

Abstract
Although most cells undergo growth arrest during hypoxia, endothelial cells and placental cytotrophoblasts proliferate in response to low O2. We demonstrate that proliferation of embryonic multilineage hematopoietic progenitors is also regulated by a hypoxia-mediated signaling pathway. This pathway requires HIF-1 (HIF-1α/ARNT heterodimers) because Arnt−/− embryoid bodies fail to exhibit hypoxia-mediated progenitor proliferation. Furthermore, Arnt−/− embryos exhibit decreased numbers of yolk sac hematopoietic progenitors. This defect is cell extrinsic, is accompanied by a decrease in ARNT-dependent VEGF expression, and is rescued by exogenous VEGF. Therefore, “physiologic hypoxia” encountered by embryos is essential for the proliferation or survival of hematopoietic precursors during development.
Keywords: ARNT, HIF, hypoxia, hematopoiesis, VEGF, embryoid body, ES cells
Low O2 tension (hypoxia) is a condition commonly associated with pathology. Mammals adapt to hypoxia on an organismal level by increasing expression of erythropoietin (EPO) resulting in increased red blood cell production (Semenza et al. 1991), and vascular endothelial growth factor (VEGF) to promote increased vascularization of affected tissues (Shweiki et al. 1992; Forsythe et al. 1996). On a cellular level, mammals adapt to decreased O2 by increasing expression of glycolytic enzymes (Bunn and Poyton 1996; Semenza et al. 1996; Wenger and Gassmann 1997) and glucose transporters (Bashan et al. 1992) for increased anaerobic respiration.
Whereas the growth and division of many cell types is suppressed until O2 tensions return to normoxic levels (20% O2) (Graeber et al. 1996; Carmeliet et al. 1998), certain cell types must grow and proliferate in response to decreased O2. These include placental cytotrophoblasts (Genbacev et al. 1997), which form the maternal–fetal interface in the womb (Rodesch et al. 1992; Fischer and Bavister 1993), and vascular endothelial cells, which proliferate to form new capillaries in hypoxic tissues (Phillips et al. 1995). A critical component of the hypoxic response machinery is the bHLH–PAS transcription factor complex hypoxia-inducible factor 1 (HIF-1). HIF-1 activates the expression of genes involved in a broad spectrum of adaptive responses to oxygen deprivation ranging from basic metabolism to angiogenesis and erythropoiesis (Bunn and Poyton 1996; Wenger and Gassmann 1997; Maltepe and Simon 1998), and may impact cell-cycle regulation by interacting with and stabilizing the tumor supressor protein p53 (An et al. 1998). HIFs are obligate heterodimers comprised of the bHLH–PAS proteins HIF-1α (or HIF-2α) and the arylhydrocarbon receptor nuclear translocator (ARNT; HIF-1β). All components are constitutively expressed, although HIF-1α and HIF-2α are quickly degraded under normoxia and stabilized under hypoxia (Salceda and Caro 1997; Wiesener et al. 1998), allowing for HIF activity specifically under hypoxic conditions.
Before establishment of a circulatory system capable of delivering oxygenated blood to the embryo, mammalian development occurs in an environment exhibiting O2 concentrations within the hypoxic range (Rodesch et al. 1992; Fischer and Bavister 1993). In E8.5–E18 mouse embryos, HIF-1α protein is detectable, demonstrating the presence of a hypoxic environment (Iyer et al. 1998). In embryonic stem (ES) cells lacking HIF subunits, the hypoxic transcriptional response is ablated, and animals lacking HIF-1α or ARNT exhibit an embryonic lethality by E9.5 or E10.5, respectively (Kozak et al. 1997; Maltepe et al. 1997; Carmeliet et al. 1998; Iyer et al. 1998; Ryan et al. 1998). Specifically, Arnt−/− embryos exhibit defects in blood vessel formation in the yolk sac, branchial arches, and placenta (Kozak et al. 1997; Maltepe et al. 1997). These results suggest that HIF-1-mediated hypoxic gene regulation is important for proper vascular development.
Vascular endothelial cells and hematopoietic stem cells are thought to arise from a bipotential hemangioblast (Choi et al. 1998), based on spatial and temporal association during development, as well as common expression of many cytokines, cytokine receptors, and transcription factors (Ferrara et al. 1996; Shalaby et al. 1997). Because endothelial cells are known to proliferate under hypoxia, we wished to determine whether the proliferation and expansion of hematopoietic progenitors is also stimulated by low O2. We report here that hematopoietic progenitors proliferate in response to hypoxia in an ARNT-dependent manner. This requirement for ARNT is cell extrinsic, in that Arnt−/− ES cells contribute competitively to all hematopoietic lineages in chimeric animals. Importantly, decreased hematopoietic progenitor numbers in Arnt−/− embryoid bodies (EBs) can be rescued with exogenous VEGF. These data implicate hypoxic VEGF production as a possible mechanism for both endothelial cell and hematopoietic progenitor cell proliferation in the developing embryo.
Results and Discussion
To determine whether hypoxia stimulates hematopoietic progenitor number, we differentiated ES cells in vitro in 3% O2 to form EBs, from which hematopoietic progenitors were enumerated in colony forming unit (CFU) assays (Keller et al. 1993; Kennedy et al. 1997) (Fig. 1a). Methylcellulose cultures were scored for erythrocyte (E), macrophage (M), granulocyte–erythrocyte–megakaryocyte–macrophage (GEMM), granulocyte–macrophage (GM), and granulocyte (G) CFU (Fig. 1b). As shown in Figure 1a, hypoxia induced a significant increase (P < 0.006 to 0.04) in most CFU types generated by wild-type EBs, similar to results seen previously with human bone marrow (Maeda et al. 1986). To test whether the observed hypoxia-induced progenitor expansion is ARNT (HIF-1) dependent, Arnt−/− ES cells were used in the same assay. In contrast to wild-type controls, no hypoxic stimulation of Arnt−/− progenitor numbers was detected (Fig. 1a). These results suggest that hypoxia, within the range encountered by a developing mammalian embryo, stimulates expansion of hematopoietic progenitors in an ARNT-dependent manner.
Figure 1.
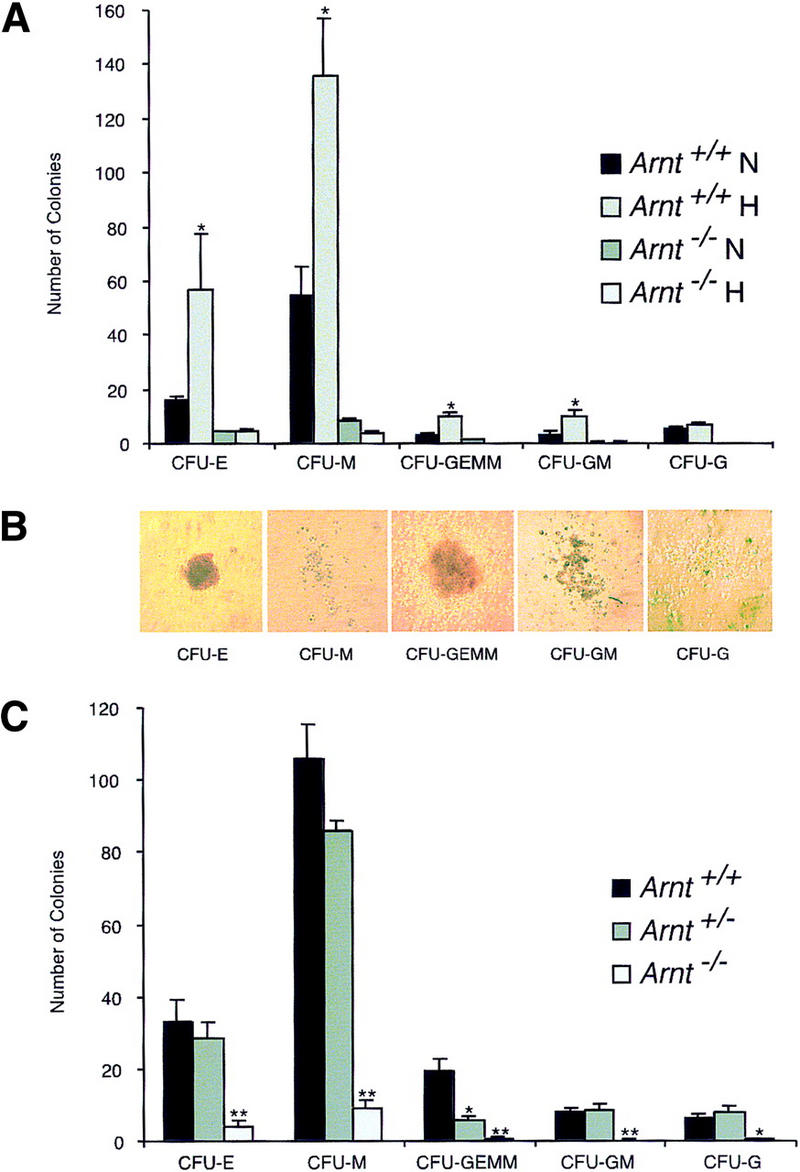
In vitro differentiation of ES cells and replating of hematopoietic progenitors. (a) Hypoxia. ES cells were differentiated for 9 days under 20% O2, 5% CO2, and 75% N2 (N) or 3% O2, 5% CO2, and 92% N2 (H) and CFU scored 6–7 days after replating under normoxic conditions. A significant increase (* P < 0.05) was seen in Arnt+/+ CFU-E, CFU-M, CFU-GEMM, and CFU-GM cultured under hypoxia. No increase was seen from Arnt−/− EBs under hypoxia. (b) Representative CFU in methylcellulose (original magnification; 10x). (c) Normoxia. ES cells were differentiated for 9 days and CFU scored 6–7 days after replating. CFU-E, CFU-M, CFU-GEMM, and CFU-GM (** P < 0.005) and CFU-G (* P <0.05) were all reduced in Arnt−/− EBs. Also, Arnt+/− CFU-GEMM showed a significant decrease (* P < 0.05) compared to Arnt+/+ (n = 4).
Normoxic progenitor levels from Arnt−/− EBs were also significantly decreased as compared to those derived from wild-type EBs (Fig. 1a). EBs are three-dimensional structures that exhibit O2 gradients due to their relatively large size (Gassmann et al. 1996). Thus, even when cultured under normoxic conditions (20% O2) EBs can contain regions of mild hypoxia. To confirm that the progenitor defect in Arnt−/− EBs was due to an inability to respond to this mild hypoxia, rather than a toxic consequence of gene targeting and subsequent selection procedures, Arnt+/− ES cells that had been subjected to the same selection protocol (Mortensen et al. 1992) as the Arnt−/− ES cells were also compared in this assay. Because Arnt+/− ES cells do not exhibit significant defects in hypoxia responses (Maltepe et al. 1997), these cells provide a control for cell viability. As shown in Figure 1c, Arnt−/− ES cells generated significantly fewer CFU-E, CFU-M, CFU-GEMM, CFU-GM, and CFU-G progenitors (P < 8.7 × 10−5 to 0.01) at 20% O2 than wild-type or heterozygous ES cells. These data suggest a primary hematopoietic defect in Arnt−/− embryoid bodies that is not simply a result of in vitro manipulation.
To ascertain whether ARNT-mediated hypoxia responses are also required for the expansion of hematopoietic progenitors in vivo, Arnt+/− animals were mated to yield Arnt+/+, Arnt+/− , and Arnt−/− offspring. Because Arnt−/− animals exhibit embryonic lethality by E10.5 (Kozak et al. 1997; Maltepe et al. 1997), E9.5 embryos were analyzed. The production of extraembryonic yolk sac blood islands marks the beginning of hematopoiesis and vasculogenesis in the mouse embryo. Many E9.5 Arnt−/− embryos lack blood-filled vitelline vessels, suggesting a possible deficiency in blood cell maturation in addition to the previously described vascular defects (Maltepe et al. 1997). Hematopoietic colony formation assays performed with Arnt+/+, Arnt+/−, and Arnt−/− E9.5 yolk sacs are shown in Figure 2a. A statistically significant decrease in the number of CFU-E, CFU-M, CFU-GEMM, CFU-GM, and CFU-G progenitors was observed in Arnt−/− yolk sacs (P < 2.2 × 10−5 to 0.005). Interestingly, Arnt+/− mice also generated significantly fewer (∼50%) CFU-GEMM and CFU-GM than Arnt+/+ animals (P < 0.02 to 0.03). This finding was reminiscent of the 50% reduction in CFU-GEMM observed after in vitro differentiation of the Arnt+/− EBs (see Fig. 1c). To ensure that the hematopoietic defects seen in the Arnt−/− embryos were not due to the stunted growth and apoptotic cell death observed in some E9.5 Arnt−/− embryos (data not shown), we performed yolk sac progenitor assays on E8.5 embryos that appear morphologically indistinct from their age-matched Arnt+/+ and Arnt+/− littermates. Only CFU-E could be properly enumerated at this earlier developmental stage as other CFU progenitors are produced at very low numbers (Olson et al. 1995). Like E9.5 embryos, the E8.5 Arnt−/− embryos showed a significant decrease in hematopoietic progenitor number (Fig. 2b; P < 0.0005). These data indicate that ARNT is required for the production of normal numbers of yolk sac hematopoietic progenitors in vivo. Furthermore, the in vitro differentiation data confirm the primary nature of this hematopoietic defect, as placental or other vascular defects are circumvented during in vitro EB culture, and therefore, do not induce secondary defects, as may occur in E9.5 Arnt−/− embryos.
Figure 2.


Yolk sac hematopoietic colony formation assays. (a) E9.5 yolk sac colonies from Arnt+/+ (n = 12), Arnt+/− (n = 22), and Arnt−/− (n = 11) embryos are shown. All Arnt−/− CFU showed significant decreases (** P < 0.005) when compared to Arnt+/+ CFU, as did the CFU-GEMM and CFU-GM of Arnt+/− yolk sacs (* P < 0.05). (b) E8.5 yolk sac colonies from Arnt+/+ (n = 16), Arnt+/− (n = 36), and Arnt−/− (n = 12) embryos are depicted. Arnt−/− CFU-E showed significant decreases (** P < 0.0005) when compared to Arnt+/+ or Arnt+/− CFU. Non-CFU-E contain cells of the myeloid lineages, and Mixed contain both erythroid and myeloid cells.
To establish whether the ARNT-mediated hematopoietic defect was intrinsic to CFU progenitors, we injected Arnt+/− and Arnt−/− ES cells (129 strain) into wild-type C57BL/6 blastocysts and assayed chimeric animals for CFU formation in adult bone marrow (Robb et al. 1996; Wang et al. 1998) (Fig. 3a). The degree of ES cell contribution was initially estimated by coat color, and more precisely determined by Southern blot analysis of bone marrow DNA. Although the progenitors represent a small subset of total bone marrow cells, this assessment of ES cell contribution is the most technically feasible. Femur and tibial bone marrow was extracted from 6-week-old animals and cultured in methylcellulose in the presence or absence of G418, which selects for neomycin resistance (neor) introduced during the original ES cell targeting. Thus, the number of neor clones derived from these animals reflects the relative contribution of the Arnt+/− or Arnt−/− hematopoietic progenitors. Arnt+/− ES cells behave like Arnt+/+ ES cells, with the exception of a modest but significant decrease in CFU-GEMM (Fig. 1c), indicating that Arnt+/− ES cells should contribute to progenitor populations like Arnt+/+ ES cells in chimeras. As expected, the number of G418-resistant (G418r) Arnt+/− CFUs correlated well with the degree of Arnt+/− ES cell contribution to chimeric animals (Table 1). If the Arnt−/− progenitor deficiency were due to a cell-intrinsic defect, few to no G418r CFUs should appear in cultures of bone marrow cells derived from Arnt−/− chimeric animals. Interestingly, the number of G418r CFUs derived from these animals correlated with the percent chimerism of each animal, indicating a cell-extrinsic defect (Table 1). Southern blot analysis of G418r colonies confirmed they were exclusively derived from Arnt−/− ES cells (Fig. 3b).
Figure 3.

Assay of ES-cell-derived G418r CFU in Arnt/wild-type chimeras. (a) G418 selection strategy to distinguish wild-type or ES-derived CFU from chimeric animals. (b) Southern blot analysis of Arnt−/− chimera bone marrow CFU cultured in either 0, 1.0, or 1.5 mg/ml G418. Only the targeted Arnt allele (mt) remains after G418 selection, indicating the presence of solely Arnt−/−-derived G418r CFU.
Table 1.
Arnt+/−and Arnt−/−ES cells contribute to bone marrow and yolk sac hematopoietic progenitors
| Tissue
|
ES cell genotype
|
Chimera
|
Percent ESa
|
CFUb
|
||
|---|---|---|---|---|---|---|
| total
|
G418r
|
%G418r
|
||||
| Bone marrow | 1 | 0 | 249 | 0 | 0 | |
| 2 | 0 | 303 | 0 | 0 | ||
| +/− | 3 | 50 | 294 | 62 | 21 | |
| 4 | 50 | 165 | 58 | 35 | ||
| 5 | 60 | 297 | 152 | 51 | ||
| 1 | 70 | 315 | 195 | 62 | ||
| 2 | 70 | 352 | 266 | 76 | ||
| −/− | 3 | 70 | 385 | 314 | 82 | |
| 4 | 70 | 338 | 240 | 71 | ||
| 5 | 75 | 329 | 259 | 79 | ||
| Yolk sac | 1 | 40 | 732 | 352 | 48 | |
| 2 | 40 | 816 | 326 | 40 | ||
| −/− | 3 | 55 | 804 | 482 | 60 | |
| 4 | 55 | 1016 | 440 | 43 | ||
| 5 | 80 | 644 | 578 | 90 | ||
Percentage of ES cell contribution was determined by Southern blot analysis of bone marrow DNA or embryo DNA (for yolk sac analyses).
G418r CFU were selected in 1.5 mg/ml G418.
There is considerable evidence to suggest yolk sac and bone marrow progenitor populations are derived from distinct mesodermal progenitor cells (Medvinsky and Dzierzak 1996). Because Arnt−/− embryos display a yolk sac progenitor defect, we wanted to determine whether this population, in addition to the bone marrow progenitor population, exhibits a cell-extrinsic defect. Toward this end, we generated chimeric animals with Arnt−/− ES cells and harvested E10.5 yolk sacs. The level of chimerism of each animal was determined by Southern blot analysis of the embryo proper, and yolk sac cells grown in duplicate methylcellulose cultures in the presence and absence of G418. Similar to results observed from the chimeric bone marrow assays, the number of yolk sac G418r CFUs correlated with the degree of chimerism, demonstrating a cell extrinsic defect of the yolk sac progenitor population (Table 1). Taken together these studies demonstrate that the Arnt−/− hematopoietic defect is not intrinsic to hematopoietic progenitors, but instead due to a specific defect in other cell populations.
One potential explanation that might account for these findings is that Arnt−/− yolk sacs produce insufficient levels of extracellular cytokines essential for hematopoietic progenitor proliferation (Wu et al. 1995; Lin et al. 1996). Both EPO and VEGF are critical hematopoietic cytokines (Wu et al. 1995; Ferrara et al. 1996; Lin et al. 1996; Shalaby et al. 1997)and direct targets of ARNT transcriptional activity (via HIF-1) (Bunn and Poyton 1996; Wood et al. 1996; Maltepe et al. 1997; Wenger and Gassmann 1997). To determine whether these cytokines were dysregulated in our in vitro cultures, RNA extracted from Arnt+/+ and Arnt−/− EBs differentiated under both normoxic and hypoxic conditions was analyzed by Northern blot. Normoxically cultured Arnt+/+ EBs produced threefold more VEGF mRNA (including multiple splice variants) than Arnt−/− EBs (Fig. 4a), likely due to an inability of the Arnt−/− cells to respond to the intrinsic hypoxia of EBs. When Arnt+/+ EBs were cultured in 3% O2, they displayed a 4.3-fold increase in VEGF production over normoxic levels. In contrast, and consistent with previous studies, Arnt−/− EBs showed only a slight increase (twofold) in VEGF expression in response to hypoxia (Fig. 4a). We were unable to detect EPO expression in any of the EBs with this assay (data not shown). To determine whether inadequate VEGF levels contributed to the Arnt−/− progenitor defect, we added exogenous VEGF to EB cultures. As indicated in Figure 4b, exogenous VEGF restored CFU-E, CFU-M, CFU-GEMM, and CFU-GM progenitor numbers to wild-type levels in Arnt−/− EBs. In direct contrast, addition of EPO failed to increase progenitor number in the Arnt−/− EBs, demonstrating that inadequate EPO production is not responsible for the multilineage hematopoietic defect seen in Arnt−/− EBs (Fig. 4c).
Figure 4.



Cytokine rescue experiments. (a) Northern analysis of poly(A)-selected RNA (5 μg per lane) derived from Arnt+/+ and Arnt−/− EBs, differentiated for 9 days under normoxia (20% O2) or hypoxia (3% O2). 3′-UTR probes for VEGF (recognizing multiple splice variants) and HIF-1α (as a loading control) were used. (b) Exogenous VEGF (5 ng/ml), when added to the primary differentiation medium, was able to rescue the number of CFU produced by Arnt−/− EBs to Arnt+/+ levels (n = 3). (c) Exogenous EPO (2 U/ml) was not able to rescue progenitor number in Arnt−/− EBs, indicating its deficiency does not lead to the observed phenotype (n = 3).
Other cytokines, including GM-CSF, IL-1, IL-3, and SCF, are not known to be direct targets of ARNT transcriptional activity, but were also tested in the methylcellulose CFU assay. All failed to rescue a variety of CFU progenitors (data not shown), although SCF did partially rescue the CFU-E precursors (data not shown). Thus, we conclude that VEGF plays a specific and unique role in the proliferation or survival of a broad spectrum of hematopoietic progenitors during EB formation. Low levels of VEGF mRNA detected in Arnt−/− EBs is consistent with previous data obtained from Arnt−/− embryos exhibiting decreased VEGF mRNA expression (Maltepe et al. 1997). It has been suggested recently that signaling through Flk-1, a known VEGF receptor, is important for hematopoietic progenitor proliferation, but not initial progenitor formation (Schuh et al. 1999), and the requirement for Flk-1 signaling may be dependent on the hematopoietic microenvironment (Hidaka et al. 1999). We propose that ARNT-mediated hypoxic induction of VEGF is a critical developmental signal acting through the Flk-1 receptor to promote proliferation of both vascular endothelial and hematopoietic progenitor cells.
Cytokines, cytokine receptors, and transcription factors represent well-established determinants of hematopoiesis. However, the studies described in this report demonstrate for the first time that hypoxia, a condition normally associated with pathology, is a critical regulator of the proliferation, expansion, or survival of hematopoietic precursors during embryogenesis. Just as embryoid bodies contain mild regions of hypoxia due to the limitations of diffusion, so too, would a developing embryo. These data suggest that subsequent to E8.5, oxygen sensing and responsiveness become critical to the continued expansion and maturation of tissues involved in oxygen and nutrient delivery. Interestingly, this is precisely when Arnt−/− embryos begin to show stunted development. Therefore, we propose a molecular pathway in which the hypoxia-inducible factor HIF-1 activates the transcription of Vegf, the expression of which stimulates the proliferation or survival (Katoh et al. 1998) of hematopoietic progenitors. The decreased levels of a broad spectrum of hematopoietic progenitors in Arnt−/− embryos suggests a very early defect, possibly involving the hemangioblast, hematopoietic stem cell, or lineage-committed progenitor.
In addition to increasing our understanding of the molecular pathways that regulate embryonic hematopoiesis, our results may have therapeutic relevance. Cytotoxic chemotherapy treatments often leave patients with a decreased ability to regenerate short-lived erythroid and myeloid cells, as hematopoietic stem cells are particularly vulnerable to these agents (Domen and Weissman 1999). Such deficiencies often lead to worsening prognoses, requiring the use of stem cell transplantation. Current transplant protocols are severely curtailed by difficulties in maintaining and expanding hematopoietic stem cells in culture. The hypoxic culture conditions or exogenous VEGF administration discussed in this paper may represent simple and efficient methods of promoting hematopoietic progenitor cell expansion in vitro, thereby enhancing the feasibility of this treatment.
Materials and methods
Generation of ES cells, chimeras, and mice
Arnt+/− and Arnt−/− R1 ES cell clones were generated and genotyped by Southern blot analysis as described (Maltepe et al. 1997). Arnt+/− cells were selected in G418 during the production of Arnt−/− clones (Mortensen et al. 1992). Both heterozygous- and homozygous-targeted ES cells were used to generate chimeric mice, and a colony of Arnt+/+ and Arnt+/− animals was maintained by standard Arnt+/− by Arnt+/− crosses. E10.5 yolk sacs or 6-week-old bone marrow were collected for hematopoietic progenitor assays. Chimerism was assessed by agouti coat-color contribution in the adult mice and by Southern blot analysis of the bone marrow, tail, and liver DNA (or E10.5 embryo proper DNA).
Hematopoietic progenitor assays
E8.5, E9.5, and E10.5 yolk sacs were dissected free of the embryo and incubated in 0.25% collagenase (Sigma) in PBS supplemented with fetal bovine serum for 1 hr, followed by mechanical shearing with a 22-gauge needle and syringe. Bone marrow from 6-week-old chimeras was obtained from the femur and tibia. Cells (total number from yolk sac, 2 × 104 from bone marrow) were then plated into methylcellulose medium (Stem Cell Technologies) supplemented with 15% fetal bovine serum, 1% BSA, 10 μg/ml Insulin, 200 μg/ml transferrin (iron-saturated), 10−4 m 2-mercaptoethanol, 2 mm l-glutamine, 10 ng/ml rmIL-3, 10 ng/ml rhIL-6, 5 ng/ml rmSCF, 0.5 ng/ml GM-CSF, and 3 U/ml rhEPO. Cultures of cells from chimeric mice were also supplemented with 0, 1.0, or 1.5 mg/ml G418 for selection of Arnt+/− or Arnt−/− progenitors. Hematopoietic colonies were scored by morphology after 6–7 days and confirmed by cytological staining (Olson et al. 1995).
In vitro differentiation and replating of ES cells
Gelatin-adapted R1 ES cells were cultured in medium consisting of highglucose DMEM (GIBCO-BRL) supplemented with 15% fetal calf serum (Hyclone), penicillin–streptomycin (GIBCO-BRL), MEM nonessential amino acids (GIBCO-BRL) and leukemia inhibitory factor. ES cells (∼3 × 103) were plated in methylcellulose containing 10% serum, 500 U/ml rhIL-1, 5 ng/ml rmIL-3, 10 μg/ml insulin, 200 μg/ml transferrin, and 10−4 m α-monothioglycerol, and allowed to differentiate under normoxic (20% O2, 5% CO2, 75% N2) or hypoxic (3% O2, 5% CO2, 92% N2) conditions. rmVEGF (5 ng/ml), rhEPO (2 U/ml), rmSCF (5 ng/ml), or GM-CSF (0.5 ng/ml) were added as described. After 9 days, cystic EBs were washed free of methylcellulose with PBS and disaggregated with trypsin (GIBCO-BRL) and mechanical shearing, using a 21-gauge needle and syringe. Cells were replated into a secondary methylcellulose medium identical to that used for the hematopoietic progenitor assays and incubated under normoxic conditions. The number of cells plated was equal to the number of cells in 50 embryoid bodies derived from wild-type ES cells differentiated under normoxic conditions. Hematopoietic colonies were scored after 6–7 days.
Northern analysis
Embryoid bodies from ES cells differentiated under 20% and 3% O2 for 9 days were washed with PBS and RNA was extracted with TRIzol reagent (GIBCO-BRL) according to the manufacturer's instructions. mRNA was isolated using a poly(A)+ selection kit (Invitrogen). The probes for VEGF and HIF-1α were generated with RT–PCR using 3′-UTR-specific primers (Maltepe et al. 1997).
Acknowledgments
We thank Cynthia Clendinin, Michele Hadhazy, Kirsten Sigrist, and Donna Fackenthal for technical assistance; Navdeep Chandel for help with hypoxic cell culture; Jeffrey Leiden and Brian Keith for critically reviewing the manuscript; Cheryl Small for secretarial assistance; and Denise Wiler for preparing the illustrations. D.M.A. is a fellow of the Medical Scientist Training Program. E.M. is a Nathan and Francis Goldblatt Society Fellow. M.C.S. is an investigator of the HHMI.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL csimon@medicine.bsd.uchicago.edu; FAX (215) 746-5511.
References
- An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-iducible factor 1 alpha. Nature. 1998;392:405–408. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- Bashan N, Burdett E, Hundal HS, Klip A. Regulation of glucose transport and GLUT1 glucose transporter expression by O2 in muscle cells in culture. Am J Physiol. 1992;262:C682–690. doi: 10.1152/ajpcell.1992.262.3.C682. [DOI] [PubMed] [Google Scholar]
- Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 1996;76:839–885. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshet E. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumor angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G. A common precursor for hematopoietic and endothelial cells. Development. 1998;125:725–832. doi: 10.1242/dev.125.4.725. [DOI] [PubMed] [Google Scholar]
- Domen J, Weissman IL. Self-renewal, differentiation or death: Regulation and manipulation of hematopoietic stem cell fate. Mol Med Today. 1999;5:201–208. doi: 10.1016/S1357-4310(99)01464-1. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- Fischer B, Bavister BD. Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J Reprod Fertil. 1993;99:673–679. doi: 10.1530/jrf.0.0990673. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann M, Fanrey J, Bichet S, Wartenberg M, Marti HH, Bauer C, Wenger RH, Acker H. Oxygen supply and oxygen-dependent gene expression in differentiating embryonic stem cells. Proc Natl Acad Sci. 1996;93:2867–2872. doi: 10.1073/pnas.93.7.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of humna placental development by oxygen tension. Science. 1997;277:1669–1672. doi: 10.1126/science.277.5332.1669. [DOI] [PubMed] [Google Scholar]
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- Hidaka M, Stanford WL, Bernstein A. Conditional requirement for the flk-1 receptor in the in vitro generation of early hematopoietic cells. Proc Natl Acad Sci. 1999;96:7370–7375. doi: 10.1073/pnas.96.13.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes & Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh O, Takahashi T, Oguri T, Kuramoto K, Mihara K, Kobayashi M, Hirata S, Watanabe H. Vascular endothelial growth factor inhibits apoptotic death in hematopoietic cells after exposure to chemotherapeutic drugs by inducing MCL1 acting as an antiapoptotic factor. Cancer Res. 1998;58:5565–5569. [PubMed] [Google Scholar]
- Keller G, Kennedy M, Papayannopoulou T, Wiles MV. Hematopoietic commitment during embryonic stem cell differentiation in culture. Mol Cell Biol. 1993;13:472–486. doi: 10.1128/mcb.13.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M, Firpo M, Choi K, Wall C, Robertson S, Kabrun N, Keller G. A common precursor for primitive erythropoiesis and definitive haematopoiesis. Nature. 1997;386:488–493. doi: 10.1038/386488a0. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Abbott B, Hankinson O. ARNT-deficient mice and placental differentiation. Dev Biol. 1997;191:297–305. doi: 10.1006/dbio.1997.8758. [DOI] [PubMed] [Google Scholar]
- Lin CS, Lim SK, D'Agati V, Costantini F. Differential effects of an erythropoietin receptor gene disruption on primitive and definitive erythropoiesis. Genes & Dev. 1996;10:154–164. doi: 10.1101/gad.10.2.154. [DOI] [PubMed] [Google Scholar]
- Maeda H, Hotta T, Yamada H. Enhanced colony formation of human hemopoietic stem cells in reduced oxygen tension. Exp Hematol. 1986;14:930–934. [PubMed] [Google Scholar]
- Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprovation in mice lacking the protein ARNT. Nature. 1997;386:403–407. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- Maltepe E, Simon MC. Oxygen, genes, and development: An analyis of the role of hypoxic gene regulation during murine vascular development. J Mol Med. 1998;76:391–401. doi: 10.1007/s001090050231. [DOI] [PubMed] [Google Scholar]
- Medvinsky A, Dzierzak E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell. 1996;86:897–906. doi: 10.1016/s0092-8674(00)80165-8. [DOI] [PubMed] [Google Scholar]
- Mortensen RM, Conner DA, Chao S, Geisterfer-Lowrance AA, Seidman JG. Production of homozygous mutant ES cells with a single targeting construct. Mol Cell Biol. 1992;12:2491–2395. doi: 10.1128/mcb.12.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MC, Scott EW, Hack AA, Su GH, Tenen DG, Singh H, Simon MC. PU.1 is not essential for early myeloid gene expression but is required fro terminal myeloid differentiation. Immunity. 1995;3:703–714. doi: 10.1016/1074-7613(95)90060-8. [DOI] [PubMed] [Google Scholar]
- Phillips PG, Birnby LM, Narendran A. Hypoxia induces capillary network formation in cultured bovine pulmonary microvessel endothelial cell. Am J Physiol. 1995;268:L789–800. doi: 10.1152/ajplung.1995.268.5.L789. [DOI] [PubMed] [Google Scholar]
- Robb L, Elwood NJ, Elefanty AG, Kontgen F, Li R, Barnett LD, Begley CG. The sc1 gene product is required for the generation of all hematopoietic lineages in the adult mouse. EMBO J. 1996;15:4123–4129. [PMC free article] [PubMed] [Google Scholar]
- Rodesch F, Simon P, Donner C, Jauniaux E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol. 1992;80:283–285. [PubMed] [Google Scholar]
- Ryan HE, Lo J, Johnson RS. HIF-1a is required for solid tumor formation and embyonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- Schuh AC, Faloon P, Hu QL, Bhimani M, Choi K. In vitro hematopoietic and endothelial potential of flk-1(−/−) embryonic stem cells and embryos. Proc Natl Acad Sci. 1999;96:2159–2164. doi: 10.1073/pnas.96.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci. 1991;88:5680–5684. doi: 10.1073/pnas.88.13.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- Shalaby F, Ho J, Stanford WL, Fischer KD, Schuh AC, Schwartz L, Bernstein A, Rossant J. A requirement for Flk1 in primitive and definitive hematopoiesis and vasculogenesis. Cell. 1997;89:981–990. doi: 10.1016/s0092-8674(00)80283-4. [DOI] [PubMed] [Google Scholar]
- Schweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Wang LC, Swat W, Fujiwara Y, Davidson L, Visvader J, Kuo F, Alt FW, Gilliland DG, Golub TR, Orkin SH. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes & Dev. 1998;12:2392–2402. doi: 10.1101/gad.12.15.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger RH, Gassmann M. Oxygen(es) and the hypoxia-inducible factor-1. Biol Chem. 1997;378:609–616. [PubMed] [Google Scholar]
- Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, Ratcliffe PJ, Maxwell PH. Induction of endothelial PAS domain protein-1 by hypoxia: Characterization and comparison with hypoxia-inducible factor-1alpha. Blood. 1998;92:2260–2268. [PubMed] [Google Scholar]
- Wood SM, Gleadle JM, Pugh CW, Hankinson O, Ratcliffe PJ. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression. Studies in ARNT-deficient cells. J Biol Chem. 1996;271:15117–15123. doi: 10.1074/jbc.271.25.15117. [DOI] [PubMed] [Google Scholar]
- Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83:59–67. doi: 10.1016/0092-8674(95)90234-1. [DOI] [PubMed] [Google Scholar]