-Jun amino-terminal kinase (JNK)/stress-activated protein kinase (SAPK), and p38. In contrast to p42 and p44 MAP kinases, which are frequently downstream effectors of Ras and are central elements mediating cell proliferation by a variety of growth factors, JNK and p38 are activated most potently by cellular stresses and inflammatory cytokines (Kyriakis and Avruch 1996).
Genetic epistasis analysis in yeast as well as studies in mammalian cells have indicated that Ste20 related kinases function upstream of MKKKs to regulate the JNK MAPK module, and Ste20 kinases have been considered to be MAP kinase kinase kinase kinases (MKKKK) (Herskowitz 1995; Hu et al. 1996; Pombo et al. 1995; Su et al. 1997). Two families of protein kinases, which are closely related to Ste20 in their kinase domain, have been identified based on their structure and regulation. The first family includes the yeast Ste20 protein kinase and the mammalian and Drosophila p21-activated protein kinases (PAKs) (Manser et al. 1994; Martin et al. 1995; Harden et al. 1996). Kinases in this group contain a conserved p21Rac- and Cdc42-binding domain in their amino terminus (Burbelo et al. 1995; Martin et al. 1995) and are activated by binding GTP-bound Cdc42 and Rac (Manser et al. 1994; Bagrodia et al. 1995; Herskowitz 1995; Martin et al. 1995). The second family lacks p21Rac- and Cdc42-binding domains and is named for the yeast SPS1 protein kinase (Friesen et al. 1994). In addition, in contrast to PAKs that contain an amino-terminal regulatory and a carboxy-terminal kinase domain, members of this second family contain an amino-terminal kinase domain and a carboxy-terminal regulatory region. Several SPS1 family kinases have been identified in mammalian cells and include mammalian Ste20-like kinase 1 (MST1) and MST2, germinal center kinase (GCK), NCK-interacting kinase (NIK), Ste20/oxidant stress response kinase (SOK), hematopoietic progenitor kinase (HPK1), and GCK-like kinase (GLK) (Creasy and Chernoff 1995; Pombo et al. 1995; Hu et al. 1996; Diener et al. 1997; Su et al. 1997).
Although a clear pathway has been mapped out between cell surface receptors and the activation of p42 and p44 MAP kinase, the pathways that connect a specific extracellular stimulus to activation of the JNK pathway are not well established. A number of MKKKs or Ste11 related kinases have been found to activate JNK when overexpressed transiently in mammalian cells, but little is known about how these kinases are regulated or which of these kinases couple specific upstream signals to JNK activation. In addition, little is known about the regulation or function of Ste20 kinases; in fact, no mammalian Ste20 kinase has yet been shown to link a specific upstream signal to JNK activation. The ability of several mammalian SPS1 family members, such as GCK, NIK, GLK, and HPK1, to activate JNK when overexpressed transiently in mammalian cells is the strongest evidence that these kinases might activate JNK in response to upstream signals (Pombo et al. 1995; Hu et al. 1996; Diener et al. 1997; Su et al. 1997). However, these studies may not be conclusive because JNK activation has been measured under conditions in which the Ste20 kinases are expressed at very high levels and therefore, could have nonphysiological effects.
Genetic studies in lower organisms, such as Drosophila and Caenorhabditis elegans, have been particularly helpful in unraveling complex signaling pathways (Dickson and Hafen 1994). For example, studies of these organisms proved invaluable in furthering our understanding of the Ras–Raf–MAP kinase signaling pathway. Because of the difficulty in studying Ste20 kinases in mammalian cells, placement of a Ste20 kinase on a genetic pathway in Drosophila or C. elegans would greatly facilitate our understanding of the normal physiological functions of these kinases. Recently, we have identified a Drosophila homolog of NIK (a member of the mammalian SPS1 family of Ste20 kinases), called misshapen (msn). msn was identified in a screen for genes that are regulated by the photoreceptor transcription factor Glass. Its name derives from the finding that msn is required for the normal shape of Drosophila photoreceptor cells. However, msn is also required for normal embryogenesis, as null mutations are embryonic lethal. A Drosophila JNK, basket (bsk), and JNK kinase, hemipterous (hep), have also been shown to be essential for normal embryonic development (Glise et al. 1995; Riesgo-Escovar et al. 1996; Sluss et al. 1996). Loss of either kinase is embryonic lethal and causes a “dorsal open” phenotype. Dorsal closure of the Drosophila embryo involves changes in cell shape leading to elongation and migration of the lateral epithelial sheets (Campos-Ortega and Hartenstein 1985; Young et al. 1993). This coordinated movement of the lateral epithelia functions to internalize the amnioserosa and connect the two sides of the embryo. The finding that the mammalian homolog of msn, NIK, activates the JNK MAPK module, coupled with the finding that msn and JNK are required for normal embryonic development in Drosophila, led us to test whether msn is upstream of JNK activation in the Drosophila embryo.
In this report, we provide biochemical and genetic evidence that msn functions upstream of the JNK MAPK module to direct dorsal closure in Drosophila. To our knowledge, this is the first demonstration that an SPS1 family Ste20 kinase activates the JNK MAPK module under normal physiological circumstances. Understanding the upstream and downstream components of this signaling pathway should provide insights into the regulation of Ste20 kinases.
Results
msn is the Drosophila homolog of NIK
The Drosophila msn and C. elegans mig-15 proteins are highly homologous to mammalian NIK, an activator of the JNK module (Fig. 1A). These three proteins share the same overall structure, containing an amino-terminal kinase domain and a carboxy-terminal putative regulatory domain. Moreover, these three proteins are highly conserved within both the kinase domain and the carboxy-terminal regulatory domain. The high conservation within these two domains suggested that msn and mig-15 would activate the JNK MAPK module, because previously we have shown that both of these domains are essential for NIK to fully activate JNK in mammalian cells (Su et al. 1997). To determine whether msn activates JNK, 293 cells were transfected with msn together with an epitope-tagged JNK, and kinase activity assays were performed on JNK precipitates. Overexpression of either msn or NIK led to about a four- to fivefold increase in JNK kinase activity as assessed by in vitro kinase reaction (Fig. 1B). In agreement with previous studies using NIK, a mutation abolishing msn’s kinase activity markedly reduced msn’s ability to activate JNK. The ability of msn to activate JNK was confirmed by examining its effect on an activated transcription factor 2 (ATF2)-stimulated luciferase reporter gene; JNK has been shown to phosphorylate and activate ATF2 (Gupta et al. 1995). Overexpression of msn in 293 cells led to about a 10-fold increase in the transcriptional activity of ATF2 (Fig. 1C). These findings indicate that msn and NIK are both structurally and functionally similar and suggests that msn may function to activate JNK in Drosophila.
Figure 1.


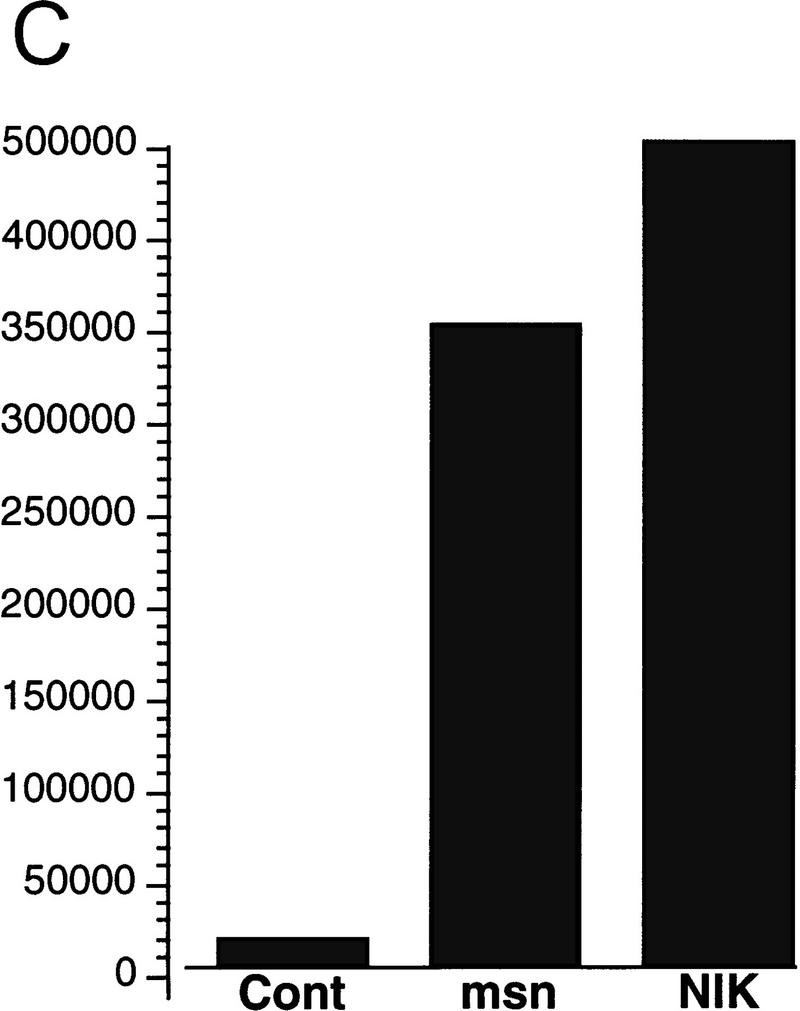
(A) Amino acid alignment of the deduced amino acid sequence of msn, NIK, and mig-15. Identical amino acids are shown in black. The amino-terminal kinase domain and the carboxy-terminal regulatory domain show a high degree of identity between the three proteins. (B,C) Activation of JNK by msn and NIK. (B) msn, NIK, and msn (D160N), which is a kinase inactive form of msn with asparagine substituted for aspartic acid at position 160, were transfected into 293 cells together with 1 μg of GST-tagged JNK. JNK activity in the transfected cell lysates was then determined as described in Materials and Methods. Equal amounts of GST–JNK were precipitated between samples (data not shown). (C) msn (0.2 μg) was cotransfected into 293 cells together with 10 ng of a plasmid expressing a fusion protein consisting of ATF2 and GAL4 DNA binding domain and 5 μg of a plasmid espressing a GAL4–luciferase reporter (Su et al. 1997). Transfection efficiency was assessed by cotransfecting 1 μg of a plasmid expressing β-galactosidase under the control of an SV40 promoter. Luciferase activity is expressed in arbitrary units after being standardized to β-galactosidase activity.
Mutations in msn impair dorsal closure in the Drosophila embryo
Drosophila embryos mutant for JNK (bsk), JNK kinase (hep), or Djun display a dorsal open phenotype, indicating that activation of this pathway is essential for normal embryonic development (Glise et al. 1995; Riesgo-Escovar et al. 1996; Sluss et al. 1996; Hou et al. 1997; Kockel et al. 1997). To test whether msn activates the JNK MAPK module in Drosophila, we determined whether embryos zygotically mutant for msn also display a dorsal open phenotype. Two inversion alleles of msn, msn102 and msn172, were used in this analysis (Treisman et al. 1997). Embryos homozygous for either msn allele or transheterozygous for the two alleles display a defect in dorsal closure, resembling embryos zygotically mutant for bsk (Fig. 2). The observed defect in dorsal closure was observed at a frequency of ∼15%, which is similar to that found for bsk1 (data not shown). Expression of a msn cDNA in the epidermis rescues the dorsal closure defect in msn mutant embryos, allowing survival of all homozygotes to the pupal stage; this demonstrates that the phenotype is attributable to loss of msn function. GAL4 driven by the ectoderm-specific promoter at 69B (Brand and Perrimon 1993) was used to direct the expression of UAS–msn in msn mutant embryos. msn mutant pupae that are rescued because of ectopic expression of msn are longer as the result of the lack of the dominant Tubby marker on the balancer. We found that 47 of 126 pupae obtained from a cross between UAS–msn; msn102/SM6.TM6B and 69B–GAL4, msn102/TM6B were longer; the percentage of long pupae (36%) is very close to the 33% that is predicted if ectopic expression of msn rescued msn homozygous embryos.
Figure 2.

Effect of msn on dorsal closure. Cuticle preparations from embryos of the genotypes indicated. The hole in the dorsal cuticle is indicated with an arrow. Dorsal views are shown in which anterior is up.
msn regulates dpp expression in the leading edge cells surrounding the amnioserosa
The role of DJNK in dorsal closure is to phosphorylate and activate Djun, resulting in transcriptional activation of the dpp gene at the leading edge of the dorsal epidermis (Glise and Noselli 1997; Hou et al. 1997; Riesgo-Escovar and Hafen 1997a). Binding of dpp to its receptors thick veins (tkv) and punt (put) on the ventrally adjacent epithelial cells in turn induces reorganization of the cytoskeleton, leading to epithelial cell elongation and subsequent closure over the amnioserosa (Glise and Noselli 1997; Hou et al. 1997; Kockel et al. 1997; Riesgo-Escovar and Hafen 1997a). This is supported by the findings that dpp expression is decreased in the dorsal-most epithelial cells in embryos lacking bsk and hep, and that expression of activated forms of Djun or tkv rescues embryos zygotically mutant for bsk or Djun (Glise and Noselli 1997; Hou et al. 1997; Kockel et al. 1997; Riesgo-Escovar and Hafen 1997a). Therefore, if msn functions to initiate dorsal closure by activating DJNK, dpp expression in the leading edge epithelial cells should be decreased in msn mutant embryos. In agreement with the idea that msn functions upstream of bsk, we observed that dpp expression in leading edge cells surrounding the amnioserosa was decreased in embryos lacking msn to a comparable degree to bsk1 embryos (Fig. 3; data not shown). We found that ∼20% of embryos derived from the msn102/+; msn102/+ cross displayed a decrease in dpp expression in the dorsal leading edge. The decrease in dpp staining in msn mutant embryos is limited to the dorsal rim cells; dpp expression in the visceral mesoderm and lateral ectoderm is normal in these embryos and serves as a control for dpp staining (Fig. 3).
Figure 3.
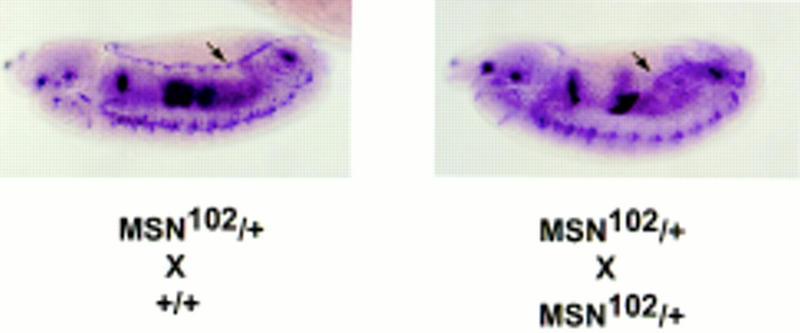
dpp expression in embryos lacking msn. Embryos (5 to 11 hr old) from a cross between msn102/+ and msn102/+ or between msn102/+ and +/+ flies were hybridized with a dpp antisense probe. About 20% of embryos derived from the msn102/+; msn102/+ cross displayed a decrease in dpp expression in cells at the dorsal leading edge (arrow).
Genetic evidence that msn functions upstream of the JNK MAP kinase pathway in Drosophila
The above findings suggest that msn and bsk act in the same pathway and that msn functions upstream of DJNK activation. Further evidence that msn and bsk function in the same pathway comes from the observation that some embryos doubly heterozygous for msn and bsk displayed a dorsal open phenotype. We found that ∼10% of embryos derived from a cross between msn/+ and bsk/+ flies exhibited a dorsal open phenotype (Fig. 4; Table 1). The defect in dorsal closure in embryos doubly heterozygous for msn and bsk is not a dominant effect of either gene; a defect in dorsal closure was very rarely observed when msn/+ or bsk/+ flies were crossed with wild-type flies (Table 1). The presence of such defects in doubly heterozygous flies strongly suggests that the genes function in the same pathway. Moreover, the severity of the phenotype correlated with the strength of the bsk allele with which msn/+ flies were crossed (Fig. 4; data not shown); Df(2L)flp 147E > bsk2 > bsk1 (Riesgo-Escovar et al. 1996; Sluss et al. 1996). To confirm genetically that msn also functions upstream of hep, we examined embryos doubly heterozygous for msn and hep. hep is on the X chromosome, and both maternal and zygotic hep contribute to dorsal closure. To obtain flies doubly heterozygous for msn and hep, msn/+ males were crossed with hepr75/+ females. We found that ∼35% of the flies obtained from this cross displayed a defect in dorsal closure. This finding is very close to the predicted frequency of 37% of embryos with a reduction in both the maternal and zygotic dosage of hep and the zygotic dosage of msn (Fig. 4; Table 1). A defect in dorsal closure is not observed when hep/+ females are crossed with +/Y males, indicating that embryos with the dorsal closure defect carry the msn mutation. Reduced dosage of both zygotic and maternal hep was required for the zygotic lethality of msn/+ embryos; +/+; msn/+ flies, which contain only a reduced dosage of maternal hep, emerged at the predicted frequency of 25% (26 of 117, 22%). In contrast, the viability of msn/+ flies lacking one copy of zygotic hep was reduced by >80%.
Figure 4.

msn interacts genetically with bsk and hep. Cuticle preparations from embryos obtained from the crosses shown. Crossing msn/+ flies to wild-type flies (+/+) did not lead to a defect in dorsal closure (see Table 1). In contrast to the normal embryos obtained from crossing msn/+ flies with wild-type flies (+/+), crossing msn/+ flies with three different bsk alleles or with hep led to defects in dorsal closure. Note that embryos derived from a cross between Df(2L)flp147/+, which is a complete loss-of-function mutant of bsk, and msn/+ flies display the most severe defect in dorsal closure. (Top) Dorsal views with anterior up; (bottom) lateral views with anterior up.
Table 1.
Embryos with defects in dorsal closure
|
|
No./total no. (%)
|
|---|---|
| +/+ × msn102/+ | 1/143 (0.7) |
| +/+ × bsk1/+ | 1/108 (0.9) |
| bsk1/+ × msn102/+ | 15/137 (10.9) |
| bsk2/+ × msn102/+ | 20/198 (10.1) |
| flp147/+ × msn102/+ | 39/388 (10.1) |
| hep1/+ × msn102/+ | 31/88 (35.2) |
| hepr75/+ × msn103/+ | 22/61 (36.0) |
Embryos were collected and aged for 24 hr. Cuticles were prepared from unhatched embryos, and the number of embryos with an aberrant dorsal cuticle was determined. The total number of hatched and unhatched embryos was determined by resuspending all of the larvae and embryos in Hoyer’s solution and counting under a microscope.
We then tested whether a constitutively active form of Djun could rescue the dorsal open phenotype in msn mutant embryos. Previous studies have shown that activated Djun rescues the bsk phenotype, indicating that one of the main functions of JNK is to phosphorylate and activate Jun (Hou et al. 1997; Riesgo-Escovar and Hafen 1997a). A constitutively active form of Djun has been made by replacing the JNK phosphorylation sites with acidic residues (Treier et al. 1995). To test whether this activated Djun rescues the dorsal open phenotype in msn mutant embryos, we expressed it under the control of the hsp70 heat shock promoter in the msn mutant background (Hou et al. 1997). Expression of activated Djun rescued the dorsal open phenotype in most of the msn mutant embryos (Fig. 5); heat shock decreased the number of embryos with a dorsal open phenotype from ∼50% (36 of 76) to <10% (6 of 65). In addition, we found that expression of an activated form of tkv, tkvQ253D (Nellen et al. 1996), also rescued the dorsal open phenotype in msn mutant embryos. GAL4 driven by the ectoderm-specific promoter at 69B (Brand and Perrimon 1993) was used to direct the expression of UAS–tkvQ253D in msn mutant embryos. This expression of activated tkv rescued partially the dorsal open phenotype caused by msn; it also had a dorsalizing effect on the ventral ectoderm of the embryos related to the earlier function of dpp in establishing the dorsoventral axis, which served to mark embryos expressing activated tkv (Fig. 5). Thus, these findings provide genetic evidence that msn functions upstream of the JNK MAP kinase module in leading edge cells.
Figure 5.
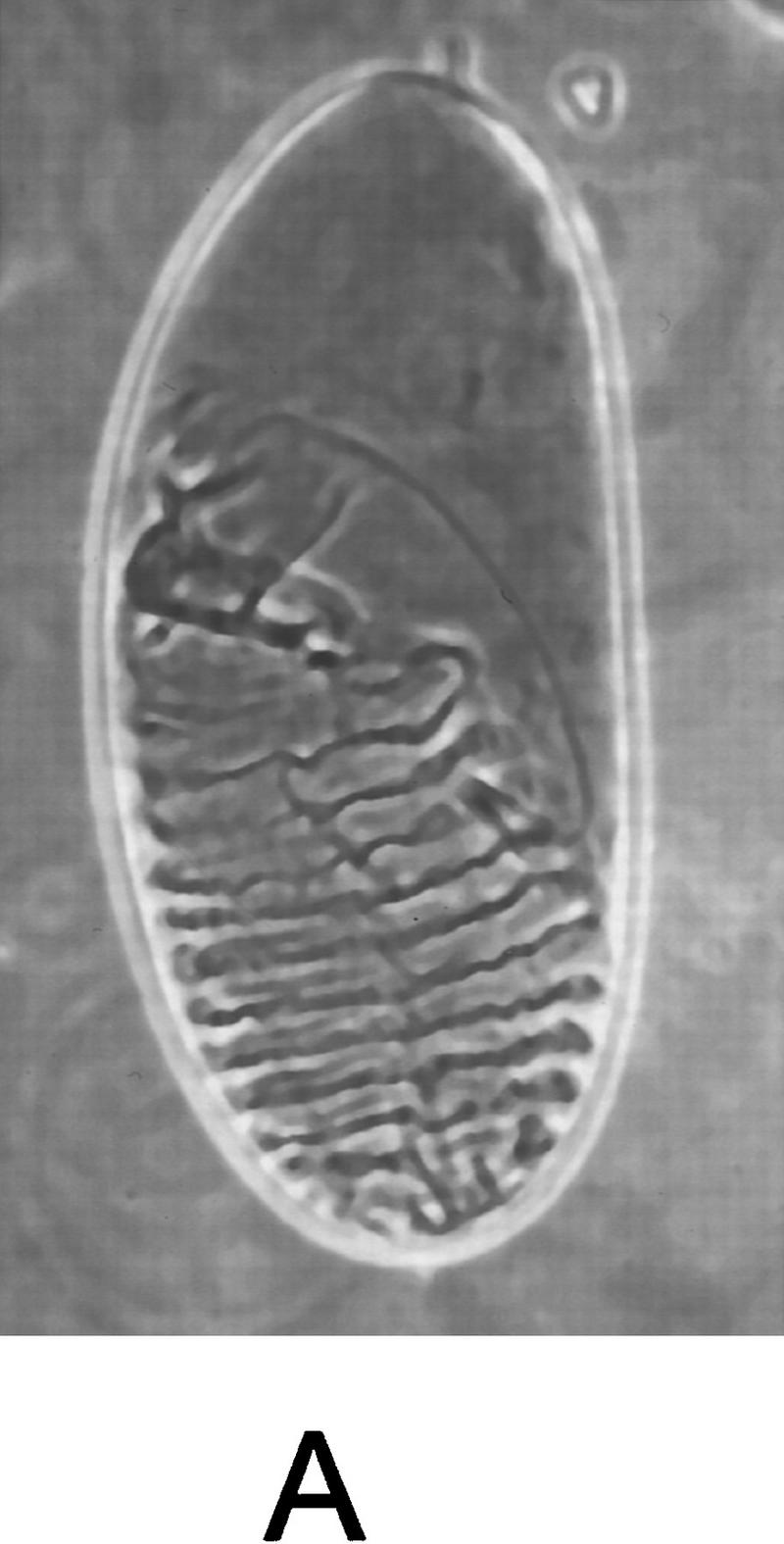
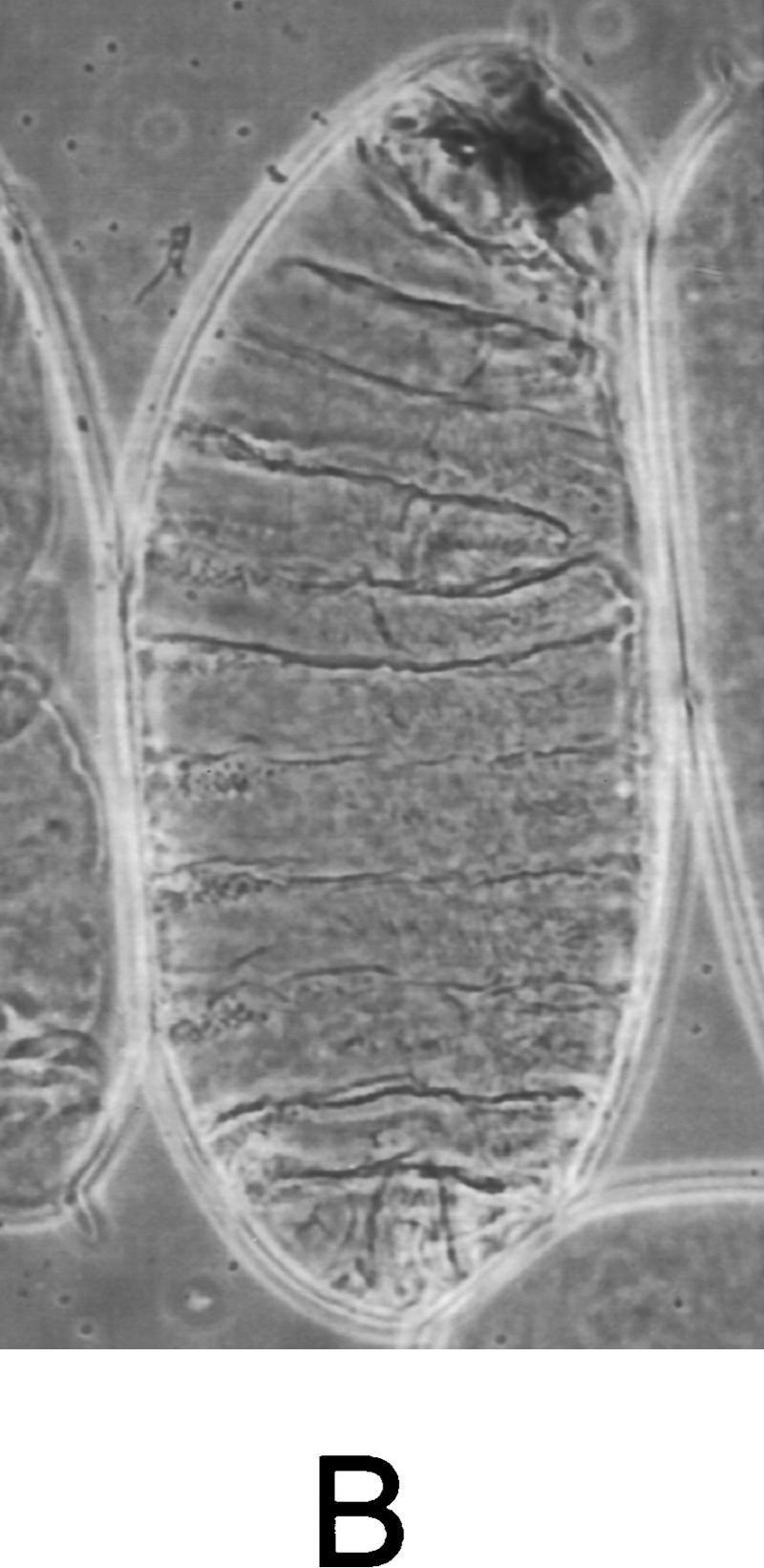
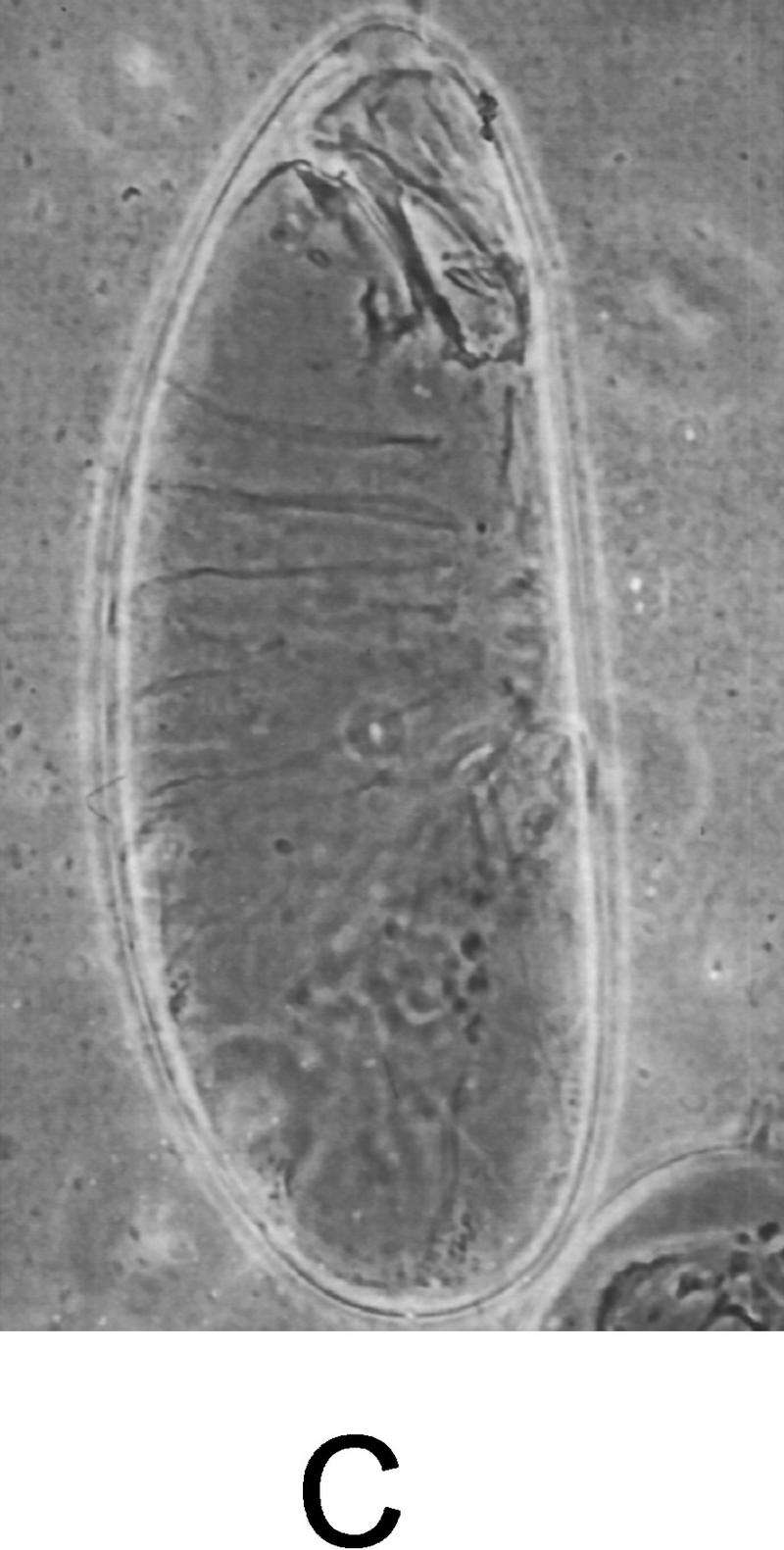
Expression of activated Djun or tkv rescues the dorsal open phenotype in msn mutant embryos. (A) msn mutant embryo containing hs-DjunAsp and not exposed to heat shock displays a severe defect in dorsal closure. (B) msn mutant embryos derived from the same cross and exposed to heat shock for 45 min (msn102/msn172; hs-SEJunAsp). The dorsal cuticle is mostly closed in msn mutant embryos expressing activated Djun. (C) msn mutant embryo expressing activated tkv. The defect in dorsal closure is partially rescued by activated tkv (msn102/msn172; UAS–tkvQ252D/69B GAL4 embryo). Lateral views are shown, with anterior up.
GTP–Rac is required for msn and NIK to fully activate JNK
Rac activation is thought to be important for stimulating dorsal closure because expression of dominant negative forms of Rac (DN Rac) or Cdc42 inhibit dorsal closure in the Drosophila embryo (Harden et al. 1995; Riesgo-Escovar et al. 1996). The finding that activated Djun rescues the defect in dorsal closure induced by expression of DN Rac indicates that Rac probably functions upstream of JNK activation to stimulate dorsal closure (Hou et al. 1997). To begin to address the mechanism whereby Rac and msn cooperate to activate JNK, 293 cells were transfected with msn or NIK together with DN Rac and an epitope-tagged JNK, and kinase activity assays were performed on JNK precipitates. Although overexpression of NIK or msn led to a four- to fivefold increase in JNK activation, coexpression of DN Rac decreased markedly JNK activation (Fig. 6).
Figure 6.

DN Rac inhibits JNK activation by msn and NIK. 293 cells were transfected with 2 μg of msn or NIK cDNA together with 1 μg of GST-tagged JNK with or without 2 μg of DN Rac. JNK activity in the transfected cell lysates was then determined as described in Fig. 1. To ensure that equal amounts of JNK were immunoprecipitated between samples, one-half of the precipitate was immunoblotted with antibodies to GST (JNK).
The PAK family of Ste20 kinases contain a conserved p21Rac and Cdc42-binding domain and are activated by binding GTP-bound Cdc42 and Rac (Burbelo et al. 1995; Martin et al. 1995). To determine whether Rac may regulate directly msn or NIK in a similar manner to the way it activates PAK, we assessed whether msn or NIK bound activated Rac in vitro. Although msn and NIK do not contain a Rac-binding domain, other proteins without a Rac-binding motif have been found recently to bind Rac (Fanger et al. 1997). We were unable to detect binding of activated Rac to either msn or NIK using an overlay assay with GTP-bound Rac or by using the yeast two hybrid system, whereas binding to a positive control, mammalian PAK1, or Drosophila p21-activated protein kinase (DPAK), was easily detected (data not shown). These findings suggest that although Rac cooperates with both NIK and msn to activate JNK, Rac is unlikely to activate directly msn or NIK.
Discussion
At least two distinct MAPK modules have now been identified in Drosophila. Genetic evidence has established that these pathways function independently and are essential for the regulation of distinct developmentally regulated processes (Glise et al. 1995; Riesgo-Escovar et al. 1996; Sluss et al. 1996). The first MAPK module to be described is activated by several receptor tyrosine kinases including the sevenless, Drosophila epidermal growth factor (DEGF), and torso receptors (for review, see Dickson and Hafen 1994; Perrimon 1994). These receptors signal through DRas to activate DRaf (MKKK), which in turn phosphorylates and activates DSor1 (MKK), which phosphorylates and activates the Drosophila MAP kinase rolled (DERK). Activation of this MAPK module is essential for the sevenless receptor to specify the fate of R7 photoreceptors, the DEGF receptor to specify embryonic dorsoventral polarity or the development of other photoreceptors, and torso to specify embryonic termini. The second MAPK module to be described in Drosophila is the JNK pathway (Glise et al. 1995; Riesgo-Escovar et al. 1996; Sluss et al. 1996). Activation of this pathway stimulates changes in the cytoskeleton that enable lateral ectodermal cells to spread over the amnioserosa and join in the dorsal midline. Failure to activate the JNK MAP kinase module leads to a dorsal open phenotype and embryonic lethality. The JNK module is also activated during the Drosophila immune response to lipopolysaccharide (LPS) (Sluss et al. 1996). Unlike the Ras–Raf–MAPK module for which the upstream signals are well known, very little is known about the receptors or signaling molecules that mediate activation of the JNK MAPK module in either Drosophila or mammalian cells under normal biological conditions. Our findings indicate that the Ste20-related kinase msn functions upstream of the JNK MAPK module and is essential for initiating changes in cell shape that regulate dorsal closure of the Drosophila embryo. We found that msn mutant embryos exhibit a defect in dorsal closure that is similar to the defect in embryos lacking components of the JNK pathway. Furthermore, the finding that flies doubly heterozygous for msn and either bsk or hep display a defect in dorsal closure, coupled with genetic epistasis analysis showing that activated Djun and tkv at least partially rescue the defect in dorsal closure in msn mutant embryos, indicates that msn and bsk function in the same pathway and that msn functions upstream of bsk (JNK) activation.
Although several members of the SPS family of Ste20-related kinases have been shown to be specific and potent activators of the JNK pathway when overexpressed transiently in mammalian cells (Pombo et al. 1995; Hu et al. 1996; Su et al. 1997), little is known about the function or regulation of these kinases under normal physiological circumstances. Our finding that msn functions upstream of the JNK MAP kinase module in a genetically defined system indicates that kinases in this family are important activators of JNK under physiologically relevant conditions. The data put to rest the idea that activation of the JNK MAPK module observed in transient transfection assays by SPS1 Ste20 family members is an experimental artifact related to misexpressing these proteins at high levels.
As a result of several recent studies, a number of signaling molecules that are critical for stimulating dorsal closure can now be ordered on a signaling pathway. Genetic epistasis analysis in yeast, as well as studies in mammalian cells, lead us to predict that msn functions as a MKKKK in Drosophila and activates the JNK pathway by activating a yet to be defined Drosophila MKKK (Fig. 7). Therefore, msn becomes the most proximal molecule identified so far on this signaling pathway. Studies over the past several years have clarified which signaling molecules acting downstream of this putative Drosophila MKKK are important in stimulating dorsal closure. A Drosophila MKKK would be likely to phosphorylate and activate the JNK kinase hep, which in turn phosphorylates and activates JNK (bsk) (Glise et al. 1995; Riesgo-Escovar et al. 1996; Sluss et al. 1996). DJNK mediates its changes in the cytoskeleton primarily by regulating the expression of target genes; DJNK phosphorylates and activates DJun, which in turn cooperates with DFos to stimulate transcription of dpp, a member of the transforming growth factor-β (TGF-β) family (Glise and Noselli 1997; Hou et al. 1997; Riesgo-Escovar and Hafen 1997a,b; Noselli 1998). Signaling of dpp through its receptors tkv and put then stimulates changes in cell shape, thereby enabling the lateral epidermal cells to stretch over and subsequently close over the amnioserosa dorsally (Affolter et al. 1994; Brummel et al. 1994; Terracol and Lengyel 1994; Letsou et al. 1995; Ruperte et al. 1995).
Figure 7.

Schematic diagram showing the function of msn, NIK, and mig-15 (see text for details). mig-15 has not yet been shown to activate C. elegans JNK (SAK1) or JNK kinase (SEK). In addition, neither SAK nor SEK have yet been shown to function upstream of Q-neuroblast migration or muscle arm targeting in C. elegans.
Currently very little is known about the downstream MKKKs/MEKKs that are activated by Ste20 kinases or the mechanisms whereby these MKKKs are activated. The identification of a large number of kinases in mammalian cells that are capable of functioning as MKKKs, coupled with the finding that many of these kinases are constitutively active when overexpressed in cells, has hindered the identification of the specific downstream targets of Ste20 kinases or the mechanisms by which they activate MKKKs (Kyriakis and Avruch 1996). Recent evidence has suggested that direct interaction of Ste20 kinases with specific downstream MKKKs may be a critical component of their regulation (Hu et al. 1996; Su et al. 1997); we have found that the carboxy-terminal domain of NIK, which is highly conserved between NIK, msn, and mig-15, is critical for NIK to activate the JNK pathway and to associate with MEKK1 in cells (Su et al. 1997).
Our findings also indicate that Rac cooperates with SPS1 family Ste20 kinases to mediate activation of JNK. Previous studies have suggested that Rac functions upstream of the DJNK MAPK module in Drosophila; expression of DN Rac or Cdc42 results in a dorsal open phenotype (Harden et al. 1995; Riesgo-Escovar et al. 1996) and the defect in dorsal closure induced by expression of DN Rac can be reversed by expressing an activated form of Djun (Hou et al. 1997). Because PAK family Ste20 kinases are activated by GTP-bound Cdc42 and Rac, it had been assumed that this family of Ste20 kinases rather than an SPS1 Ste20 kinase family member would cooperate with Rac to activate JNK (Noselli 1998). Thus, these findings have led us to consider new paradigms for how Rac functions to activate JNK. We do not think that Rac activates msn directly. Unlike PAK family members, msn does not contain a consensus Rac-binding motif and we did not detect binding of msn to activated Rac in vitro (data not shown). Rather, we favor the hypothesis that Rac cooperates with msn to activate a downstream MKKK. MKKKs of the mixed lineage kinase family as well as MEKK1 and MEKK4 have been shown to bind GTP-bound Cdc42 or Rac (Teramoto et al. 1996; Fanger et al. 1997). Thus, Rac may cooperate with msn to regulate a downstream MKKK in a manner similar to the way Ras cooperates with a yet to be defined kinase to activate RAF. In this model, binding of a MKKK to activated Rac would facilitate interaction of this MKKK with msn, thereby enabling its activation by msn (Fig. 7). However, we cannot exclude the possibility that Rac and msn activate parallel pathways converging on JNK activation.
It is intriguing that the C. elegans homolog of msn, mig-15, is also an essential gene in development and, like msn, functions to regulate processes that undoubtedly require changes in the cytoskeleton and cell shape in developing worms (E. Hedgecock, pers. comm.). mig-15 mutants have a variety of developmental defects including defects in Q-neuroblast migration and muscle arm targeting. Although it is not yet clear whether any or all of the phenotypes apparent in worms lacking mig-15 are attributable to defective activation of the C. elegans JNK, these findings suggest a common theme in which JNK activation plays a central role in a variety of developmental processes by coordinating changes in cell shape and the cytoskeleton (Fig. 7). It is likely, however, that some of the phenotypes observed in embryos lacking these Ste20 kinases are independent of their effect on JNK activation. In addition to defects in dorsal closure, some embryos mutant for msn displayed a ventral defect (data not shown). Moreover, although msn, like bsk and Djun, is not required for specifying the fate of photoreceptor cells, clones of msn mutant photoreceptor cells display an abnormal shape (Riesgo-Escovar et al. 1996; Hou et al. 1997; Treisman et al. 1997). These defects are never observed in embryos mutant for bsk and therefore indicate that msn has other essential functions that are independent of JNK activation (Riesgo-Escovar et al. 1996). We were unable to evaluate the maternal contribution of msn to dorsal closure because of its requirement for oogenesis (Treisman et al. 1997).
Our findings also support the idea that the regulation of Ste20 kinases in mammalian cells is likely to be more complex than previously recognized. Several mammalian Ste20 kinases related to msn and NIK, which specifically activate the JNK pathway, such as GC kinase and HPK1, have been identified (Pombo et al. 1995; Hu et al. 1996; Su et al. 1997). It has not been clear whether the function served by these kinases is redundant or whether each may function only under specific circumstances. Although the full repertoire of Ste20 kinases in Drosophila is not known, our results support the idea that members of this family are subject to different modes of regulation and, for at least some functions, are not redundant with other family members. Studying msn and mig-15 in defined genetic systems will be a critical tool in the effort to unravel these complex pathways in mammalian cells.
The placement of msn, mig-15, and NIK on a MAP kinase pathway that is likely to be conserved between worms, flies, and mammals is reminiscent of the better studied Ras–Raf–MAPK module. Genetic analysis of this MAP kinase pathway in both Drosophila and C. elegans was critical in eventually elucidating key components of this pathway. Despite the large number of Ste20 kinases that have now been identified in mammalian cells, virtually nothing is known about either the upstream regulation of this family of kinases or their downstream targets. Thus, the identification of other components of this pathway in Drosophila and C. elegans will undoubtedly provide valuable insights into the signals that activate Ste20 kinases and their subsequent effects on cells.
Materials and methods
Constructs, mutagenesis, and JNK assays
All constructs for tissue culture were myc-epitope tagged in their carboxyl terminus and subcloned into the vector pRK5 (Marcusohn et al. 1995; Su et al. 1997). To assess JNK activation, 2 μg of NIK, msn, or vector control were transfected into 293 cells together with 2 μg of GST-tagged JNK. GST–JNK was precipitated from 500 μg of cell lysates using glutathione agarose and subjected to an in vitro kinase assay using GST–Jun (Su et al. 1997). Reaction products were separated by SDS-PAGE and visualized by autoradiography. To make a kinase inactive form of msn, the msn cDNA was subcloned into the vector pALTER (Promega) and site directed mutagenesis was performed according to the manufacturer’s specifications. The kinase inactive msn contained a substitution of N for D at position 160 (msn D160N). Transactivation of ATF2 was assessed by transfecting a fusion protein consisting of ATF2 (amino acids 1–505) and the GAL4–DNA-binding domain together with a vector control or msn and a reporter plasmid containing 5× GAL4–DNA-binding domains. All transfections were standardized by cotransfecting a control plasmid expressing β-galactosidase (Promega). Dominant negative N17 Rac was expressed using the vector RK5 as described (Marcusohn et al. 1995).
Fly strains and genetic analysis
msn102 and msn172 are x-ray-induced inversions with breakpoints within the msn gene (Treisman et al. 1997). Both inversions break within the coding region, although msn102 causes a larger disruption of the msn gene. The Df(2L)flp 147E line, a deletion of part of the bsk-coding region, was obtained from K. Beckingham (Rice University, Houston, TX). The bsk1 and bsk2 lines were obtained from the Nusslein-Volhard laboratory by way of T. Ip (University of Massachusetts Medical School, Worcester, MA). The hep1 and hep2 lines were obtained from S. Noselli [Centre de Biologie du Development, Centre National de la Recherche Scientifique (CNRS), Toulouse, France]. The activated Djun line has been described previously (Treier et al. 1995).
Genetic interactions
To test for genetic interaction between msn and bsk, or between msn and hep, embryos were collected from a cross between msn/+ flies with either +/+, bsk/+, or hep/+ flies. Embryos were collected for 6 hr and aged for an additional 16 hr and the total number of embryos with a dorsal open phenotype was determined.
To test whether activated Jun (hs-SEJunAsp) rescues msn mutant embryos, activated Djun was introduced into the msn102 mutant background using the compound balancer SM6.TM6B to generate msn102; hs-SEJunAsp/SM6.TM6B. To induce expression of hs-SEJunAsp, 5- to 7-hr-old embryos were heat-shocked for 45 min. Cuticle preparations were then performed 16 hr after heat shock.
To target expression of activated tkv, the activated type 1 dpp receptor, to the dorsal ectoderm we used the Gal4/UAS system (Brand and Perrimon 1993). A UAS–tkvQ253D transgene on the third chromosome was recombined with msn102 to generate msn102, UAS–tkvQ253D/TM6B (Hoodless et al. 1996). To induce ectodermal expression of TKVQ252D, msn102, UAS–tkvQ253D/TM6B flies were crossed to 69B–GAL4, msn102/TM6B.
Ecptopic expression of msn
The Gal4/UAS system was also used to test whether ectopic expression of msn rescues the dorsal defect in msn mutant embryos. The msn cDNA was cloned into the vector pUAST (Brand and Perrimon 1993). Germ-line transformations were then performed using standard methods following injection of pUAS–msn with a helper plasmid turbo δ2-3 (Spradling and Rubin 1982). Transgenic lines containing UAS–msn on the second chromosome were crossed to msn102 to generate UAS–msn; msn102/SM6.TM6B. To induce ectodermal expression of msn, UAS–msn; msn102/SM6.TM6B were crossed to 69B–GAL4, msn102/TM6B.
Cuticle preparations
Embryos were collected on yeasted agar plates, dechorionated in 100% bleach, rinsed with water, and then fixed for 10 min at 65°C in a solution containing acetic acid and glycerol at a 3:1 ratio. Embryos were then mounted in Hoyer’s medium and incubated for 24 hr at 65°C.
In situ hybridization
In situ hybridization to embryos was performed using digoxigenin-labeled antisense dpp RNA probe according to the method of Tautz and Pfeifle (1989) as modified by Ronchi et al. (1993).
Acknowledgments
We thank Tony Ip of The University of Massachusetts Medical School for helpful discussions and for kindly providing the dpp cDNA and the bsk1 and bsk2 stocks. We thank S. Noselli for hep fly stocks and Gary Struhl for UAS–tkvQ2530. We thank X. Steven Hou and Norbert Perrimon of Harvard Medical School for helpful discussions and hsSEJunAsp fly stocks, and Kate Beckingham of Rice University for providing the Df(2L)flp 147E stocks. We thank Hongzhi Liu for performing the yeast two-hybrid analysis of activated DRac and msn. The manuscript was improved by the critical comments of Corinne Zaffran. This work was supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and American Diabetes Association to E.Y.S.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Skolnik@saturn.med.nyu.edu; FAX (212) 263-5711.
E-MAIL Treisman@saturn.med.nyu.edu; FAX (212) 263-7760.
References
- Affolter M, Nellen D, Nussbaumer U, Basler K. Multiple requirements for the receptor serine/threonine kinase thick veins reveals a novel function of TGFβ homologs during Drosophila development. Development. 1994;120:3105–3117. doi: 10.1242/dev.120.11.3105. [DOI] [PubMed] [Google Scholar]
- Bagrodia S, Derijard B, Davis RJ, Cerione RA. Cdc42 and PAK-mediated signaling leads to Jun kinase and p38 mitogen-activated protein kinase activation. J Biol Chem. 1995;270:27995–27998. doi: 10.1074/jbc.270.47.27995. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeting gene expression as a mean of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brummel TJ, Twombly G, Marques G, Wrana JL, Newfeld SJ, Attisano L, Massague J. Characterization and relationship of Dpp receptors encoded by the saxophone and thick veins genes in Drosophila. Cell. 1994;78:251–261. doi: 10.1016/0092-8674(94)90295-x. [DOI] [PubMed] [Google Scholar]
- Burbelo PD, Drechsel D, Hall A. A conserved binding motif defines numerous candidate target proteins for both Cdc42 and Rac GTPases. J Biol Chem. 1995;270:29072–29074. doi: 10.1074/jbc.270.49.29071. [DOI] [PubMed] [Google Scholar]
- Campos-Ortega J, Hartenstein V. The embryonic development of Drosophila melanogaster. Berlin, Germany: Springer-Verlag; 1985. [Google Scholar]
- Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- Creasy CL, Chernoff J. Cloning and characterization of human protein kinase with homology to Ste20. J Biol Chem. 1995;270:21695–21700. doi: 10.1074/jbc.270.37.21695. [DOI] [PubMed] [Google Scholar]
- Davis RJ. MAPKs: New JNK expands the group. Trends Biochem Sci. 1994;19:470–473. doi: 10.1016/0968-0004(94)90132-5. [DOI] [PubMed] [Google Scholar]
- Dickson B, Hafen E. Genetics of signal transduction in invertebrates. Curr Opin Genet Dev. 1994;4:64–70. doi: 10.1016/0959-437x(94)90092-2. [DOI] [PubMed] [Google Scholar]
- Diener K, Wang XS, Chen C, Meyer CF, Keesler G, Zukowski M, Tan TH, Yao Z. Activation of the c-Jun N-terminal kinase pathway by a novel protein kinase related to human germinal center kinase. Proc Natl Acad Sci. 1997;94:9687–9692. doi: 10.1073/pnas.94.18.9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanger GR, Johnson NL, Johnson GL. MEK kinases are regulated by EGF and selectively interact with Rac/Cdc42. EMBO J. 1997;16:4961–4972. doi: 10.1093/emboj/16.16.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen H, Lunz R, Doyle S, Segall J. Mutation of Sps1-encoded protein kinase of Sacchromyces cerevisiae leads to defects in transcription and morphology during spore formation. Genes & Dev. 1994;8:2162–2175. doi: 10.1101/gad.8.18.2162. [DOI] [PubMed] [Google Scholar]
- Glise B, Noselli S. Coupling of Jun amino-terminal kinase and Decapentaplegic signaling pathways in Drosophila morphogenesis. Genes & Dev. 1997;11:1738–1747. doi: 10.1101/gad.11.13.1738. [DOI] [PubMed] [Google Scholar]
- Glise B, Bourbon H, Noselli S. Hemipterous encodes a novel Drosophila MAP kinase kinase required for epithelial cell sheet movement. Cell. 1995;83:451–461. doi: 10.1016/0092-8674(95)90123-x. [DOI] [PubMed] [Google Scholar]
- Gupta S, Campbell D, Derijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- Harden N, Loh HY, Chia W, Lim L. A dominant inhibitory version of the small GTP binding protein Rac disrupts cytoskeletal structures and inhibits developmental cell shape changes in Drosophila. Development. 1995;121:930–914. doi: 10.1242/dev.121.3.903. [DOI] [PubMed] [Google Scholar]
- Harden NJ, Lee J, Loh HY, Ong YM, Tan I, Leung T, Manser E, Lim L. A Drosophila homolog of the Rac and Cdc42-activated serine/threonine kinase PAK is a potential focal adhesion and focal complex protein that colocalizes with dynamic actin structures. Mol Cell Biol. 1996;16:1896–1908. doi: 10.1128/mcb.16.5.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz I. MAP kinase pathways in yeast: For mating and more. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- Hoodless PA, Haerry T, Abdollah S, Stapleton M, O’Connor MB, Attisano L, Wrana JL. MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell. 1996;85:489–500. doi: 10.1016/s0092-8674(00)81250-7. [DOI] [PubMed] [Google Scholar]
- Hou XS, Goldstein ES, Perrimon N. Drosophila Jun relays the Jun amino-terminal kinase signal transduction pathway to the Decapentaplegic signal transduction pathway in regulating epithelial cell sheet movement. Genes & Dev. 1997;11:1728–1737. doi: 10.1101/gad.11.13.1728. [DOI] [PubMed] [Google Scholar]
- Hu MC, Qiu WR, Wang XR, Meyer CF, Tan T-H. Human HPK1, a novel hematopoietic progenitor kinase that activates the JNK/SAPK kinase cascade. Genes & Dev. 1996;10:2251–2264. doi: 10.1101/gad.10.18.2251. [DOI] [PubMed] [Google Scholar]
- Kockel L, Zeitlinger J, Staszewski LM, Moldzik M, Bohmann D. Jun in Drosophila development: Redundant and nonredundant functions and regulation by two MAPK signal transduction pathways. Genes & Dev. 1997;11:1748–1758. doi: 10.1101/gad.11.13.1748. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Sounding the alarm: Protein kinase cascades activated by stress and inflammation. J Biol Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- Letsou AK, Arora K, Wrana JL, Simin K, Twombly V, Jamal J, Staehling-Hampton MB, Gelbert WM, Massague J, O’Connor MB. Drosophila dpp signaling is mediated by the punt gene product: A dual ligand binding type II receptor for the TGFβ receptor family. Cell. 1995;80:899–908. doi: 10.1016/0092-8674(95)90293-7. [DOI] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Zhao Z, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- Marcusohn J, Isakoff SI, Rose E, Symons M, Skolnik EY. The GTP-binding protein Rac does not couple PI3-kinase to insulin-stimulated glucose transport. Curr Biol. 1995;5:1296–1302. doi: 10.1016/s0960-9822(95)00256-9. [DOI] [PubMed] [Google Scholar]
- Martin GA, Bollag G, McCormick F, Abo A. A novel serine kinase activated by rac1/CDC42Hs-dependent autophosphorylation is related to PAK65 and STE20. EMBO J. 1995;14:1970–1978. doi: 10.1002/j.1460-2075.1995.tb07189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nellen D, Burke R, Struhl G, Basler K. Direct and long-range action of a DPP morphogen gradient. Cell. 1996;85:357–368. doi: 10.1016/s0092-8674(00)81114-9. [DOI] [PubMed] [Google Scholar]
- Noselli S. JNK signaling and morphogenesis in Drosophila. Trends Genet. 1998;14:33–38. doi: 10.1016/S0168-9525(97)01320-6. [DOI] [PubMed] [Google Scholar]
- Perrimon N. Signaling pathways initiated by receptor protein tyrosine kinases in Drosophila. Curr Opin Cell Biol. 1994;6:260–266. doi: 10.1016/0955-0674(94)90145-7. [DOI] [PubMed] [Google Scholar]
- Pombo CM, Kehrl JH, Sanchez I, Katz P, Avruch J, Zon LI, Woodgett JR, Force T, Kyriakis JM. Activation of the SAPK pathway by the human STE20 homologue germinal centre kinase. Nature. 1995;377:750–754. doi: 10.1038/377750a0. [DOI] [PubMed] [Google Scholar]
- Pombo CM, Bonventre JV, Molnar A, Kyriakis J, Force T. Activation of a human Ste20-like kinase by oxidant stress defines a novel stress response pathway. EMBO J. 1996;15:4537–4536. [PMC free article] [PubMed] [Google Scholar]
- Riesgo-Escovar JR, Hafen E. Drosophila Jun kinase regulates expression of decapentaplegic via the ETS-domain protein Aop and the AP-1 transcription factor DJun during dorsal closure. Genes & Dev. 1997a;11:1717–1727. doi: 10.1101/gad.11.13.1717. [DOI] [PubMed] [Google Scholar]
- ————— Common and distinct roles of DFos and DJun during Drosophila development. Science. 1997b;278:669–671. doi: 10.1126/science.278.5338.669. [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar JR, Jenni M, Fritz A, Hafen E. The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes & Dev. 1996;10:2759–2768. doi: 10.1101/gad.10.21.2759. [DOI] [PubMed] [Google Scholar]
- Ronchi E, Treisman JE, Drostatni N, Struhl G, Desplan C. Down-regulation of the Drosophila morphogen Bicoid by the Torso receptor mediated signal transduction cascade. Cell. 1993;74:347–355. doi: 10.1016/0092-8674(93)90425-p. [DOI] [PubMed] [Google Scholar]
- Ruperte ET, Nellen MD, Affolter M, Basler K. An absolute requirement for both type II and type I receptors, punt and thick veins, for dpp signaling in vivo. Cell. 1995;80:890–898. doi: 10.1016/0092-8674(95)90292-9. [DOI] [PubMed] [Google Scholar]
- Sluss KK, Han Z, Barrett T, Davis RJ, Ip YT. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes & Dev. 1996;10:2745–2758. doi: 10.1101/gad.10.21.2745. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Rubin GM. Transposition of cloned P-elements into Drosophila germline chromosomes. Science. 1982;218:341–347. doi: 10.1126/science.6289435. [DOI] [PubMed] [Google Scholar]
- Su Y, Han J, Xu S, Cobb M, Skolnik EY. NIK is a new Ste20-related kinase that binds NCK and MEKK1 and activates the SAPK/JNK cascade via a conserved regulatory domain. EMBO J. 1997;16:1279–1290. doi: 10.1093/emboj/16.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98:81–95. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Teramoto H, Coso OA, Miyata H, Igishi T, Miki T, Gutkin JS. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. J Biol Chem. 1996;271:27225–27228. doi: 10.1074/jbc.271.44.27225. [DOI] [PubMed] [Google Scholar]
- Terracol R, Lengyel JA. The thick veins gene of Drosophila is required for dorsoventral polarity of the embryo. Genetics. 1994;138:165–178. doi: 10.1093/genetics/138.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treier M, Bohman D, Mlodzik M. Jun cooperates with the ETS domain protein Pointed to induce photoreceptor R7 fate in the Drosophila eye. Cell. 1995;83:753–760. doi: 10.1016/0092-8674(95)90188-4. [DOI] [PubMed] [Google Scholar]
- Treisman JE, Ito N, Rubin GM. misshapen encodes a protein kinase involved in cell shape control in Drosophila. Gene. 1997;186:119–125. doi: 10.1016/s0378-1119(96)00694-4. [DOI] [PubMed] [Google Scholar]
- Young PE, Richman AM, Ketchum AS, Kiehart DP. Morphogenesis in Drosophila requires nonmuscle myosin heavy chain function. Genes & Dev. 1993;7:29–41. doi: 10.1101/gad.7.1.29. [DOI] [PubMed] [Google Scholar]