

Abstract
The RNA degradosome is a multiprotein complex required for the degradation of highly structured RNAs. We have developed a method for reconstituting a minimal degradosome from purified proteins. Our results demonstrate that a degradosome-like complex containing RNase E, PNPase, and RhlB can form spontaneously in vitro in the absence of all other cellular components. Moreover, ATP-dependent degradation of the malEF REP RNA by the reconstituted, minimal degradosome is indistinguishable from that of degradosomes isolated from whole cells. The Rne protein serves as an essential scaffold in the reconstitution process; however, RNase E activity is not required. Rather, Rne coordinates the activation of RhlB dependent on a 3′ single-stranded extension on RNA substrates. A model for degradosome-mediated degradation of structured RNA is presented with its implications for mRNA decay in Escherichia coli.
Keywords: RNA degradosome, 3′ exonuclease, E. coli, DEAD box, RNA helicase
Many important cellular processes, including DNA replication, RNA biogenesis, and protein synthesis, are carried out by large macromolecular complexes or machines (Alberts 1998). To date, RNA decay and processing machines have been isolated from bacteria (Carpousis et al. 1994), plants (Hayes et al. 1996), and yeast (Margossian et al. 1996; Mitchell et al. 1997) and are inferred in mammalian cells (Mitchell et al. 1997). These findings suggest that multiple ribonucleases and other RNA modifying enzymes could act coordinately and/or simultaneously against a single RNA and may explain the concerted ‘all or none’ nature of mRNA decay exhibited by most mRNAs (for review, see Coburn and Mackie 1999).
The RNA degradosome from Escherichia coli is a high-molecular-weight complex of four major and three minor protein components. The major components include Rne/Ams, the major endoribonuclease, and polynucleotide phosphorylase (PNPase), a 3′ exonuclease involved in the terminal stages of mRNA decay (Carpousis et al. 1994; Py et al. 1994). The degradosome also contains stoichiometric amounts of RhlB, a putative DEAD-box RNA helicase, and enolase, a glycolytic enzyme with no known significance in mRNA decay (Carpousis et al. 1994; Py et al. 1994, 1996; Miczak et al. 1996). RhlB, enolase, and PNPase interact with distinct, discrete regions in the carboxy-terminal third of the Rne protein (Vanzo et al. 1998). It is unknown whether Rne, RhlB, PNPase, and enolase cycle between free and complexed forms. However, only 5%–10% of the total cellular enolase appears to copurify with the degradosome (Py et al. 1996). Substoichiometric components of the degradosome include GroEL, DnaK, and polyphosphate kinase (Miczak et al. 1996; Blum et al. 1997). Although the two former proteins may play a role in the assembly of the degradosome complex in vivo, neither RNase E nor PNPase requires GroEL or DnaK for activity (Cormack et al. 1993; Coburn and Mackie 1998). The function of polyphosphate kinase in mRNA decay, if any, remains unclear (Blum et al. 1997).
Members of the superfamily of proteins that contain the signature DEAD-box motif (Asp-Glu-Ala-Asp) are involved in pre-tRNA processing, pre-mRNA splicing, translation, and ribosomal biogenesis (Schmid and Linder 1992). The association of a putative RNA helicase with the degradosome is particularly exciting because both RNase E and PNPase are specific for single-stranded RNA and are impeded by RNA secondary structure. Interestingly, two putative DEVH-box RNA helicases, Ski2p and Mtr4p/Dob1p (Anderson and Parker 1998; de la Cruz et al. 1998), functionally interact with the yeast exosome complex demonstrating a conserved role for RNA helicase proteins in mRNA degradation and processing machines. One model to rationalize the role of RhlB in the RNA degradosome suggests that RhlB would unwind secondary structure to permit access by RNase E to cleavage sites that are normally occluded (Miczak et al. 1996). Alternatively, RhlB would alleviate structural impediments to PNPase, a model supported by a requirement for ATP by the degradosome in the degradation of REP (repetitive extragenic palindrome) stem–loop structures (Py et al. 1996). Moreover, antibodies raised against the RhlB protein inhibit PNPase-mediated degradation through the malEF REP stem–loop structure (Py et al. 1996). Unfortunately, there is no definitive assay for RhlB, and strains containing a deletion of the rhlB gene are apparently inviable (cited in Py et al. 1996). Furthermore, polyadenylation of bacterial mRNAs complicates understanding the role of RhlB within the degradosome. In particular, polyadenylation of RNA1 or the rpsT mRNA promotes the degradation of highly structured 3′ termini by PNPase, independent of the action of RhlB or, indeed, the presence of the degradosome (Xu and Cohen 1995; Coburn and Mackie 1998).
We have developed a method for reconstituting a minimal degradosome from purified proteins that is active against well-defined RNA substrates, including the malEF intercistronic region and the 3′ end of the rpsT mRNA. Our data prompt a model in which Rne coordinates the activation of RhlB dependent on a single-stranded 3′ extension on RNA substrates, independently of the endoribonucleolytic activity of the Rne protein.
Results
Physical reconstitution of a minimal RNA degradosome
Although many proteins copurify with degradosomes (Carpousis et al. 1994; Py et al. 1994, 1996; Miczak et al. 1996; Blum et al. 1997), we elected to reconstitute these particles from the Rne, RhlB, and Pnp proteins whose functional importance in vitro has been demonstrated (Xu and Cohen 1995; Py et al. 1996; Coburn and Mackie 1998). (Rne, Pnp, and RhlB refer to the respective products of the rne, pnp, and rhlB genes.) We have purified Rne, RhlB, and Pnp from overexpressing strains (Fig. 1A, lanes 4,6,8) and have assayed whether these proteins would associate in vitro, using coimmunoprecipitation experiments (see Materials and Methods). The presence of the Rne, Pnp and RhlB proteins in complexes was detected by Western blotting.
Figure 1.


Physical reconstitution of a minimal RNA degradosome. (A) Purified degradosome complexes and recombinant proteins used throughout this study. The following samples were denatured, separated through SDS-PAGE, and stained with Coomassie brilliant blue: (lane 1) protein molecular weight markers (Bio-Rad); (lane 2) wild-type degradosomes isolated from a wild-type strain (15 μg); (lane 3) pnp-7 degradosomes isolated from a strain containing the pnp-7 allele (12 μg); (lane 4) Rne protein (1.5 μg); (lane 5) RneΔN208 protein (1.5 μg); (lane 6) Pnp protein (1.5 μg); (lane 7) Rnb protein (1.5 μg); (lane 8) RhlB protein (1.5 μg). (B) Combinations of purified Rne, Pnp, and RhlB proteins (1 μg each) were incubated in a 40-μl volume for 20 min at 30°C. Complex formation was assayed by coimmunoprecipitation with anti-Rne, anti-Pnp, or preimmune antisera and detection by Western blotting with the appropriate antibodies as described in Materials and Methods. (Lane 1) Recombinant protein markers (100–200 ng); (lane 2) mock immunoprecipitation of the Rne/Pnp/RhlB complex with preimmune sera; (lane 3) coimmunoprecipitation of the Rne/Pnp/RhlB complex with anti-Rne antibodies; (lane 4) coimmunoprecipitation of an Rne/Pnp subcomplex with anti-Rne antibodies; (lane 5) coimmunoprecipitation of an Rne/RhlB subcomplex with anti-Rne antibodies; (lanes 6,7) a mixture of RhlB and Pnp proteins was subjected to immunoprecipitation with anti-Rne or anti-Pnp antibodies, respectively. The stoichiometry of Rne, Pnp, and RhlB in the degradosome was estimated from Coomassie blue-stained SDS–polyacrylamide gels using known amounts of each of the three components to generate standard curves. This estimation is not consistently reflected by Western analysis due to differences in the transfer efficiency of each protein, antibody titer, and exposure times.
After incubation in reconstitution conditions, Rne, Pnp, and RhlB can be coimmunopurified by antibodies raised against the Rne protein (Fig. 1B, lane 3). This result shows that the purified recombinant proteins are able to reassociate into a degradosome-like complex. The three proteins were not detected in ‘mock’ immunoprecipitates with preimmune serum (Fig. 1B, lane 2) demonstrating that the precipitation of the complex was dependent on the anti-Rne antibodies. Formation of Rne/Pnp and Rne/RhlB subcomplexes in vitro was also assessed by coimmunoprecipitation with anti-Rne antibodies and Western blotting as described above. Subcomplexes between the Rne protein and Pnp or between Rne and RhlB can form in the absence of any of the other components of the degradosome (Fig. 1B, lanes 4,5). These results clearly indicate that each of the degradosome components can assemble onto the Rne scaffold spontaneously in the absence of any of the other components in agreement with data obtained in vivo (Kido et al. 1996; Vanzo et al. 1998). Based on Coomassie-blue staining of SDS–polyacrylamide gels, we have estimated the relative molar ratio of the Rne/Pnp/RhlB proteins in purified degradosomes to be 1:1.5:1. Taking into account the differing titers of the antibodies and efficiency of detection (see the legend to Fig. 1B), the ratio of Rne:Pnp:RhlB in immunoprecipitated complexes is in good agreement with this estimate.
To determine whether the Rne protein is required for formation of minimal degradosome complexes, we attempted reconstitution experiments in its absence. The products formed in such incubations were assayed by coimmunoprecipitation with antibodies raised against either the Rne or Pnp protein. The RhlB antiserum cross-reacts with PNPase (A.J. Carpousis, pers. comm.) and was of insufficient titer to perform immunoprecipitations. Only Pnp can be recovered from a partial reconstitution containing Pnp and RhlB using anti-Pnp antibodies (Fig. 1B, lane 7). There was no detectable nonspecific binding of either RhlB or Pnp to anti-Rne antibodies (Fig. 1B, lane 6).
Functional reconstitution of a minimal RNA degradosome
To assess the activity of complexes formed in vitro, we assayed their ability to degrade structured RNA substrates using either a fragment spanning the REP sequence of the malEF intercistronic region (Fig. 2) or the 3′ end of the rpsT mRNA (see below). Initially, we tested the ATP dependence of the complex formed in a total reconstitution performed with Rne, Pnp, and RhlB. The 375-nucleotide malEF RNA was incubated with a reconstituted complex containing purified Rne, Pnp, and RhlB proteins (see Fig. 1B, lane 3) in the absence of ATP (Fig. 3A). The malEF RNA was rapidly digested to generate a previously characterized intermediate (∼305 nucleotides), referred to as the RSR (REP stabilized RNA) (McLaren et al. 1991; see Fig. 2). This intermediate is stable and remains fully resistant to digestion for 60 min (Fig. 3A). A second, faint intermediate (∼340 nucleotides), denoted by an asterisk (*) in Py et al. (1996), also appears transiently within the first 5 min of the assay (Fig. 3A). Although the structure of the malEF RNA has yet to be determined empirically, the formation of the two degradative intermediates is presumed to be the result of PNPase stalling at the base of stable stem–loop structures as shown in Figure 2. This behavior fully mimics that of native degradosomes (Py et al. 1996; see Fig. 5A, below).
Figure 2.

Secondary structure of the malEF REP RNA. The 375-nucleotide malEF intragenic spacer region was folded with RNAdraw (http:///rnadraw.base8.se). The first 207 nucleotides of the malEF RNA are depicted with a line. The large REP stem–loop structure spans residues 211–298. Previously characterized intermediates generated by the stalling of exonucleases 3′ to the base of stable stem–loop structures are denoted by the asterisk (*) and RSR (McLaren et al. 1991; Py et al. 1996).
Figure 3.

Functional reconstitution of a minimal RNA degradosome. The 375-nucleotide malEF RNA substrate (see Fig. 2) was incubated with reconstituted complexes containing recombinant Rne (750 ng/ml), Pnp (1.5 μg/ml), and/or RhlB (750 ng/ml) as indicated above each panel. As described in Materials and Methods, all incubations were performed at 30°C in the presence of 10 mm sodium phosphate and 3 mm ATP unless otherwise specified (see margins above each panel). Aliquots were removed at the times indicated (in minutes). The positions of the malEF substrate and the two major intermediates, * and RSR, are indicated with arrowheads in the margin at right of each panel. (A–C) The complex reconstituted with Rne, RhlB, and Pnp was incubated with the malEF RNA alone in the absence of ATP (A) or in the presence of ATP (B) or in the presence of 3 mm ATPγS (C). (D) The reconstituted complex was incubated with the malEF RNA in the absence of exogenous phosphate. (E–G) Subcomplexes of Rne and RhlB (E) or Rne and Pnp (F) were incubated with the malEF RNA in the presence of ATP. A mixture of RhlB and Pnp was incubated with the malEF RNA substrate in the presence of ATP (G).
Figure 5.

3′ exonucleases must function in cis with the Rne/RhlB RNA helicase. Degradosomes purified from a wild-type strain (wt degradosomes) or from a strain containing the pnp-7 allele of the gene encoding PNPase (pnp-7 degradosomes) were assayed in the presence of ATP against the malEF RNA substrate at 30°C as described in Materials and Methods. The products of the reaction were analyzed as described in the legend to Fig. 3. The positions of the malEF substrate and the two major intermediates, * and RSR, are indicated with arrowheads in the margin at right of each panel. (A) Kinetics of digestion of the malEF RNA by the wild-type degradosomes (5 μg/ml). (B) Kinetics of digestion of the malEF RNA by the pnp-7 degrado somes (5 μg/ml). (C) Digestion of themalEF RNA by purified PNPase (1.5 μg/ml). (D) Digestion of the malEF RNA by pnp-7 degradosomes (5 μg/ml) supplemented with recombinant PNPase (1.5 μg/ml). (E) Digestion of the malEF RNA by a partially reconstituted complex containing Rne (750 ng/ml) and RhlB (750 ng/ml) supplemented with recombinant RNase II (Rnb; 750 ng/ml).
In an otherwise identical assay containing ATP, the malEF RNA was initially digested to generate the * and RSR intermediates as before (Fig. 3B). However, digestion continued in the presence of ATP resulting in complete degradation of the RSR intermediate by the reconstituted degradosome after 20 min (cf. the 60- and 30-min time points in Fig. 3, A and B, respectively). Furthermore, in the presence of the nonhydrolyzable ATP analog, ATPγS, the RSR intermediate remained largely intact and resistant to degradation over 60 min of incubation (Fig. 3C). Thus, ATPγS cannot substitute for ATP in the decay of the RSR intermediate catalyzed by the reconstituted complex. In this respect, the ATP requirement of the reconstituted complex is identical to that of native degradosomes for degradation of the malEF REP sequence (Py et al. 1996). Moreover, these results indicate that decay of the malEF RNA can be functionally reconstituted by a minimal number of degradosome components. In particular, the Rne, Pnp, and RhlB proteins alone are necessary and sufficient for complete decay of the REP sequence. We term this complex formed during reconstitution with these proteins a ‘minimal degradosome.’
To assess the interdependence of one enzyme on another in reconstituted minimal degradosomes, partial reconstitution experiments were performed with various combinations of the Rne, Pnp, and RhlB proteins. The essential role of PNPase in the degradation of the REP sequences was confirmed in two ways. First, a complete reconstitution was performed and the activity of the complex was assayed with the 375-nucleotide malEF RNA substrate in the absence of exogenous phosphate. Under such conditions, the full-length malEF RNA is fully resistant to degradation and is stable for at least 60 min (Fig. 3D). Second, the malEF RNA is also resistant to degradation during a 60-min incubation with a complex reconstituted with the Rne and RhlB proteins alone (Fig. 3E). These results demonstrate that formation of the RSR intermediate and its subsequent ATP-activated degradation depends directly on the presence of PNPase in the reconstitution and on its activity in the subsequent assay.
To test a requirement for the RhlB protein, the 375-nucleotide substrate was incubated with a reconstituted complex formed from the Rne and Pnp proteins alone (Fig. 3F). The full-length RNA is rapidly converted into the RSR intermediate. This intermediate remains largely resistant to further digestion as no more than 50% of the RSR is degraded over 60 min. When the RhlB and Pnp proteins were incubated together under conditions favoring reconstitution and assayed with the full-length malEF RNA in the presence of ATP, the malEF RNA was shortened by PNPase to generate the RSR intermediate as before (Fig. 3G). This intermediate remained stable for at least 60 min despite the presence of ATP and RhlB. These results demonstrate that PNPase alone or complexed to Rne can initiate exonucleolytic attack on the malEF substrate to generate the RSR intermediate but that RhlB and Rne are essential for the subsequent ATP-activated degradation of the RSR intermediate.
RNase E activity is not required for RhlB-activated decay of the malEF REP sequence by PNPase
The endonucleolytic activity of RNase E resides in the amino-terminal half of the Rne protein (Taraseviciene et al. 1995; McDowall and Cohen 1996). The RneΔN208 and RneΔN408 proteins, which initiate at codons 208 and 408, respectively, lack the putative S1 RNA-binding domain (residues 35–125) and exhibit greatly reduced endonuclease activity. RneΔN208 cleaves the 9S RNA precursor >100-fold slower than the full-length Rne protein, whereas RneΔN408 is catalytically inactive against the 9S RNA substrate (X. Miao et al., unpubl.).
Amino-terminally truncated Rne proteins were substituted for the full-length Rne protein in reconstitution experiments. Portions of each mixture were assayed for complex formation by coimmunoprecipitation (data not shown) and for activity against the malEF RNA substrate (see below). In assays containing the RneΔN208, Pnp, and RhlB proteins in the absence of ATP, the full-length malEF REP RNA is rapidly digested to generate the RSR intermediate as observed previously for minimal degradosomes containing the full-length Rne protein (cf. Figs. 3A and 4A). This intermediate remains fully intact and resistant to degradation after 60 min of digestion (Fig. 4A). In otherwise identical assays containing ATP, the RSR intermediate is rapidly degraded once formed (Fig. 4B). Similar results were obtained in reconstitutions containing the RneΔN408, RhlB, and Pnp proteins (Fig. 4C). In both cases, the kinetics of formation and disappearance of the RSR were similar to those observed with reconstituted complexes containing the full-length Rne protein (cf. Fig. 4B,C with Fig. 3B). These results demonstrate that neither the putative S1 domain nor RNase E activity are required for the ATP-dependent 3′ degradation of the RSR intermediate.
Figure 4.

RNase E activity is not required for ATP-activated degradation of the malEF RNA. The 375-nucleotide malEF RNA substrate was incubated with a reconstituted complex containing recombinant RneΔN208 (750 ng/ml), Pnp (1.5 μg/ml), and RhlB (750 ng/ml) in the absence of ATP (A) or in the presence of ATP (B). A reconstituted complex containing RneΔN408 (750 ng/ml), Pnp (1.5 μg/ml), and RhlB (750 ng/ml) was incubated with the malEF RNA in the presence of ATP (C). The products of the reaction were analyzed as described in the legend to Fig. 3. The positions of the malEF substrate and the two major intermediates, * and RSR, are indicated with arrowheads in the margin at right of each panel.
3′ exonucleases cannot function in trans with the RhlB helicase
Because PNPase exhibits 3′ exonuclease activity independent of the degradosome, we have tested whether it or another 3′ exonuclease, RNase II, can function in RhlB-dependent degradation of the malEF REP sequence without being physically associated with the degradosome. Degradosomes prepared from a strain harboring the pnp-7 allele that abolishes the 3′ exonucleolytic activity of the Pnp protein (Reiner 1969) display no difference in the quality, composition, or protein stoichiometry from wild-type degradosomes as judged by Coomassie-blue staining of SDS–polyacrylamide gels (Fig. 1A; cf. lanes 2 and 3). In particular, there is a full complement of the mutant Pnp-7 protein in the pnp-7 degradosomes. Although their RNase E activity is normal (data not shown), pnp-7 degradosomes are unable to digest the full-length 375-nucleotide malEF RNA substrate that remains unaltered after 60 min of incubation in the presence of ATP (Fig. 5B). Wild-type degradosomes assayed in parallel behaved as expected (Fig. 5A).
Assays using pnp-7 degradosomes were supplemented with purified recombinant Pnp protein at the same concentrations used in reconstitution experiments. This amount of Pnp alone can degrade the malEF RNA rapidly, within the first 5 min, to generate the RSR intermediate (Fig. 5C). Likewise, purified Pnp, in the presence of pnp-7 degradosomes, catalyzed the rapid formation of the RSR intermediate (Fig. 5D). The RSR intermediate so formed remained relatively resistant to digestion as no more than 55% of this intermediate was completely degraded after 60 min of incubation. This result clearly contrasts with that obtained for the wild-type RNA degradosome (Fig. 5A) or that obtained for the reconstituted minimal degradosome (Fig. 3B). In their simplest interpretation, these results suggest that PNPase cannot function efficiently in trans with the Rne/RhlB complex and that PNPase does not readily cycle between a free and complexed form. Alternatively, the pnp-7 form of PNPase may either be unable to respond to RhlB or dissociate from the degradosome, in agreement with our data. A novel pnp allele (G507D) behaves as if its binding to the degradosome is tighter than wild type, consistent with the latter explanation (García-Mena et al. 1999).
In a second experiment, the unfractionated products of a reconstitution performed with purified Rne, RhlB, and RNase II were assayed against the full-length malEF RNA in the presence of ATP. After 60 min of digestion, ∼50% of the 375-nucleotide substrate was digested by RNase II to generate the * intermediate that remained completely stable throughout the assay (Fig. 5E). Only very small amounts of the RSR intermediate were generated by RNase II (Fig. 5E). Thus, RNase II cannot substitute for PNPase under conditions in which the Rne/RhlB ATPase would be functionally active, suggesting that once a substrate is activated by RhlB, it must be passed in cis to PNPase.
RhlB-activated degradation is dependent on a single-stranded 3′ end
The major product of RNase E cleavage of the rpsT mRNA encoding ribosomal protein S20 releases a 147-nucleotide fragment coterminal with the 3′ end of the molecule (Mackie 1991). The decay of this fragment requires PNPase and poly(A) polymerase I both in vivo and in vitro (Mackie 1989; Coburn and Mackie 1996b, 1998). In this regard it differs from the malEF REP RNA that contains a largely single-stranded 3′ terminus (Fig. 2) and whose degradation is independent of polyadenylation in vitro. We have investigated whether both polyadenylation and RhlB activity are required for complete degradation of the 3′ end of the rpsT mRNA by a minimal degradosome under the assay conditions used by Blum et al. (1999).
The substrate rpsT(268–447) mimics the 147-nucleotide fragment generated by RNase E (Mackie et al. 1997; Coburn and Mackie 1998). We incubated this substrate with the minimal reconstituted degradosome (Rne, RhlB, and Pnp) in the presence of ATP. The rpsT(268–447) RNA substrate remains largely resistant to degradation, as 50% of the starting material is intact after 60 min of digestion (Fig. 6A). To test the effect of polyadenylation, we added a single 30-nucleotide poly(A) tail to the 3′ end of the substrate to form rpsT[268–447–poly(A)30]. In the absence of ATP, the reconstituted minimal RNA degradosome rapidly removes this poly(A) tail from the substrate to generate a relatively stable intermediate that terminates 3′ to the base of the rpsT terminator stem–loop (Fig. 6B). At least 50% of the intermediate persists after 60 min of incubation (Fig. 6B). In contrast, the rpsT[268–447–poly(A)30] RNA is rapidly digested in assays containing the reconstituted minimal RNA degradosome supplemented with ATP (Fig. 6C). In this case, however, almost no intermediate is formed and almost all the substrate is completely degraded within 20 min of incubation (Fig. 6, cf. the 20-min time points in C with those in B). These results show that efficient degradation of the rpsT mRNA by PNPase requires both polyadenylation of the substrate and activation by RhlB and ATP. Moreover, the requirement for polyadenylation (Fig. 6, cf. A with C) demonstrates that RhlB-activated degradation of structured RNA is dependent on a single-stranded 3′ end.
Figure 6.

A single-stranded 3′ end stimulates significantly the action of the minimal reconstituted degradosome. Derivatives of the rpsT mRNA were incubated with a reconstituted complex containing Rne (750 ng/ml), Pnp (575 ng/ml), and RhlB (750 ng/ml) in the presence of 3 mm ATP and 10 mm sodium phosphate as described in Materials and Methods. The products of the reaction were analyzed as described in the legend to Fig. 3. The positions of the RNA substrates and an intermediate corresponding to the removal of the poly(A)30 tail are indicated with arrowheads in the margin at right of each panel. A schematic diagram of the substrates and intermediates is also shown in the margin at right of each panel. (A) Kinetics of digestion in the absence of a poly(A) tail. The substrate, rpsT(268–447), encompasses residues 268–447 of the rpsT mRNA terminating with a flush stem–loop structure at the natural Rho-independent terminator (see text). (B) Kinetics of digestion of a polyadenylated substrate, rpsT(268–447)–poly(A)30, in the absence of ATP. The rpsT(268–447) substrate contains an additional 30 adenylate residues at its 3′ end as described in Materials and Methods. (C) Kinetics of digestion of a polyadenylated substrate in the presence of ATP. The substrate is identical to that in B.
Discussion
Reconstitution of a minimal RNA degradosome complex
Our work demonstrates that a physical complex containing the Rne, RhlB, and Pnp proteins can form spontaneously in vitro. This complex, which we term the minimal RNA degradosome, assembles and is active in the absence of any other cellular cofactors including enolase and the minor components of the degradosome: polyphosphate kinase, DnaK, and GroEL. The minimal reconstituted degradosome is fully functional as the ATP-dependent degradation of the malEF REP sequence by the reconstituted minimal degradosome is indistinguishable from that of degradosomes isolated from whole cells. These results clearly demonstrate that a degradosome-like complex containing Rne, RhlB, and Pnp alone is necessary and sufficient for the ATP-activated degradation of the malEF REP sequence. The Rne protein is an essential scaffold in the reconstitution process, but its activity is not required. Reconstitution shows that the Rne protein is bifunctional (as an endonuclease and an organizing platform for 3′ decay), a finding fully consistent with other data (Taraseviciene et al. 1995; Kido et al. 1996; McDowall and Cohen 1996; Lopez et al. 1999). The development of a simple method of reconstitution as described here should open the way to systematic structure-function investigations of the essential components of this macromolecular complex. Additionally, the reconstituted system provides a basis to assess the potential role of auxiliary proteins that may regulate the activity of the degradosome.
RhlB-activated RNA degradation by PNPase
The presence of a putative RNA helicase is a common feature of macromolecular complexes for RNA processing and decay. Extensive kinetic and structural investigations of DNA helicases provide an excellent framework for modelling the mechanism of action of RhlB-mediated RNA degradation by the degradosome (shown in Fig. 7). Both models below predict that ATP binding and hydrolysis drive conformational changes within the RhlB helicase that result in differential affinities for single-stranded and double-stranded RNA.
Figure 7.
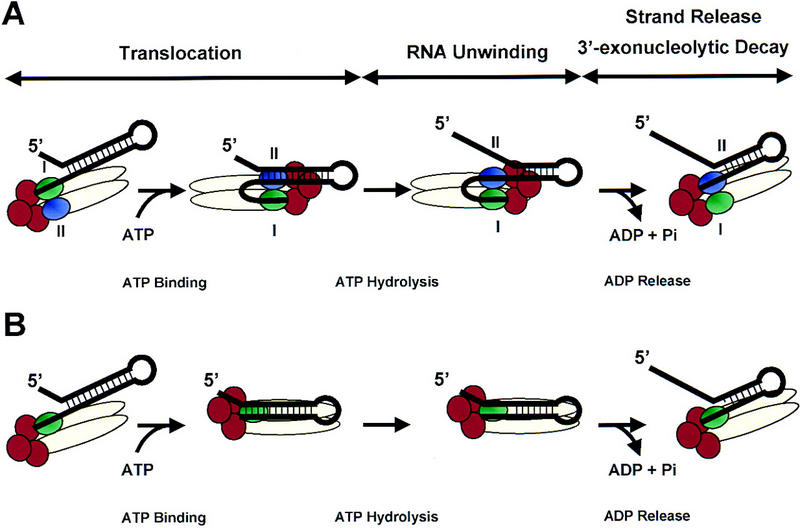
Two potential mechanisms for RhlB-activated degradation of structured RNA by the degradosome. (A) A model prompted by the active, rolling model for DNA unwinding and translocation by the Rep DNA helicase (Wong and Lohman 1992; Korolev et al. 1997). The Rne protein (beige oval) serves as a scaffold to coordinate the action of the RhlB helicase (subunit I, green oval; subunit II, purple oval) with PNPase (red circles). This allows single-stranded RNA generated by the RNA helicase to be passed directly to the 3′ exonuclease PNPase (see text). The nucleotide-bound state of RhlB would result in a cycle of conformational and RNA affinity changes that result in translocation, RNA unwinding, and strand release/exonucleolytic degradation. (B) A second model based on the inchworm mechanism proposed for the PcrA DNA helicase (Velanker et al. 1999). In this case, RhlB acts as a monomeric protein (shown in green) that can bind both single- and double-stranded RNA. Degradation of single-stranded RNA by PNPase (red circles) takes place during the translocation step.
One model, prompted by the active, rolling model for DNA unwinding and translocation by the E. coli Rep DNA helicase (Wong and Lohman 1992; Korolev et al. 1997), implies that RhlB is present as a dimer or higher oligomer in the degradosome (Fig. 7A). Single-stranded 3′ ends, generated either by endonucleolytic cleavage downstream of a stem–loop or by the addition of a poly(A) tail, provide a platform for subunit I (shown in green) of the non-nucleotide bound form of RhlB to load onto an RNA substrate. During translocation, ATP binding permits subunit II of RhlB (purple) to bind to the RNA duplex. Hydrolysis of ATP accompanies the breakage of several hydrogen bonds within the stem, resulting in an overall weakening of the stem–loop. In the ADP-bound form, both subunits I and II remain bound to single-stranded RNA. Release of ADP permits subunit I to pass single-stranded RNA directly to the PNPase (shown in red) for its degradation. If the stability of the stem–loop structure has been weakened below a specific threshold, PNPase may proceed through the remaining stem–loop structure. If, however, the stem–loop remains resistant to degradation, then additional rounds of ATP-activated RNA unwinding may be required.
A second model is based on the inchworm mechanism for DNA unwinding (Yarranton and Gefter 1979) recently proposed for the PcrA DNA helicase (Velanker et al. 1999). As in the active, rolling model, ATP binding permits RhlB to bind to the RNA duplex, whereas ATP hydrolysis results in destabilization of the stem–loop (Fig. 7B). RNA degradation takes place during the translocation cycle as RhlB ‘inches’ ahead of the 3′ exonuclease exposing new single-stranded RNA. In contrast to the former model that predicts cooperativity between two subunits of a dimeric protein, the inchworm mechanism suggests that each RhlB monomer can interact with single-stranded and double-stranded RNA simultaneously. Nonetheless, the inchworm mechanism is also consistent with an oligomeric enzyme structure. Conceivably, RNA-binding domains within Rne or Pnp may contribute to the RNA-unwinding mechanism.
Alternative models can be imagined in which RhlB plays a more subtle role in RNA degradation by mediating global RNA structural changes or by disrupting protein–RNA and/or protein–protein interactions within the degradosome. Rearrangements within the degradosome–RNA complex mediated by RhlB and ATP hydrolysis may be required for recycling the degradosome or in switching the activity of the degradosome from a 5′-dependent endonuclease (Mackie 1998) to a highly processive 3′ dependent exonuclease.
RNA unwinding and polyadenylation
Degradation of the highly structured 3′ end of the rpsT mRNA shows a strong dependence on polyadenylation in vivo (Coburn and Mackie 1998). Moreover, the action of PAP I and PNPase alone is necessary and sufficient for complete degradation of the 3′ 147-nucleotide fragment generated by RNase E. Under our previous assay conditions, RhlB was not required for degradation of the 3′ end of the rpsT mRNA (Coburn and Mackie 1998). Subsequently, Blum et al. (1999) have shown that addition of a single poly(A) tail to RNA substrates can significantly enhance the action of the degradosome.
These results can be reconciled readily. The conditions chosen by Blum et al. (1999) promote RhlB dependence by retaining stalled PNPase–RNA enzyme–substrate complexes for sufficient times for RhlB to act (data not shown). The higher ionic strengths in our previous assay conditions result in the rapid dissociation of PNPase at stem–loop barriers and readenylation of the substrate to permit reassociation of PNPase. In vivo, we believe that both RhlB activity and polyadenylation of 3′ ends are required for efficient degradation of the most structured mRNA fragments. Polyadenylation facilitates loading of degradative enzymes (Xu and Cohen 1995; Coburn and Mackie 1998; Blum et al. 1999), whereas RhlB increases the processivity of PNPase, thereby minimizing the number of adenylation/deadenylation cycles and conserving ATP. The machinery required for 3′-end degradation, including an RNA helicase, one or more exonucleases, an organizing platform, and, in some cases, polyadenylation, is unexpectedly dynamic. Nonetheless, the paradigm of the degradosome is reflected by comparable molecular machines in chloroplasts (Hayes et al. 1996), yeasts (Margossian et al. 1996; Mitchell et al. 1997), and higher eukaryotes (Mitchell et al. 1997).
Materials and methods
Bacterial strains and plasmids
The E. coli strain CF881 [F− lac argA trp recB1009 (xthA–pnc) Δrna] was obtained from Dr. M. Cashel, strain RD100 (lacZ met pnp-7 relA1 rna trpD9778) from Dr. M. Pearson, strain MV1190 [(Δlac–pro) thi supE Δ(srl–recA)306::Tn10 (tetR) (F′ traD36 proAB lacIqZΔM15)] from Bio-Rad, whereas plasmids pET-11 and pET-24b and the host strain BL21(DE3) were obtained from Novagen. Plasmid pCH77 (McLaren et al. 1991) containing the intergenic spacer region of the malE–malF mRNA under the control of a T7 promoter was provided by Dr. C.F. Higgins. Plasmid pGM102, containing the ams/rne/hmp-1 gene (Cormack et al. 1993); pJG175, encompassing residues 268–447 of the rpsT gene; and plasmid pJG175p(A), a derivative of pJG175 that contains a 30-bp poly(A)–poly(T) tract located immediately 3′ to the base of the Rho-independent rpsT terminator have been described previously (Mackie and Genereaux 1993; Mackie et al. 1997; Coburn and Mackie 1998).
Construction of overexpressing strains
The following oligonucleotide primers were synthesized based on the previously published sequence of the rhlB gene (accession no. X56310; Kalman et al. 1991): F(rhlB), 5′-GGCGCGGATCCAGGAGGTCCACACTATGAGCAAAACACAT-3′ and R(rhlB), 5′-GGCGCGGATCCTTAACCTGAACGACGACGATT-3′. The forward primer F(rhlB) contains a Shine–Dalgarno sequence 5′ to the rhlB start codon such that the rhlB gene could be subsequently overexpressed using the T7 RNA polymerase encoded by BL21(DE3) (Studier et al. 1990). The predicted coding sequence (nucleotides 233–1378) of the rhlB gene (mmrA) from E. coli was amplified from the genomic DNA of strain MV1190 by the polymerase chain reaction (Sambrook et al. 1989). The amplified products were cleaved with the restriction enzyme BamHI (underlined above) and ligated into the unique BamHI site of pET-11 to generate plasmid pGC300. The orientation of the 1.1-kb fragment in the recombinant plasmid was verified by restriction mapping and DNA sequencing. Plasmid pGC300 was used to transform BL21(DE3) to yield strain GC300.
Purification of the recombinant RhlB protein
Cultures of GC300 were grown in 500 ml of LB media (Sambrook et al. 1989) at 30°C with good aeration. Induction, harvest, and cell lysis were performed as described previously for PNPase (Coburn and Mackie 1998). The cell lysate was clarified by centrifugation at 30,000g to generate the S-30 fraction. The overexpressed protein was largely insoluble in low salt at pH 7.5, presumably due to isoelectric precipitation (calculated pI = 7.5). Thus, purification of the recombinant protein was performed at pH ≥8.5. Approximately 60 mg of the S-30 fraction was loaded onto a 0.75 × 10-cm column of Affi-Gel blue agarose (Bio-Rad) equilibrated with five column volumes of buffer A (25 mm Tris-HCl at pH 8.5, 5% glycerol, 0.1 mm EDTA, 1 mm DTT) containing 100 mm NaCl. After the column had been washed with five column volumes of Buffer A, RhlB was eluted from the column with a 30-ml gradient of KCl (0–3 m KCl) at ∼1.7 m KCl. Pooled fractions containing RhlB were concentrated. A portion was subjected to gel filtration on a 1 × 48-cm column of Bio-Gel A (0.5 m; Bio-Rad) in buffer A containing 50 mm NaCl. Pooled fractions containing RhlB were concentrated and loaded onto a 0.75 × 10-cm column of Affi-Gel heparin (Bio-Rad) equilibrated with five column volumes of buffer A containing 50 mm NaCl. After the column had been washed with five volumes of starting buffer, RhlB was eluted with a 30-ml gradient of NaCl (0–1 m NaCl) at 500 mm NaCl. Fractions containing RhlB were pooled and concentrated before being loaded at 1 ml/min onto a 1-ml Resource-Q column (Amersham-Pharmacia) equilibrated with several volumes of starting buffer. RhlB was eluted from the column with a 30-ml gradient of NaCl (50–500 mm NaCl) at 120–150 mm NaCl. Fractions containing RhlB were pooled and stored at −70°C. The presence of RhlB in various fractions was monitored qualitatively by SDS–polyacrylamide gels.
Other enzymes
The Rne, RneΔN208, and RneΔN408 proteins were purified by preparative gel electrophoresis followed by electroelution as described previously with slight modifications (Cormack et al. 1993). Following renaturation from 6 m guanidine–HCl, proteins were dialyzed extensively into 25 mm HEPES-NaOH (pH 7.6), 100 mm NaCl, 1 mm EDTA, and 1 mm DTT and concentrated with an Ultra-Free centrifugal filter device (Millipore). Protein concentrations were estimated by comparison with a BSA standard on a Coomassie blue-stained SDS–polyacrylamide gel.
The recombinant RNase II and Pnp (PNPase) proteins were purified by conventional chromatographic techniques as described previously (Coburn and Mackie 1996a, 1998). The recombinant proteins were ∼95% homogeneous, with only some minor contaminating bands, as judged by Coomassie blue-stained SDS–polyacrylamide gels. Degradosomes were prepared from strain CF881 (wild-type degradosomes) or from strain RD100 (pnp-7 degradosomes) as described previously (Coburn and Mackie 1998). Protein concentrations were determined by the Bradford protein assay (Bio-Rad) using BSA as a standard.
Preparation of RNA substrates and enzyme assays
The 375-nucleotide malEF REP RNA was synthesized from pCH77 linearized with EcoRI. Synthesis of two RNA substrates derived from the 3′ end of the rpsT mRNA, rpsT(268–447) RNA and rpsT[(268–447–poly(A)30 RNA], was directed from pJG175 or pJG175p(A) that had been linearized previously with DraI or XbaI, respectively. Transcription was performed with T7 RNA polymerase in the presence of [α-32P]CTP as described (Cormack and Mackie 1992; Mackie and Genereaux 1993).
Assays were assembled in a 40-μl reaction containing 0.8 pmoles (20 nm) of RNA in a buffer containing 20 mm Tris-HCl (pH 7.5), 1.5 mm DTT, 1 mm MgCl2, 20 mm KCl, and 10 mm sodium phosphate as described previously (Py et al. 1994). ATP was added to a final concentration of 3 mm. The purified recombinant Rne, Pnp, and RhlB proteins were added last to final concentrations specified in the figure legends, and the reactions were performed at 30°C. Samples were withdrawn at various times and quenched with three volumes of loading buffer containing 90% formamide, 22 mm Tris, 22 mm boric acid, 0.5 mm EDTA, 0.1% xylene cyanol FF, and 0.1% bromophenol blue. The reaction products were separated electrophoretically through 6% polyacrylamide gels containing 8 m urea and visualized by autoradiography or with a Molecular Dynamics PhosphorImager system.
Immunoprecipitations and protein blotting
Immunoprecipitation experiments were performed essentially as described (Vanzo et al. 1998). Anti-Rne or anti-Pnp rabbit polyclonal antisera (1 μl) were cross-linked to protein A beads (GIBCO BRL) in CLB buffer (20 mm Na2HPO4, 5 mm NaH2PO4, 0.2 m NaCl, 0.5 mm EDTA) containing 1% glutaraldehyde. The cross-linked beads were washed three times with 10 volumes of CLB and an additional three times with IP buffer (100 mm Tris-HCl at pH 8.5, 0.25 m NaCl, 0.05% genapol X-080, 0.1 mm EDTA).
Various combinations of the purified Rne, RhlB, and Pnp proteins (1 μg each) were incubated in a 40-μl volume at 30°C under the assay conditions described above. After, 20 min, half of the reaction was diluted 10-fold into IP buffer and incubated with anti-Rne or anti-Pnp beads for 2 hr at 4°C. The beads were washed three times with IP buffer, and the immunopurified complexes were released by heating at 55°C for 15 min in SDS sample buffer (120 mm Tris-HCl, 3% SDS, 10% glycerol) lacking DTT. The eluted proteins were subsequently boiled in SDS sample buffer containing 50 mm DTT for 5 min and were subjected to analysis by Western blotting as described previously (Rouleau et al. 1994). Blotted proteins were detected with rabbit polyclonal anti-Rne antibodies (1:20,000), anti-Pnp (1:10,000), or anti-RhlB antibodies (1:1000). Visualization of the primary antibody was performed with goat anti-rabbit antibodies conjugated to horseradish peroxidase (GIBCO BRL) using the ECL Plus detection system (Amersham-Pharmacia) and exposure to Kodak X-Omat AR film.
Acknowledgments
We thank Dr. C.F. Higgins for plasmid pCH77 and Dr. A.J. Carpousis for antisera directed against the RhlB protein. This work was funded by an operating grant (to G.A.M.) from the Medical Research Council of Canada. G.A.C. received a fellowship from the U.B.C. Killam Endowment.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gamackie@unixg.ubc.ca; FAX (604) 822-5227.
References
- Alberts B. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell. 1998;92:291–294. doi: 10.1016/s0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- Anderson JSJ, Parker R. The 3′ to 5′ degradation of yeast mRNA is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J. 1998;17:1497–1506. doi: 10.1093/emboj/17.5.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum E, Py B, Carpousis AJ, Higgins CF. Polyphosphate kinase is a component of the Escherichia coli RNA degradosome. Mol Microbiol. 1997;26:387–398. doi: 10.1046/j.1365-2958.1997.5901947.x. [DOI] [PubMed] [Google Scholar]
- Blum E, Carpousis AJ, Higgins CF. Polyadenylation promotes degradation of 3′-structured RNA by the Escherichia coli mRNA degradosome in vitro. J Biol Chem. 1999;274:4009–4016. doi: 10.1074/jbc.274.7.4009. [DOI] [PubMed] [Google Scholar]
- Carpousis AJ, Van Houwe G, Ehretsmann C, Krisch HM. Copurification of E. coli RNase E and PNPase: Evidence for a specific association between two enzymes important for RNA processing and degradation. Cell. 1994;76:889–900. doi: 10.1016/0092-8674(94)90363-8. [DOI] [PubMed] [Google Scholar]
- Coburn GA, Mackie GA. Overexpression, purification and properties of Escherichia coli ribonuclease II. J Biol Chem. 1996a;271:1048–1053. doi: 10.1074/jbc.271.2.1048. [DOI] [PubMed] [Google Scholar]
- ————— Differential sensitivities of portions of the mRNA for ribosomal protein S20 to 3′-exonucleases dependent on oligoadenylation and RNA secondary structure. J Biol Chem. 1996b;271:15776–15781. doi: 10.1074/jbc.271.26.15776. [DOI] [PubMed] [Google Scholar]
- ————— Reconstitution of the degradation of the mRNA for ribosomal protein S20 with purified enzymes. J Mol Biol. 1998;279:1061–1074. doi: 10.1006/jmbi.1998.1842. [DOI] [PubMed] [Google Scholar]
- ————— Degradation of mRNA in Escherichia coli: An old problem with some new twists. Prog Nucleic Acids Res Mol Biol. 1999;62:55–108. doi: 10.1016/s0079-6603(08)60505-x. [DOI] [PubMed] [Google Scholar]
- Cormack RS, Mackie GA. Structural requirements for the processing of Escherichia coli 5S ribosomal RNA by RNase E in vitro. J Mol Biol. 1992;228:1078–1090. doi: 10.1016/0022-2836(92)90316-c. [DOI] [PubMed] [Google Scholar]
- Cormack RS, Genereaux JL, Mackie GA. RNase E activity is conferred by a single polypeptide: Overexpression, purification and properties of the ams/rne/hmp-1 gene product. Proc Natl Acad Sci. 1993;90:9006–9010. doi: 10.1073/pnas.90.19.9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz J, Kressler D, Tollervey D, Linder P. Dob1p (Mtr4p) is a putative ATP-dependent RNA helicase required for 3′ end formation of 5.8S rRNA in Saccharomyces cerevisiae. EMBO J. 1998;17:1128–1140. doi: 10.1093/emboj/17.4.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Mena J, Das A, Sánchez-Trujillo A, Portier C, Montañez C. A novel mutation in the KH domain of polynucleotide phosphorylase affects autoregulation and mRNA decay in Escherichia coli. Mol Microbiol. 1999;33:235–248. doi: 10.1046/j.1365-2958.1999.01451.x. [DOI] [PubMed] [Google Scholar]
- Hayes R, Kudla J, Schuster G, Gabay L, Maliga P, Gruissem W. Chloroplast mRNA 3′-end processing by a high molecular weight protein complex is regulated by nuclear encoded RNA-binding proteins. EMBO J. 1996;15:1132–1141. [PMC free article] [PubMed] [Google Scholar]
- Kalman M, Murphy H, Cashel M. rhlB, a new Escherichia coli K-12 gene with an RNA helicase-like protein sequence motif, one of at least five such possible genes in a procaryote. New Biologist. 1991;3:886–895. [PubMed] [Google Scholar]
- Kido M, Yamanaka K, Mitani T, Niki H, Ogura T, Hiraga S. RNase E polypeptides lacking the carboxy-terminal half suppress a mukB mutation in Escherichia coli. J Bacteriol. 1996;177:491–496. doi: 10.1128/jb.178.13.3917-3925.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolev S, Hsieh J, Gauss GH, Lohman TM, Waksman G. Major domain swiveling revealed by the crystal structure of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell. 1997;90:635–647. doi: 10.1016/s0092-8674(00)80525-5. [DOI] [PubMed] [Google Scholar]
- Lopez PJ, Marchand I, Joyce SA, Dreyfus M. The C-terminal half of RNase E, which organizes the Escherichia coli degradosome, participates in mRNA degradation but not rRNA processing in vivo. Mol Microbiol. 1999;33:188–199. doi: 10.1046/j.1365-2958.1999.01465.x. [DOI] [PubMed] [Google Scholar]
- Mackie GA. Stabilization of the 3′ one-third of Escherichia coli ribosomal protein S20 mRNA in mutants lacking polynucleotide phosphorylase. J Bacteriol. 1989;171:4112–4120. doi: 10.1128/jb.171.8.4112-4120.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Specific endonucleolytic cleavage of the mRNA for ribosomal protein S20 of Escherichia coli requires the product of the ams gene in vivo and in vitro. J Bacteriol. 1991;173:2488–2497. doi: 10.1128/jb.173.8.2488-2497.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Ribonuclease E is a 5′-end-dependent endonuclease. Nature. 1998;395:720–723. doi: 10.1038/27246. [DOI] [PubMed] [Google Scholar]
- Mackie GA, Genereaux JL. The role of RNA structure in determining RNase E-dependent cleavage sites in the mRNA for ribosomal protein S20 in vitro. J Mol Biol. 1993;234:998–1012. doi: 10.1006/jmbi.1993.1654. [DOI] [PubMed] [Google Scholar]
- Mackie GA, Genereaux JL, Masterman SK. Modulation of the activity of RNase E in vitro by RNA sequences and secondary structures 5′ to cleavage sites. J Biol Chem. 1997;272:609–616. [PubMed] [Google Scholar]
- Margossian SP, Li H, Zassenhaus HP, Butow HP. The DexH box protein Suv3p is a component of a yeast mitochondrial 3′ to 5′ exonuclease complex that suppresses group I intron toxicity. Cell. 1996;84:199–209. doi: 10.1016/s0092-8674(00)80975-7. [DOI] [PubMed] [Google Scholar]
- McDowall KJ, Cohen SN. The N-terminal domain of the rne gene product has RNase E activity and is non-overlapping with the arginine-rich RNA-binding site. J Mol Biol. 1996;255:349–355. doi: 10.1006/jmbi.1996.0027. [DOI] [PubMed] [Google Scholar]
- McLaren RS, Newbury SF, Dance GSC, Causton HC, Higgins CF. mRNA degradation by processive 3′-exoribonucleases in vitro and the implication for mRNA decay in vivo. J Mol Biol. 1991;221:81–95. [PubMed] [Google Scholar]
- Miczak A, Kaberdin VR, Wei CL, Lin-Chao S. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc Natl Acad Sci. 1996;93:3865–3869. doi: 10.1073/pnas.93.9.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P, Petfalski E, Shevchenko A, Mann M, Tollervey D. The exosome: A conserved eukaryotic RNA processing complex containing multiple 3′-5′ exoribonucleases. Cell. 1997;91:457–466. doi: 10.1016/s0092-8674(00)80432-8. [DOI] [PubMed] [Google Scholar]
- Py B, Causton H, Mudd EA, Higgins CF. A protein complex mediating mRNA turnover in Escherichia coli. Mol Microbiol. 1994;14:717–729. doi: 10.1111/j.1365-2958.1994.tb01309.x. [DOI] [PubMed] [Google Scholar]
- Py B, Higgins CF, Carpousis AJ. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature. 1996;381:169–172. doi: 10.1038/381169a0. [DOI] [PubMed] [Google Scholar]
- Reiner AM. Isolation and mapping of polynucleotide phosphorylase mutants of Escherichia coli. J Bacteriol. 1969;97:1431–1436. doi: 10.1128/jb.97.3.1431-1436.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau M, Smith RJ, Bancroft JB, Mackie GA. Purification, properties, and subcellular localization of foxtail mosaic potexvirus 26-kDa protein. Virology. 1994;204:254–265. doi: 10.1006/viro.1994.1530. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. In: Molecular cloning: A laboratory manual. Ford N, Nolan C, Ferguson M, editors. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schmid SR, Linder P. D-E-A-D protein family of putative RNA helicases. Mol Microbiol. 1992;6:283–292. doi: 10.1111/j.1365-2958.1992.tb01470.x. [DOI] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to express cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Taraseviciene L, Björk GR, Uhlin BE. Evidence for an RNA-binding region in the Escherichia coli processing endoribonuclease RNase E. J Biol Chem. 1995;270:26391–26398. doi: 10.1074/jbc.270.44.26391. [DOI] [PubMed] [Google Scholar]
- Vanzo NF, Li YS, Py B, Blum E, Higgins CF, Raynal LC, Krisch H, Carpousis AJ. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes & Dev. 1998;12:2770–2781. doi: 10.1101/gad.12.17.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velanker SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- Wong I, Lohman TM. Allosteric effects of nucleotide cofactors on Escherichia coli REP helicase-DNA binding. Science. 1992;256:350–355. doi: 10.1126/science.256.5055.350. [DOI] [PubMed] [Google Scholar]
- Xu F, Cohen SN. RNA degradation in Escherichia coli is regulated by 3′-adenylation and 5′-phosphorylation. Nature. 1995;374:180–183. doi: 10.1038/374180a0. [DOI] [PubMed] [Google Scholar]
- Yarranton GT, Gefter ML. Enzyme-catalyzed DNA unwinding: Studies on Escherichia coli rep DNA helicase. Proc Natl Acad Sci. 1979;76:1658–1662. doi: 10.1073/pnas.76.4.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]