

Abstract
Background
Except for terminal deletions that lead to Jacobsen syndrome, interstitial deletions involving the long arm of chromosome 11 are not frequently reported. A clinically distinct phenotype is usually observed in these cases, and no clear genotype-phenotype correlation is proposed.
Results
Here we present a case study of a 5-year-old girl with de novo submicroscopic deletion of chromosome 11q22.3 with mild mental retardation and facial dysmorphism. A standard cytogenetic analysis did not reveal any structural aberrations. In contrary, array-CGH analysis indicated a small deletion of 11q22.3.
Discussion
To our knowledge, this is the smallest 11q22.3 deletion reported in literature, containing nine RefSeq genes. Although none of the deleted genes are obvious candidates for the features observed in our patient, genes CUL5 and SLN could play a key role in the features described.
Keywords: 11q22.3 deletion, mild mental retardation, facial dysmorphism
Background
Interstitial deletions of the long arm of chromosome 11 are not often reported in literature. Usually, the terminal deletions that cause Jacobsen syndrome are well-described, whereas interstitial aberrations are less well-defined due to the band pattern similarity in this region and limited resolution offered by classical cytogenetic methods [1,2].
The clinical phenotype in 11q deletion patients usually varies depending on the size and position of the deletion. The same trend can be observed when 11q22.3 deletions are compared. No clear genotype-phenotype correlation can be observed, mainly because the deletions encompassing this region also include other bands [1-12]. The most common phenotypic features observed in these patients are mild to severe mental retardation, developmental delay, a high arched or cleft palate (in some cases with a cleft lip), and hypertelorism. A delay in speech and walking are usually present in all cases [1,2,8,10,11]. Additional features such as trigonocephaly [10,11], small hands and feet [8,11], syndactyly [1], simian crease [2,10], and others are also observed. To the best of our knowledge a case with a deletion limited to 11q22.3 has so far not been reported.
Here we present a girl with de novo submicroscopic deletion of 11q22.3, detected by array-CGH analysis (aCGH). Despite the fact that only much larger deletions have been described previously, we propose a genotype-phenotype correlation for this region. Hopefully, the discovery of new patients with microdeletions in 11q22.3 will lead to a better understanding of the roles that the deleted genes play in the clinical outcome.
Case presentation
An infant girl was born at 36 weeks gestation to a healthy 23-year-old mother and 28-year-old father. Her birth weight was 2690 g (50th centile for gestational age), her birth length was 47 cm (25th centile), her head circumference was 32.5 cm (25th centile) and her Apgar score was 9/9. After the birth there were no major concerns, although patent ductus arteriosus and haemodynamically significant atrial septal defect (ASD) had been diagnosed, and pronounced dysmorphic features had been observed. She had upward and slanted palpebral fissures with ptosis of both. This was more pronounced in the left upper palpable. Hypertelorism was also observed and the nose was small in size and saddle-shaped. She had a prominent frontal part of the skull, giving the false impression of macrocrania. Her ears were low-set, asymmetrical, triangular and slightly protuberant. Both lips were accentuated in shape and sometimes stretched in an asymmetrical manner (Figure 1). A high-arched palate was also observed. There was evidence of general muscular hypotonia and her developmental milestones were delayed. She sat up without help at 12 months and started walking at 18 months. Her speech development was also late, and only developed after she was three years old. Her social and psychical development were surprisingly good. A magnetic resonance imaging (MRI) scan of the brain showed hyperintensive changes in the white matter and hypoplastic corpus callosum. The electroencephalogram (EEG) was normal and there were no convulsions.
Figure 1.

The facial features observed in our patient include ptosis, hypertelorism, low-set dysplastic ears and a prominent forehead.
At 12 months, she suffered from serious acute pyelonephritis, and bilateral vesicoureteral reflux (VUR) was diagnosed thereafter. The ultrasound kidney volumes were normal with no major dilatation of the renal pyelon. The endoscopic treatment of the VUR was performed on two occasions. Lastly, after classical surgical treatment, the VUR was successfully treated. At that time, her neurodevelopment was equivalent to that of a 9-month-old infant.
Assessments of the child are being carried out every six months. With the exception of one case of pneumonia and one case of acute laryngitis, no additional health problems had been reported. There were no recurrent urinary tract infections.
The girl is currently 5 years old. She weighs 15.5 kg (10th centile), has a height of 110 cm (50th centile) and a head circumference of 49 cm (10th centile). The dysmorphic features are still prominent. She has lower basic muscle tone, especially in the shoulder muscles and the pelvic girdle. Her muscle strength is fairly good and her reflexes are normal. There are no major problems with fine motorics and there are no abnormal movements or contractures. Her palms, especially hypothenar, are hypoplastic and her fingers are thin. Her foot arches are prominent. Hips incline to internal rotation with knees adduction and inward feet rotation, which has no major impact on her gait. Both renal function and blood pressure are normal. At present, she is preparing for the correction of a congenital heart defect.
She is managed with a team of experts and receives special logopaedic, psychological, and defectological assistance. The girl is motivated, cooperates and strives for success and progress. She is pleasant, friendly, good-natured and no emotional or behavioural problems have been observed. Despite her relatively normal development, a psychological examination (Goodenough Draw, Gessell Drawing Test, Brunet-Lesine scale, Vineland Social Maturity Scale, Wechsler Intelligence Scale for Children - WISC) reveals some cognition deficits and borderline intellectual functioning, requiring specialised help and causing delays in her formal education. She follows and understands simple instructions, speaks in sentences, but her pronunciation is unclear. Her global intelligence quotient is 76 (WISC test). Her social maturity meets the requirements of a 5-year-old child (Vineland Social Maturity Scale). According to the Brunet-Lezine scale, her mental age is that of a 4.6 year old child.
Results
As conventional karyotyping of the proband did not reveal any structural rearrangements, the karyotype was therefore determined to be 46, XX. Molecular karyotyping was performed on the Agilent 60K platform. Data analyses indicated a 743 kb deletion of 11q22.3 (Figure 2). An aCGH analysis of the patients' parents did not show any abnormalities; thus deletion has been determined to be de novo. Confirmation of the deletion was performed by fluorescent in situ hybridisation (FISH) using BAC probe RP11-56J3. A single red signal can be seen on the normal chromosome 11 and in the nucleus, whereas the signal is absent in the chromosome with the 11q22.3 deletion (Figure 3).
Figure 2.
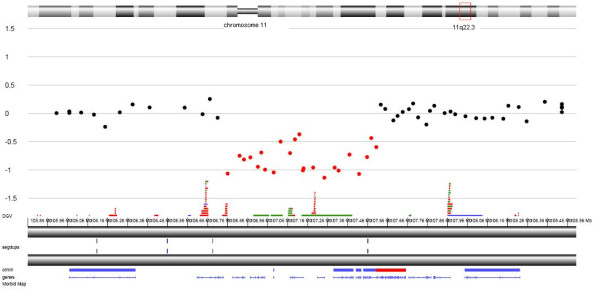
An aCGH analysis of the patient revealed a deletion of the 11q22.3 region. The breakpoints of the deletion were therefore determined to be arr 11q22.3(107349817-108093259)x1 dn (hg19; NCBI build 37).
Figure 3.
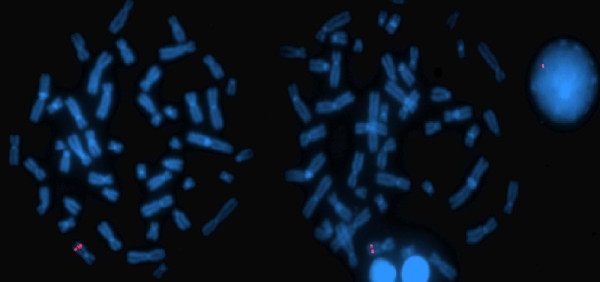
A FISH analysis of the patient was carried out using BAC probe RP11-56J3 (labelled red) encompassing the 11q22.3 region. A single red signal can be seen on the normal chromosome 11 and in the nucleus, whereas the signal is absent in the chromosome with the 11q22.3 deletion.
Discussion
In the relevant literature, deletions involving the long arm of chromosome 11 are usually associated with Jacobsen syndrome, a condition with multiple anomalies caused by the terminal deletion of this chromosome [13]. In contrast, interstitial deletions of 11q are less common and often not fine-mapped, due to the similarity between two band patterns (11q14 and 11q22) when conventional karyotyping is performed [1,2]. Furthermore, no clear genotype-phenotype correlations have been proposed in reported patients. Here, we present a new patient with de novo submicroscopic deletion of chromosome 11q22.3. The deletion was determined by aCGH (Figure 2) and confirmed by a FISH analysis (Figure 3). The clinical phenotype in our patient includes mild mental, developmental and speech delay, without emotional and behavioural difficulties. She has prominent facial dysmorphisms, ptosis, hypertelorism, low-set dysplastic ears and a prominent forehead (Figure 1). Hypoplastic corpus callosum, VUR and ASD were also diagnosed.
Much larger deletions, overlapping this region, have been described previously. To our knowledge, none of the reported cases had deletion limited only to 11q22.3; therefore, this is the smallest deletion of 11q22.3 reported in the literature. Most of the patients with an overlapping deletion of this region had mild to severe mental retardation and developmental delay, usually depending on the size and position of the deletion. A high arched or cleft palate was observed in all cases. Dysmorphic features such as trigonocephaly, microcephaly, hypertelorism, eye anomalies, hypotonia, speech delay and others were also observed [1-12].
Apart from unspecific features, such as mental and developmental delay, and eye and palate anomalies, no specific common features were observed in our case study or those previously described. Interestingly, the Decipher patient 950 with a small inherited duplication of this region (arr 11q22.3(107792125-107953547)x3 pat (hg19; NCBI build 37)) has concordant phenotypic features with our patient, including strabismus, kidney anomalies, and hypoplastic corpus callosum, all of which have not been observed in other cases.
The deletion interval in our case contains nine RefSeq genes: ALKBH8, ELMOD1, LOC643923, SLN, SLC35F2, RAB39, CUL5, ACAT1 and NPAT. The ALKBH8 gene encodes the ALKBH8 protein, which belongs to a group of human homologues of AlkB protein in E. coli. ALKBH8 is involved in the repair of methylation-induced DNA damage by oxidative demethylation [14]. Its role was described in the prevention of mutation and carcinogenesis in various types of mammalian cells [15,16]. Secondly, the ELMOD1 gene encodes a GTPase-activating protein. As ELMOD2 protein, another member of ELMO family, ELMOD1 acts on the Arf family, which are members of the Ras superfamily of proteins [17]. The LOC643923 gene encodes a hypothetical protein. Another gene, SLN, encodes a small proteolipid that catalyses the ATP-dependent transport of Ca(2+) from the cytosol into the lumen of the sarcoplasmic reticulum in muscle cells [18]. Although, Sln-null mice do not exhibit any cardiac anomalies, a study from Babu et al., 2007, demonstrated that homozygous-null mice exhibit increased cardiac contractility [19]. The SLC35F2 gene encodes a member of nucleotide sugar transporters in the Golgi apparatus and the endoplasmic reticulum [20]. A member of the Ras-like family, involved in vesicle budding, docking, and fusion in endocytosis and exocytosis pathways, is encoded by the RAB39 gene [21-24]. The CUL5 gene encodes vasopressin-activated calcium-mobilising receptor 1, which is a component of a multiple SCF-like ECS E3 ubiquitin-protein ligase complex. This complex mediates the ubiquitination and subsequent proteasomal degradation of target proteins [25,26]. The enzyme encoded by the ACAT1 gene is involved in the metabolism of acetyl-CoA. An autosomal recessive beta-ketothiolase deficiency is described as an inborn error of isoleucine catabolism caused by the disruption of this gene [27]. The last, NPAT gene, encodes the nuclear protein of the ataxia telangiectasia mutated locus. This is a housekeeping gene involved in the cell cycle. Imai et al., 1997, proposed that the NPAT gene shares the same promoter region with the ATM gene, the disruption of which causes an autosomal recessive ataxia telangiectasia. A loss of heterozygosity of this gene along with others on 11q22-q24 locus has been frequently described in various types of tumours [28].
Although none of the genes described previously indicate their direct role in the development of phenotype seen in our patient, Feng et al., 2007, reported that the CUL5 gene plays an essential role in neuron migration during cortical development [29]. The haploinsufficiency of this gene has not yet been reported in patients or knockout mice; therefore, a direct genotype-phenotype correlation is not possible and further investigation should be conducted. A lower expression of the SLN gene was described in children with congenital heart defects by Vittoius et al., 2007, but not in the association with ASD [30].
Conclusion
The majority of the patients reported previously with the deletion containing the 11q22.3 region display mental retardation and facial dysmorphism, including the patient presented here. Specific features such as strabismus, kidney anomalies, and hypoplastic corpus callosum are observed only in Decipher patient 950 and our patient. Both of these probands have aberrations which are limited only to 11q22.3.
None of the deleted genes are obvious candidates for the features observed in our patient, although genes CUL5 and SLN could play a key role in the features described. The scarcity of patients with 11q22.3 microdeletions aggravates a clear genotype-phenotype correlation. Hopefully, the discovery of new patients with microdeletions in this region will lead to a better understanding of the roles that the deleted genes play in the clinical outcome.
Materials and methods
Chromosome analysis
The chromosomal analysis was performed on metaphase chromosomes using peripheral blood lymphocytes. The chromosomes were harvested and analysed in accordance with standard cytogenetic methods.
Molecular karyotyping
Molecular karyotyping was performed on DNA extracted from peripheral blood leukocytes of both, the patient and her parents. An aCGH analysis was performed using Agilent SurePrint G3 Human CGH Microarray Kit 8 × 60K (Agilent Technologies, Santa Clara, CA, USA). The assay was performed according to the manufacturers' instructions with minor modifications. The obtained data was analysed using the arrayCGHbase software tool [31]. The DECIPHER [32] and ISCA [33] databases were used for the genotype-phenotype correlations.
FISH analysis
The fluorescent in situ hybridisation (FISH) analysis was performed using BAC probe RP11-56J3. The BAC clone was selected from the human library RPCI-11 according to the UCSC Human Genome Assembly (March 2006 release) [34].
Consent
Written informed consent was obtained from the parents of the patient for publication of this case report and accompanying images. A copy of written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
DK wrote the manuscript and carried out the molecular karyotyping and data analysis; NMV performed a clinical analysis of the patient; AZ conducted the cytogenetic analysis; and NKV coordinated the study. All the authors have read and approved the manuscript.
Contributor Information
Danijela Krgovic, Email: danijela.krgovic@ukc-mb.si.
Natasa Marcun Varda, Email: natasa.marcunvarda@amis.net.
Andreja Zagorac, Email: andreja.zagorac@ukc-mb.si.
Nadja Kokalj-Vokac, Email: nadja.kokalj-vokac@ukc-mb.si.
Acknowledgements
We would like to thank Dr. Björn Menten and people responsible in Center for Medical Genetics, Ghent, Belgium for their kind help to allow us to use their laboratory and arrayCGHbase for performing initial aCHG analysis.
References
- McPherson E, Meissner L. 11q-syndrome: review and report of two cases. Birth Defects Orig Artic Ser. 1982;18(3B):295–300. [PubMed] [Google Scholar]
- Klep-de Pater JM, de France HF, Bijlsma JB. Interstitial deletion of the long arm of chromosome 11. J Med Genet. 1985;22(3):224–6. doi: 10.1136/jmg.22.3.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taki H, Kusuda S, Ohsasa Y, Hase Y, Tsuruhara T, Yoshimura A. A case report of partial deletion of long arm of chromosome 11; del(11)(q21q23) Jpn J Hum Genet. 1983;28:179–180. [Google Scholar]
- Okamura T, Sagehashi N, Tsukagoshi T. 11q - syndrome with cleft palate. A case report. J Jpn PSR. 1988;8:353–358. [Google Scholar]
- Hori T, Masuno M, Wakazono A, Takahashi E, Katakura R, Orii T. Interstitial deletion of the long arm of chromosome 11 determined by fluorescence in situ hybridization. Jpn J Hum Genet. 1993;38(2):219–24. doi: 10.1007/BF01883713. [DOI] [PubMed] [Google Scholar]
- Hertz JM, Tommerup N, Sørensen FB, Henriques UV, Nielsen A, Therkelsen AJ. Partial deletion 11q: report of a case with a large terminal deletion 11q21-qter without loss of telomeric sequences, and review of the literature. Clin Genet. 1995;47(5):231–5. doi: 10.1111/j.1399-0004.1995.tb04302.x. [DOI] [PubMed] [Google Scholar]
- Ono J, Hasegawa T, Sugama S, Sagehashi N, Hase Y, Oku K, Endo Y, Ohdo S, Ishikiriyama S, Tsukamoto H, Okada S. Partial deletion of the long arm of chromosome 11: ten Japanese children. Clin Genet. 1996;50(6):474–8. doi: 10.1111/j.1399-0004.1996.tb02715.x. [DOI] [PubMed] [Google Scholar]
- De Pater JM, Ippel PF, Bijlsma JB, Van Nieuwenhuizen O. Interstitial deletion 11q. Case report and review of the literature. Genet Couns. 1997;8(4):335–9. [PubMed] [Google Scholar]
- de Lonlay-Debeney P, de Blois MC, Bonnet D, Amiel J, Abadie V, Picq M, Lyonnet S, Sidi D, Munnich A, Vekemans M, Cormier-Daire V. Ebstein anomaly associated with rearrangements of chromosomal region 11q. Am J Med Genet. 1998;80(2):157–9. doi: 10.1002/(SICI)1096-8628(19981102)80:2<157::AID-AJMG12>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Horelli-Kuitunen N, Gahmberg N, Eeva M, Palotie A, Järvelä I. Interstitial deletion of bands 11q21-- > 22.3 in a three-year-old girl defined using fluorescence in situ hybridization on metaphase chromosomes. Am J Med Genet. 1999;86(5):416–9. doi: 10.1002/(SICI)1096-8628(19991029)86:5<416::AID-AJMG4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Syrrou M, Fryns JP. Interstitial deletion of chromosome 11 (q22.3-q23.2) in a boy with mild developmental delay. J Med Genet. 2001;38(9):621–4. doi: 10.1136/jmg.38.9.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdeva R, Sears JE, Rychwalski PJ. A novel case of bilateral high myopia, cataract, and total retinal detachment associated with interstitial 11q deletion. Ophthalmic Genet. 2010;31(2):84–8. doi: 10.3109/13816811003628833. [DOI] [PubMed] [Google Scholar]
- Grossfeld PD, Mattina T, Lai Z, Favier R, Jones KL, Cotter F, Jones C. The 11q terminal deletion disorder: a prospective study of 110 cases. Am J Med Genet A. 2004;129A(1):51–61. doi: 10.1002/ajmg.a.30090. [DOI] [PubMed] [Google Scholar]
- Shimada K, Nakamura M, Anai S, De Velasco M, Tanaka M, Tsujikawa K, Ouji Y, Konishi N. A novel human AlkB homologue, ALKBH8, contributes to human bladder cancer progression. Cancer Res. 2009;69(7):3157–64. doi: 10.1158/0008-5472.CAN-08-3530. [DOI] [PubMed] [Google Scholar]
- Spelsberg TC, Webster RA, Pikler GM. Chromosomal proteins regulate steroid binding to chromatin. Nature. 1976;262(5563):65–7. doi: 10.1038/262065a0. [DOI] [PubMed] [Google Scholar]
- Olsson M, Lindahl T. Repair of alkylated DNA in Escherichia coli. Methyl group transfer from O6-methylguanine to a protein cysteine residue. J Biol Chem. 1980;255(22):10569–71. [PubMed] [Google Scholar]
- Bowzard JB, Cheng D, Peng J, Kahn RA. ELMOD2 is an Arl2 GTPase-activating protein that also acts on Arfs. J Biol Chem. 2007;282(24):17568–80. doi: 10.1074/jbc.M701347200. [DOI] [PubMed] [Google Scholar]
- Odermatt A, Taschner PE, Scherer SW, Beatty B, Khanna VK, Cornblath DR, Chaudhry V, Yee WC, Schrank B, Karpati G, Breuning MH, Knoers N, MacLennan DH. Characterization of the gene encoding human sarcolipin (SLN), a proteolipid associated with SERCA1: absence of structural mutations in five patients with Brody disease. Genomics. 1997;45(3):541–53. doi: 10.1006/geno.1997.4967. [DOI] [PubMed] [Google Scholar]
- Babu GJ, Bhupathy P, Timofeyev V, Petrashevskaya NN, Reiser PJ, Chiamvimonvat N, Periasamy M. Ablation of sarcolipin enhances sarcoplasmic reticulum calcium transport and atrial contractility. Proc Natl Acad Sci USA. 2007;104(45):17867–72. doi: 10.1073/pnas.0707722104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida N, Kawakita M. Molecular physiology and pathology of the nucleotide sugar transporter family (SLC35) Pflugers Arch. 2004;447(5):768–75. doi: 10.1007/s00424-003-1093-0. [DOI] [PubMed] [Google Scholar]
- Simons K, Zerial M. Rab proteins and the road maps for intracellular transport. Neuron. 1993;11(5):789–99. doi: 10.1016/0896-6273(93)90109-5. [DOI] [PubMed] [Google Scholar]
- Somsel Rodman J, Wandinger-Ness A. Rab GTPases coordinate endocytosis. J Cell Sci. 2000;113(Pt 2):183–92. doi: 10.1242/jcs.113.2.183. [DOI] [PubMed] [Google Scholar]
- Gonzalez L Jr, Scheller RH. Regulation of membrane trafficking: structural insights from a Rab/effector complex. Cell. 1999;96(6):755–8. doi: 10.1016/S0092-8674(00)80585-1. [DOI] [PubMed] [Google Scholar]
- Chen T, Han Y, Yang M, Zhang W, Li N, Wan T, Guo J, Cao X. Rab39, a novel Golgi-associated Rab GTPase from human dendritic cells involved in cellular endocytosis. Biochem Biophys Res Commun. 2003;303(4):1114–20. doi: 10.1016/S0006-291X(03)00482-0. [DOI] [PubMed] [Google Scholar]
- Byrd PJ, Stankovic T, McConville CM, Smith AD, Cooper PR, Taylor AM. Identification and analysis of expression of human VACM-1, a cullin gene family member located on chromosome 11q22-23. Genome Res. 1997;7(1):71–5. doi: 10.1101/gr.7.1.71. [DOI] [PubMed] [Google Scholar]
- Burnatowska-Hledin MA, Spielman WS, Smith WL, Shi P, Meyer JM, Dewitt DL. Expression cloning of an AVP-activated, calcium-mobilizing receptor from rabbit kidney medulla. Am J Physiol. 1995;268(6 Pt 2):F1198–210. doi: 10.1152/ajprenal.1995.268.6.F1198. [DOI] [PubMed] [Google Scholar]
- Korman SH. Inborn errors of isoleucine degradation: a review. Mol Genet Metab. 2006;89(4):289–99. doi: 10.1016/j.ymgme.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Imai T, Sugawara T, Nishiyama A, Shimada R, Ohki R, Seki N, Sagara M, Ito H, Yamauchi M, Hori T. The structure and organization of the human NPAT gene. Genomics. 1997;42(3):388–92. doi: 10.1006/geno.1997.4769. [DOI] [PubMed] [Google Scholar]
- Feng L, Allen NS, Simo S, Cooper JA. Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev. 2007;21(21):2717–30. doi: 10.1101/gad.1604207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vittorini S, Storti S, Parri MS, Cerillo AG, Clerico A. SERCA2a, phospholamban, sarcolipin, and ryanodine receptors gene expression in children with congenital heart defects. Mol Med. 2007;13(1-2):105–11. doi: 10.2119/2006-00054.Vittorini. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menten B, Pattyn F, De Preter K, Robbrecht P, Michels E, Buysse K, Mortier G, De Paepe A, van Vooren S, Vermeesch J, Moreau Y, De Moor B, Vermeulen S, Speleman F, Vandesompele J. arrayCGHbase: an analysis platform for comparative genomic hybridization microarrays. BMC Bioinformatics. 2005;6:124. doi: 10.1186/1471-2105-6-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DECIPHER v5.1. http://decipher.sanger.ac.uk/
- ISCA Consortium and Public Database. https://www.iscaconsortium.org/
- UCSC Genome Browser (University of California Santa Cruz, March 2006 release) http://genome.ucsc.edu